Graphene-based materials for catalysis
Bruno F.
Machado
ab and
Philippe
Serp
*a
aLaboratoire de Chimie de Coordination, UPR CNRS 8241, Composante ENSIACET, Université de Toulouse UPS-INP-LCC, 4 allée Emile Monso, BP 44362, 31432 Toulouse Cedex 4, France. E-mail: philippe.serp@ensiacet.fr; Fax: +33 05 34 32 35 96; Tel: +33 05 34 32 35 72
bLaboratório de Catálise e Materiais (LCM), Laboratório Associado LSRE/LCM, Departamento de Engenharia Química, Faculdade de Engenharia, Universidade do Porto, Rua Dr. Roberto Frias, 4200-465 Porto, Portugal
First published on 8th November 2011
Abstract
Graphene is one of the most promising materials in nanotechnology. From a theoretical point of view, it provides the ultimate two-dimensional model of a catalytic support. Its unique physical, chemical and mechanical properties are outstanding, and could allow the preparation of composite-materials with unprecedented characteristics. Even though the use of a single graphene sheet as a catalytic support has not yet been reported, some promising results have already been obtained with few-layer graphene. In this review, we will briefly discuss the most relevant synthetic routes to obtain graphene. Then, we will focus our attention on the properties and characterization techniques of graphene that are of relevance to catalysis, with emphasis on adsorption. After presenting an overview of the most common and effective preparation methods, we will discuss the catalytic application of graphene and graphene-based composites, with particular attention on energy conversion and photocatalysis.
 Bruno F. Machado | Bruno F. Machado received is PhD in chemical and biological engineering from the University of Porto in 2009. He is currently carrying out his postdoctoral work at Laboratory of Coordination Chemistry (University of Toulouse, France) in the field of nanotechnology and heterogeneous catalysis. His research interests include the development and preparation of carbon-based nanocomposites with semiconductor nanostructures, as well as their applications in catalysis. |
 Philippe Serp | Philippe SERP is full Professor of Inorganic Chemistry at Ecole Nationale supérieure des Ingénieurs en Arts Chimiques Et Technologique Toulouse University. He was the recipient of the Catalysis Division of the French Chemical Society Award in 2004, and the APDF “Celestino da Costa/Jean Perrin” award in 2005. His current research interests in Laboratoire de Chimie de Coordination (UPR 8241 CNRS) include nanocatalysis, gas phase preparation of nanostructured catalytic materials and the understanding of homogeneous catalytic reactions, fields in which with co-workers he has published over 120 papers, among them 5 review articles, 12 book chapters and 13 patents. |
1. Introduction
The use of carbon nanomaterials in catalysis has grown in importance and has dominated advances in nanoscience and nanotechnology for the last 25 years. They are nowadays one of the most commonly used materials and can be used either as supports for immobilizing active species or as metal-free catalysts.1 This is mainly due to their unique structure and intrinsic properties including high specific surface areas, chemical and electrochemical inertness and easy surface modification.2Carbon nanotubes (CNTs) were discovered soon after the successful laboratory synthesis of fullerenes.3 Since their first observation using high resolution electron microscopy in 1991 by Iijima,4 CNTs have been the focus of materials research mainly because of their unique structural, electronic and mechanical properties. However, following the report by Novoselov et al.5 on the direct observation and characterization of mechanically exfoliated graphene, a single-atom-thick sheet of hexagonally arrayed sp2-bonded carbon atoms, there has been an exponential growth in graphene research among both the scientific and engineering communities. Not surprisingly, the importance regarding the discovery of these nanostructured carbons was recognized by the Nobel Prize committee with the award of two Nobel Prizes. In 1996, Robert F. Curl Jr., Sir Harold Kroto and Richard E. Smalley shared the Nobel Prize in Chemistry “for their discovery of fullerenes”, and in 2010, Andre Geim and Konstantin Novoselov received the Nobel Prize in Physics “for groundbreaking experiments regarding the two-dimensional material graphene”.Ideally, graphene is a single-layer material, but graphene samples with two or more layers are being investigated with equal interest. Graphene possesses unique electronic, optical, thermal, and mechanical properties. In addition, given its large specific surface area, good biocompatibility and high adsorption capacity, graphene and its derivatives can be used as valuable substrates to interact with various species. These composites can then be used over a wide range of applications, including memory devices,6–8 energy storage,9–11 catalysis,12–21 photocatalysis,22–27 solar cells,28–32 sensing platforms,33–36 Raman enhancement,37–39 molecular imaging,40,41 and even drug delivery.42
Given the massive attention inspired by the properties and potential applications of graphene-based materials, the number of publications has increased exponentially in the last few years. Despite the existence of several reviews highlighting the unique physical, chemical and mechanical properties of graphene,43–55 only a very limited number deal with the application of these materials in catalysis.56–58 Because the success of a catalytic application begins with catalyst design, it is fundamental to understand all the key aspects involved. Hence, we will briefly discuss the most common synthetic routes to obtain graphene. Afterwards, we will focus our attention on the properties and characterization techniques of graphene that are of relevance to catalysis, emphasis being given to adsorption. After presenting an overview of the most common and effective preparation methods, we will discuss the most recent advances in the catalytic application of graphene and graphene-based composites. Finally, we will present a brief summary and an outlook on what to expect regarding future applications of graphene-based composites in the field of catalysis.
2. Synthesis and properties of graphene
2.1 Synthesis
Graphene is a two-dimensional crystal that can be considered as the basic building block for carbon materials of different dimensionalities: fullerenes (0D), nanotubes (1D) or graphite (3D).49 This unique nanostructure holds great promise for potential applications in technological fields such as optical electronics, sensors, energy conversion and storage, catalysis, among many others. In that context, to realize this potential, reliable methods for producing large-area single-crystalline graphene domains are required. However, as was the case in the early days of nanotube and nanowire research, graphene faces a problem that is common to many novel materials: the absence of process for production in high yields. In order to overcome this deficiency, there are currently several methods that can be used for the production of single or few-layered graphene samples, evidencing variable degrees of success. We present here a brief overview of the most common.The existence of single-layer graphene was not considered possible until the recent achievement of the micromechanical cleavage of highly ordered pyrolytic graphite (HOPG).5,59 Unfortunately, the single layers obtained by repeated peeling are only a small portion amongst the large quantities of thin graphite flakes (few- to multi-layer graphene), and, thus is not suitable for large-scale fabrication processes.
High-quality large-area graphene sheets can be prepared by epitaxial growth on single-crystal silicon carbide (SiC). This is commonly achieved through ultrahigh vacuum annealing of the SiC surface.60–63 Since the sublimation rate of silicon is higher than that of carbon, excess carbon is left behind on the surface, which rearranges to form graphene nanosheets. An important issue within this technique is related to the interface between the graphene layer and the substrate, since it is recognized that both the structure and electronic properties of graphene are affected.64 Once there is an improved control over the growth mechanism (leading to a control in the number of layers) and interface effects, this method is set to be used industrially to produce wafer scale graphene. On the other hand, conditions for graphene growth such as high-temperature, ultra-high vacuum and single-crystal substrate will likely hinder the use of this technique for large-scale applications.65
One attractive alternative to the production of individual graphene sheets is the epitaxial growth of graphene films on metal surfaces such as Ni, Cu, Co, Pt, Ir and Ru, using chemical vapor deposition (CVD).62,66–70 This method uses the atomic structure of the metal substrate to seed the growth of graphene. The nucleation and growth of graphene usually occurs by exposure of the transition metal surface to a hydrocarbon gas under low pressure or ultra-high vacuum conditions. Large area epitaxial graphene films up to a few micrometres in size can be subsequently transferred to other substrates after etching off the metals. Epitaxial growth of graphene offers probably the only viable route towards electronic applications, and so a rapid progress in this direction is expected over the next few years.
One of the most developed methods to obtain higher yields of single-layered graphene consists of the initial oxidation of graphite to graphite oxide (GO), followed by the subsequent mechanical/chemical or thermal exfoliation of graphite oxide to graphene oxide sheets, and their eventual reduction to graphene (Fig. 1).71–73 Although the exact structure of GO is still subject to intense debate, it is believed that for GO, the aromatic lattice of graphene is interrupted by epoxide, hydroxyl, carbonyl and carboxylic groups.74 The most accepted model is the one by Lerf and Klinowski,75 where it is assumed that the heavily oxygenated graphite oxide contains hydroxyl and epoxide functional groups on the basal planes, in addition to carbonyl and carboxyl groups located at the edges. These oxygen functionalities render the graphene oxide layers of hydrophilic GO and water molecules can readily intercalate between the layers. This results in an increase of the inter-layer distance (d-spacing, d002 in Fig. 1) of GO as well as a change of hybridization of the oxidized carbon atoms from planar sp2 to tetrahedral sp3.72 Rapid heating of GO causes a rapid evaporation of the intercalated water resulting in its expansion and delamination. Even though this simple method has been applied on a large scale, if the oxidation is not sufficient it could result in incomplete exfoliation of graphite to the level of individual graphene sheets. In addition, the functionalization disrupts the electronic structure of graphene by several orders of magnitude, compared to pristine graphene. Chemical, electrochemical or thermal reduction of graphene oxide (removal of the functional groups) into graphene can partly restore its graphitic structure as well as conductivity. Although reduced graphene oxide (rGO, also called chemically converted graphene, chemically modified graphene, or simply graphene) presents considerable amount of defects, which continue to disrupt the electronic properties, it is one of the most widely used methods due to its cost, facile preparation process, large productivity and potential for functionalization.
 | ||
| Fig. 1 Illustration on the preparation of reduced graphene oxide. Reprinted with permission from ref. 56. Copyright 2011 Wiley-VCH. | ||
In this context, the preparation of high-quality 2D graphene sheets is the first and most crucial step, since the existence of residual defects (oxygenated species that cannot be fully removed by chemical treatments) will severely influence the properties (mainly electronic) of graphene and limit its applications. Thus, an efficient process for the large scale production of high quality few-layer graphene should be modelled.
2.2 Properties
Not all physicochemical properties determined for graphene are of interest for catalyst design and application. Hence, only those potentially interesting for catalysis are discussed in this section. For more details regarding other properties of graphene, the reader can refer to specific review articles.50,51,65,76,77Based on theoretical calculations for graphene as the parent material of carbon nanotubes, its properties were expected to be outstanding. With the development of new methodologies to increase both the yield and the quality of graphene samples, these estimates could be finally experimentally assessed. Unfortunately, some of the properties can only be observed at an extremely low defect concentration. Nevertheless, like in any other real material, structural defects do exist in graphene and can radically alter its properties.
In graphene, carbon atoms are arranged in a hexagonal manner, forming a 2D honeycomb structure. While the strong σ bonds work as the rigid backbone of the hexagonal structure, the out-of plane π bonds control interaction between different graphene layers. This allows delocalized π electrons to be easily conducted through the basal plane, i.e., the plane of the graphene sheets normal to the c-axis of graphite (Fig. 1). For this reason, graphene is considered a zero-bandgap semi-conductor, possessing a small overlap between the valence and conduction bands.49 Furthermore, graphene sheets exhibit a highly anisotropic behavior as the electron conduction along the c-axis is much lower (around three orders of magnitude) than that observed through the basal planes.9 The electronic properties of graphene vary both with the number of layers and the relative position of atoms in adjacent layers (stacking order). For double-layer graphene, the stacking order can be either AA (each atom on top of another atom) or AB (a set of atoms in the second layer sits on top of the empty center of a hexagon of the first layer). Other properties, such as thermal conductivity and thermal expansion also present a similar variation. Upon comparing graphene with graphene oxide, it can be observed that the latter exhibits a significant loss of conductivity (up to several orders of magnitude) due to the presence of oxygenated surface groups and defects in the basal plane. The GO sheets need to be reduced in order to restore the sp2 hybrid network and, thus, reintroduce the conductivity. Depending on the level of reduction the surface of the graphene sample can be fine-tuned to achieve different electronic and optoelectronic properties.28,78
In addition, graphene is found to possess a high optical transparency,79 due to its one-atom thickness, rendering it extremely useful in transparent conducting electrodes, used for example in touch-screens, liquid crystal displays and solar cells.
Furthermore, it is noteworthy to highlight its excellent chemical stability and mechanical strength. The mechanical properties of a defect-free monolayer of graphene were measured using a nano-indentation technique and have shown it to be one of the strongest materials ever investigated.80
Among other properties that have received considerable interest, one of the most important is adsorption. Understanding the adsorption mechanism and interaction between adsorbed species and the carbon surface is essential to fabricate graphene-based materials. In order to develop this knowledge, theoretical studies are of great interest and allow an optimization regarding the characteristics of novel materials. Regarding metal deposition, several theoretical calculations have been performed in order to provide an atomic level understanding of the interactions between adatoms (adsorbed atoms) and graphene. These investigations focused on the stable configurations of metal adatoms on graphene, embedding transition-metal atoms in graphene, charge transfer between graphene and metal adatoms, and magnetism. Commonly, authors consider the binding of the adatom over graphene on three sites of high symmetry: hollow (H) at the center of a hexagon, bridge (B) at the midpoint of a carbon–carbon bond, and top (T) directly above a carbon atom (Fig. 2). Hu et al.81 studied the adsorption of various adatoms over graphene using first-principles density-functional theory with the generalized gradient approximation. They found that H sites are the most favorable for Sc, Ti, V, Fe, Co and Ni adsorbed on graphene, while B or T sites are the most stable for transition metals that have a filled or near filled d-shell (Cu, Pd and Pt). Half-filled d-shell transition metal atoms and Au, Ag, Zn have small adsorption energies. Depending on the intensity of the adsorption energy, the favored adsorption site can indicate the nature of the chemical bond between adatoms and graphene. Hence, the adsorption of Au, Ag and Cu was considered as physisorption, whereas Co, Ni, Pt and Pd covalently bonded to graphene (chemisorption). Results obtained for Pt, Ag and Au were supported by Tang et al. in a similar work.82
 | ||
| Fig. 2 Three different high symmetry adsorption sites: hollow (H), bridge (B) and top (T). | ||
Recently, Nakada and Ishii83 studied the adsorption and migration energies for different atomic species from hydrogen to bismuth (except lanthanides and noble gases) over a graphene sheet using a first-principles band calculation technique based on density functional theory. Table 1 shows the most stable sites for each adatom (the orange, blue and white boxes represent the most stable sites T, B or H, respectively) accompanied by the corresponding adsorption energy. For transition metal elements the most commonly stable site is H; for the non-metallic elements, B site is the most stable; for H, F, Cl, Br and I, the most stable adsorption site is T. When the adsorption energy of the adatom is small, there is almost no difference between the three adsorption sites. Marked in bold in Table 1 are adatoms that cumulatively have bond distances (between graphene and adatom) smaller than 2 Å and migration energy (for the most stable sites) above 0.5 eV. For large bond distances, the adatom shows physisorption bonding; when the bond distance is short, the bond energy tends to increase and the adatom shows chemisorption bonding. The migration energy gives an indication of the ease of mobility of the adatom on the surface. Hence, the closer the adatom to the graphene sheet, the stronger it bonds to graphene and thus the highest the resistance to the adatom movement through the basal plane.

|
Computation results show that chemisorption of transition metals involves hybridization of adatom d-orbitals with the orbitals of graphene. Despite the presence of a different set of orbitals, a similar observation can also be made for hydrogen, nitrogen and oxygen adatoms. This induces a strong distortion on the carbon atom beneath the adsorbed atom, which is likely to change some of the sp2-like orbital character to a more covalently reactive sp3-like character, given the re-hybridization of the valence carbon orbitals required for the bond formation. From the adatom point of view, there is a reduction in the magnetic moment from the isolated to the adsorbed metal atom, which is thought to be related with an electronic charge transfer between adatom and graphene coupled with an electron shift between different orbitals within the adatom.81
Many researchers have also investigated the interactions between chemical or biological molecules and graphene or metal-doped graphene.84–95 The adsorption of gas-phase molecules (H2O, NH3, CO, N2O and NO) on graphene was reported by Leenaerts et al.84 They observed that the charge transfer between the adsorbates and graphene was found to be almost independent of the adsorption site, but it did depend strongly on the orientation of the adsorbate with respect to the graphene surface. Ma et al.85 investigated the adsorption of cysteine on Pt-doped graphene. Compared with graphene, Pt-doped graphene had higher binding energy value and shorter binding distance between the cysteine molecule and the graphene surface, which was caused by strong adsorption of the cysteine molecule (DOS results showed significant orbital hybridization between cysteine and Pt-doped graphene sheet). Majumder et al.86 investigated the adsorption of aromatic amino acids on graphene, and found that phenolic rings oriented preferentially parallel to the plane of graphene. This behavior is typical of low adsorbate coverage; when the coverage is higher, the molecules often tilt to the vertical position, given that it requires less space, in order to accommodate a higher number of molecules. Voloshina et al.93 focused their study on the bonding of a single pyridine molecule adsorbed on a graphene surface. They demonstrated that a H adsorption site was preferred by pyridine and that the parallel orientation was more favorable than the perpendicular one. Interaction energy between pyridine and metal substrates was found to be at least 30% stronger than that obtained between pyridine and graphene. Similar conclusions, using nitrated tyrosine, were also observed by Ding et al.94 Zhou et al.95 reported an investigation on CO oxidation catalyzed by Au8 or Pt4 clusters on defective graphene. They found that a defect greatly enhanced the reactivity of Au8 and Pt4 clusters, and reduced the reaction barrier of catalyzed CO oxidation from around 3.0 eV for the case of Au8 (0.5 eV for the case of Pt4) to less than 0.2 eV (0.13 eV for Pt4).
Hence, theoretical calculations of graphene interactions with different species can provide a huge help in establishing structures and reaction mechanisms for the chemical modifications of graphene.96–98 This could lead to the preparation of higher quality composite materials for a wide variety of applications, among which are gas sensing and storage. Gas sensing by graphene generally involves the adsorption and desorption of gaseous molecules. These act as electron donors (e.g.CO, ethanol and NH3) or acceptors (e.g.NO2, H2O and I2) on the graphene surface, which lead to a change in local carrier concentration, allowing the resistivity to be used as a convenient means of measurement. Experiments by Schedin et al.36 show that graphene-based sensors are capable of detecting individual gas molecules due to their high sensitivity to chemical doping. They found that graphene is highly sensitive to NH3, CO, H2O and especially NO2. Theoretical studies aided by DFT calculations were conducted to explain the interactions of nitrogen oxides NOX (X = 1, 2, 3) and N2O4 with graphene and graphene oxides by Tang and Cao.99 They observed that the adsorption of NOX on GO was generally stronger than that on graphene due to the presence of the active defect sites, which increase the binding energies and enhance charge transfers from NOX to GO, eventually inducing the chemisorption of gas molecules. In another case, Fowler et al.100 reported the development of practical chemical sensors from chemically converted graphene for the detection of NO2, NH3, and 2,4-dinitrotoluene. They found that the primary mechanism of the chemical response in sensors is charge transfer between the analyte and graphene, while the electrical contacts play only a limited role. Graphene-based biosensors and devices have also exhibited good sensitivity and selectivity towards the detection of glucose, hemoglobin, cholesterol, H2O2, small biomolecules, DNA, heavy metal ions, poisonous gaseous molecules, among others.101,102
The search for new hydrogen storage materials has attracted a great deal of interest due to their important role in clean energy alternatives. The ability of graphene to adsorb hydrogen makes it an excellent candidate for hydrogen storage. In a work published by Ghosh et al.,103 theoretical calculations are directly compared against experimental results for the adsorption of both H2 and CO2. Hydrogen storage reached 3.1 wt% at 100 bar and 298 K, and the uptake varied linearly with the surface area. Theoretical calculations showed that single-layer graphene could accommodate up to 7.7 wt% of hydrogen, while double- and triple-layer graphene can have an uptake of ca. 2.7 wt%. CO2 uptake of few-layer graphene at 1 atm and 195 K was around 35 wt% (theoretical calculations show that graphene could have a maximum uptake of 37.9 wt% of CO2). Ruoff et al.104 prepared a graphene-based powder sample by chemical reduction of a colloidal suspension of exfoliated GO with a BET surface area of 640 m2 g−1. The hydrogen adsorption capacity of the obtained graphene was 1.2 wt% at 77 K and 10 bar and 0.72 wt% at 100 bar and room temperature.
The properties described in this section show that graphene has an enormous potential in the catalysis field. Unfortunately, the physical properties of graphene in its powder form do not allow this material to be used in industrial reactors (e.g. fixed- and fluidized-bed). The problem arises mostly due to the low bulk density of graphene, which induces either a low catalyst mass/reactor volume ratio (fixed-bed reactor) or a difficult fluidization process (fluidized-bed reactor). However, this limitation should depend on the number of layers. For example, we have measured an apparent density of ca. 1.2 g cm−3 for few-layer graphene of low surface area (40 m2 g−1) which can be compared to the value reported for MWCNT (0.02–0.3 g cm−3) and CNF (0.3–1.4 g cm−3).1 In an attempt to overcome these difficulties, several strategies can be envisaged. One of the most relevant consists in the macroscopic shaping of graphene to form pellets. This pelletization process consists in the production of rigid porous granules by extrusion, using binders to assist the aggregation. The process was already successfully performed to pelletize CNT, and it was observed that the resulting material possessed very similar textural properties compared to those observed for CNTs in the powder form.105
In addition to the dependency regarding the number of defects, most of these properties are also dependent on the number of graphene layers present on the sample. In order to normalize the terminology used, and based on their electronic spectra, it is widely accepted that three different types of graphene can be distinguished: single-, double- and few- (3 to 10) layer graphene.49 Thicker structures should be considered as thin graphite films or flakes. In order to differentiate between these types of graphene, several characterization techniques are presented in the next section, special emphasis being given on how the analysis results are affected by the number of layers.
2.3 Characterization
The identification and counting of graphene layers is one of the major difficulties encountered during the characterization of these materials. This is mainly due to the fact that monolayers are often in great minority among ensembles of thicker crystals. Accordingly, a variety of techniques can be envisaged for the analysis of graphene and its derivative materials. Among the more frequently applied techniques are high resolution transmission electron microscopy (HRTEM), Raman spectroscopy, X-ray diffraction (XRD), atomic force microscopy (AFM), scanning tunneling microscopy (STM) and nitrogen adsorption–desorption at 77 K. Given that no single technique is able to provide all the necessary information, it is necessary to couple two, and sometimes more, of these tools to accurately characterize the morphology, texture, crystal structure and intrinsic properties of graphene-based materials.65,43–45,106 Specific information about the most relevant techniques for the characterizations of graphene-based materials is summarized below.The atomic structure of single-layer graphene can be studied by HRTEM (Fig. 3). This technique is especially important because it allows the evaluation of the crystalline character for graphene flakes based on their electron diffraction patterns. The central part of the sheets usually appears on TEM images as uniform and spotless areas (sheets often present wrinkles, identifiable as dark marks), whereas near the edges the sheets tend to roll (Fig. 3a). Such folds provide a clear TEM signature for the number of graphene layers. A folded graphene sheet is locally parallel to the electron beam and for monolayer graphene a fold exhibits a dark line. Under favorable conditions (namely sample orientation), these folds situated at the edges or within the free hanging sheets could be used to estimate the number of layers present in a sample by direct visualization. Nevertheless, this counting technique should be done very carefully, since multiple folds can give rise to several dark lines (even for monolayer graphene), as evidenced experimentally.107Electron diffraction patterns show an expected hexagonal lattice of graphene (inset, Fig. 3a). Suspended graphene also evidences “rippling” of the flat sheet. Fig. 3b evidences regions where fringes are observed and regions where they are not, which indicates that there is a curvature in the sheets. These ripples may be intrinsic to graphene as a result of the instability of two-dimensional crystals, or may be extrinsic, originating from the presence of contaminations.108
 | ||
| Fig. 3 (a) TEM with the corresponding selected area diffraction pattern in inset, and (b) HRTEM micrograph of graphene. | ||
Raman spectroscopy is a powerful non-destructive tool to characterize carbonaceous materials, particularly for distinguishing ordered and disordered crystal structures of carbon.109,110 Accordingly, it provides a quick and facile way to characterize the structure and quality of graphene. The two most intense features are the G (ca. 1580 cm−1) and the G′ band (ca. 2700 cm−1), the second most prominent peak always observed. When a certain amount of disorder or edges appear within the structure, a disorder-induced band (D-band, ca. 1350 cm−1) appears. If no D-band is observed for graphene, this indicates the absence of a significant number of defects. On the other hand, a D-band is often observed when symmetry is broken by edges or in samples with a high density of defects. Similarly to TEM analysis, Raman spectroscopy can also be used to determine the number of graphene sheets present. This technique is particularly sensitive for low number of layers. Hence, one can clearly distinguish a single layer, from a bilayer, from few layers (less than 5). For more than 5 layers the Raman spectra become hardly distinguishable from that of bulk graphite (Fig. 4).111 In bulk graphite, the G′ band includes two contributions, the intensities of which are roughly ¼ and ½ that of the G peak for the low and high shifts, respectively. For single layer graphene, the G′ band is a single sharp peak at the lower shift, with intensity roughly 4 times that of the G peak.44 As a consequence from varying number of layers, the G′ band changes its shape, width and position with increasing number of layers, whereas the G peak position shows a down-shift with number of layers (Fig. 4).69 The in-plane crystallite sizes (La) can be calculated from the Raman spectra of the graphene samples by employing the relation La = 4.4 (IG/ID).112
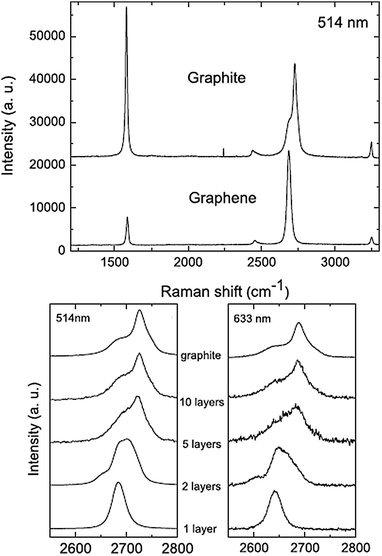 | ||
| Fig. 4 Both the G and G′ bands undergo significant changes due to the number of layers. Reprinted with permission from ref. 111. Copyright 2006 American Physical Society. | ||
The crystalline structures of pristine graphite and graphite oxides can also be evaluated by X-ray diffraction (XRD). The feature diffraction peak for both graphene and exfoliated graphene oxide is related to the A–B stacking order, corresponding to the (002) reflection. This peak appears at 2θ ≈ 26° for pristine graphite, whereas the same peak is shifted to 2θ ≈ 11° after the oxidation of the layers (Fig. 5). Using the Scherrer equation, the number of layers in graphene samples can be obtained from the corresponding line broadening by Lorentzian fitting of the (002) reflection.45 The distance between layers (d-spacing) is typically 0.335 nm for graphite. The oxidation of graphite is accompanied by the increase of the d-spacing, indicating the presence of intercalated species between the graphene layers. A sharp reflection (low full width at half maximum) in the XRD pattern indicates that the sample contains a large number of layers.
 | ||
| Fig. 5 X-Ray diffraction patterns of (a) pristine graphite, (b) exfoliated GO, (c) electrochemically reduced GO and (d) chemically reduced GO. Reprinted with permission from ref. 113. Copyright 2009 American Chemical Society. | ||
Atomic force microscopy (AFM) is currently the leading method allowing the identification of single- and few-layer crystals.59 As the tip scans across the surface, it is possible to analyze the topography of the sample, and thus, count the number of layers present by differential height measurements at the edge. The use of different AFM techniques allows the study of mechanical, electrical, magnetic, and even elastic properties of graphene flakes.51,80,114
While with AFM one can directly obtain the number of layers, scanning tunneling microscopy (STM) images are useful in determining the morphology and presence of defects on graphene. This technique allows one to obtain atomically resolved images of single-layered graphene. The characteristic features of STM images are readily interpreted in terms of the A–B stacking of the graphene planes in graphite. In bulk graphite, the carbon atoms on the surface are not equivalent to those directly beneath (Fig. 6b). Hence, half of the carbon atoms in the surface layer are located above carbon atoms in the adjacent lower layer (A-type atoms); the other half is placed over a void (B-type). This asymmetry in the surface atom electronic environment results in a threefold symmetry (“three-for-six”) pattern in which three bright or dark features can be observed for each set of six carbon atoms. This behavior also is present for graphene flakes that are two or more atomic layers thick. For single-layer sheets of graphene, this asymmetry disappears (Fig. 6a).115
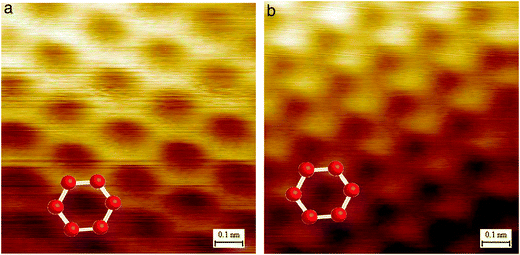 | ||
| Fig. 6 (a) STM image from a single layer of graphene, where the honeycomb structure is observed; (b) STM image of graphite showing a threefold symmetry pattern in which three bright (or dark) features can be observed for each set of six carbon atoms; reprinted with permission from ref. 115. Copyright 2007 by National Academy of Sciences of the USA. | ||
With the exception of STM, all other previously described local analysis techniques can, to some extent, enable an approximate calculation of the number of layers present in a graphene sample. Even though this is not the case for gas adsorption measurements (commonly N2adsorption-desorption at 77K), these can provide important data regarding the textural properties of bulk samples. Despite the possibility to determine the specific surface area, total pore volume, and pore size distribution for most solids, for graphene only the first is of significant importance, due to the nature of its texture. Theoretical calculations have shown that the highest surface area possible for a single layer graphene is 2630 m2 g−1.116 Unfortunately, this value would only be observed in a hypothetical case where no overlap of sheets existed. In a real system, a significant amount of surface area is not available for N2 adsorption because of the overlap of exfoliated graphene sheets. From an experimental point of view, the calculation of specific surface areas can be severely affected by the random agglomeration state of the dry powders. This often results in large surface area variations, even within the same batch. Table 2 compares the specific BET surface areas as a function of the preparation method and corresponding number of layers. As shown in Table 2, most of the surface areas published in the literature are obtained through the exfoliation of graphitic oxide. This is probably due to the ease of this technique to provide larger productivities, necessary for reliable measurements. Given the absence of textural data for graphene samples synthesized by micromechanical cleavage, epitaxial growth on single-crystal silicon carbide and chemical vapor deposition a full comparison between most common methods cannot be made.
| Preparation method | Number of layers | S BET/m2 g−1 | Reference |
|---|---|---|---|
| Thermal exfoliation of graphitic oxide | 1–3 | 700–1500 | 73 |
| Thermal exfoliation of graphitic oxide | 3–4 | 270–1550 | 103 |
| Thermal exfoliation of graphitic oxide | 3–6 | 925 | 11 |
| Thermal exfoliation of graphitic oxide | Ultrathin sheets | 737 | 117 |
| Chemical exfoliation of graphitic oxide | 1 | 705 | 118 |
| Chemical exfoliation of graphitic oxide | 3–7 | 640 | 104 |
| Microwave exfoliation of graphite oxide | Few-layered | 463 | 119 |
| SiC-derived | — | 300–950 | 120 |
3. Decoration of graphene-based materials
As mentioned in the previous section, graphene possesses a plate-like structure with a large specific surface area. This textural property, coupled with its excellent thermal, electronic and mechanical features, makes graphene an attractive substrate for the deposition of inorganic nanoparticles to produce highly dispersed composites. In addition, aggregation of graphene sheets can be partly prevented by intercalating particles within the graphene layers.Graphene sheets can be blended with various components to form functional composites. Most of the graphene-based materials have two components, although materials containing more than two components can also be produced to achieve the requirements of specific applications. The incorporation of graphene into the composites can provide unique properties and possibly induce new functions based on synergetic effects, providing a new opportunity for designing and developing next-generation catalysts. Usually, the second component can be a metal,14–15,17,121–127 metal oxide,128–134polymer (insulating and conducting),135–137 small organic compound,138,139 biomaterial,34,140,141 metal–organic framework,142–144 or even other carbon nanomaterials (carbon nanotubes or fullerenes).145–149 In this section, we will mainly focus on recent achievements dealing with the development of effective strategies for synthesizing high-quality graphene–metal and graphene–metal oxide composites.
3.1 Graphene–metal nanoparticles
Graphene sheets decorated with metal nanoparticles are an example of emerging metal–carbon composites that currently attract special research efforts due to their enhanced potential for several applications. Most types of composites reported in the literature consist of noble metal nanoparticles, including Au,14–16,88,121,122,150–169Pt,15,122,125,126,155,166–168,170–191Pd,12,13,15,17,127,155,167,168,170,181,192–202Ag,15,123,124,167–169,203–212Ru,125,167,172,180,182Rh,167 and Ir.167 In addition, metal nanoparticles of Fe,213,214Cu,201,215Ni,188Co,216Ge,217 and Sn218 were also used to produce metal–graphene composites.There are several strategies commonly used to synthesize well dispersed metal nanoparticles on the basal plane of graphene. Among those, solution-based techniques, where the liquid wets the entire surface area of graphene, are generally preferred. To synthesize nanoparticles using this approach, several factors need to be carefully controlled to obtain a narrow particle size distribution, namely, type of solvent, nature and concentration of metal precursor, the presence of a dispersing and/or reducing agent, and finally the deposition time and temperature. One common technique to produce nanostructured graphene composites, already used to cover other carbonaceous materials, consists of the chemical functionalization of the graphitic surface in order to induce anchoring sites for the metal precursor nucleation. This allows a covalent bond of the metal to the basal plane of graphene, yielding high dispersions (Fig. 7 and Table 3). Depending on the preparation method, graphene sheets can already possess a high amount of functional groups, e.g.graphene oxide (as a result of the exfoliation of graphite oxide). Then, graphene oxide and the metal precursor can be chemically reduced to form the corresponding graphene–metal composite.122,124,125,152–155,170,172–176,196–198,208,211,219 It has been found that graphene oxide is better than its reduced counterpart for in situgrowth of nanoparticles.152,163,204 An alternate reduction method involves the simultaneous reduction of both metal nanoparticles and the graphene oxide by means of microwave-irradiation.13,121,150,171,179,187,199–201,220,221 The main advantage of this method over other conventional heating methods is that the reaction mixture is heated uniformly and rapidly, allowing for large-scale and highly efficient production of graphene–metal composites.
 | ||
| Fig. 7 TEM micrograph of a Pt–Ru/G catalyst. | ||
| Composite | Metal precursor | Preparation method | Amount of metal/wt% | Particle size/nm | Application | Reference |
|---|---|---|---|---|---|---|
| *COD: 1,5-cyclooctadiene; COT: 1,3,5-cyclooctatriene. | ||||||
| Au/G | Chloroauric acid | Reduction with sodium citrate | 8.4 | 20 | Surface-enhanced Raman spectroscopy | 152 |
| Au/G | Chloroauric acid | Reduction with sodium dodecyl sulfate | 8 | 2–3 | Suzuki reaction | 16 |
| 21 | 7.5 | |||||
| Au/G | Chloroauric acid | Photochemical reduction | 1.2 | <1 | — | 162 |
| Pt–Au/G | Hexachloroplatinic acid and chloroauric acid | Reduction with sodium borohydride | 18.2 | 3.3 | Formic acid oxidation | 122 |
| Pt–Ru/G | Pt(CH3)2(COD) and Ru(COD)(COT)* | Gas-phase reduction | 5 | 2–3 | — | This work |
| Pt/G | Hexachloroplatinic acid | Reduction with ethylene glycol | 11 | 2–5.5 | Methanol oxidation | 227 |
| Pt/N–G | Hexachloroplatinic acid | Reduction with ethylene glycol | 14 | 2–3 | Methanol oxidation | 227 |
| Pt/G | Diammine dinitritoplatinum(II) | Gas-phase reduction | 20 | 0.5–2 | Hydrogen oxidation | 189 |
| Pt/G | Potassium tetrachloroplatinate(II) | Electrodeposition | — | As small as 2 | Methanol oxidation | 186 |
| Pt/G | Hexachloroplatinic acid | Gas-phase reduction | 20 | 2 | Oxygen reduction | 126 |
| Pt/G | Potassium hexachloroplatinate(IV) | Microwave synthesis | 30 | 3.31 | Methanol oxidation | 187 |
| 47 | 4.89 | |||||
| 67 | 5.81 | |||||
| Pd/G | Tetraamminepalladium(II) nitrate | Gas-phase reduction | 0.18 | 1–6 | Hydrogenation of alkynes | 17 |
| Pd/G | Potassium tetrachloropalladate(II) | Reduction with sodium borohydride | 0.19 | 2.5 | Hydrogenation of alkynes | 194 |
| Pd/G | Palladium(II) chloride | Microwave assisted reduction | 20 | 2.5 | Oxidation of methanol and ethanol | 13 |
| Pd/G | Palladium(II) acetate | Reduction with hydrazine | 7.5 | 2.0–5.6 | — | 196 |
| Pd/G | Palladium nitrate | Microwave assisted reduction | 7.9 | 7–9 | Suzuki and Heck reactions | 200 |
| Pd/GO | 6.4 | 12–15 | ||||
| Ag/G | Silver nitrate | Reduction with PVP | 65–88 | 10–30 | Surface-enhanced Raman spectroscopy | 209 |
| Ag/G | Silver nitrate | Hydrothermal synthesis | 56 | 10 | Surface-enhanced Raman spectroscopy | 207 |
| Ag/G | Silver nitrate | Reduction with sodium borohydride | — | 5–10 | SERS and antibacterial activity | 124 |
The downside to the covalent attachment of metal particles on the graphene is the disruption of the sp2 bonded carbon atoms in the basal plane which leads to reduced transport properties of graphene because of additional scattering sites.150 A possible way to circumvent the disruption of the sp2carbon atoms involves the non-covalent wrapping of the graphitic surface with a surfactant or a polymer. By this way, the nanoparticles can grow on the surfactant with a minimal chemical perturbation of the basal planes. Furthermore, the use of surfactants also allows the control of the morphology (size and shape) of the metal nanoparticles.161,174,198,222 Unfortunately, since the metal is not covalently bonded to the graphene, the liaison is much weaker and thus can be easily broken and induce a leaching effect.
Other techniques for metal nanoparticle decoration on the graphitic nanostructure include electro-deposition,186,192thermal evaporation,223,224 photochemical,162,225 and solventless bulk synthesis.226 Although these methods present some processing advantages over solution-based techniques, they can be somewhat expensive and energy consuming. Table 3 shows a comparison between common preparation methods for metal-graphene composites.
3.2 Graphene–metal oxide composites
There is currently great demand for the synthesis of graphene–semiconductor composites. The development of graphene–metal oxide composites (having more versatile and tailor-made properties with performances superior to those of the individual materials) provides an important milestone to improve the application of oxide nanomaterials in different fields such as energy harvesting, conversion and storage devices, nano-electronics, nano-optics and conductors, among others. In addition, the application of these materials in the catalysis field has grown considerably over the last few years with the use of graphene-based composites in photocatalysis (see Section 4.2.3).To date, various kinds of inorganic nanomaterials have been synthesized and supported on graphene-based templates including TiO2,10,24–27,128,228–252ZnO,129,253–266SnO2,235,267–278MnO2,130,268,279–287Fe3O4,10,131,288–301Fe2O3,291,302–306Co3O4,134,216,307–312NiO,6,268,313–316ZrO2,317SiO2,318Cu2O,253,319–321RuO2,10,132,322,323Al2O3,133,324–327MoO3,328,329ZnFeO4,330BiWO6331 and LiFePO4.332,333 Additionally, other materials like CdS,29–31,334–339CdSe,340–344 and ZnS31,334 have also been used to fabricate graphene-based composites.
One important obstacle in producing graphene–metal oxide nanomaterials is the difficulty of homogeneously dispersing the oxide over the graphene, since aggregation reduces the electrical, optical and magnetic properties of the resulting composite. In order to overcome this limitation, several deposition techniques have been developed with varying degrees of success. The synthetic methods for preparation of these graphene–semiconductor nanomaterial composites include sol–gel, hydrothermal/solvothermal process, electrochemical deposition, microwave-assisted growth, among others.
The direct growth approach is the most commonly used to prepare graphene–metal oxide composites. Usually, the metal precursor is mixed with GO and then converted to the corresponding oxide. After reduction of GO, graphene–metal oxide composites are finally obtained.25,229,235,242,243,265,335,345,346 When reducing GO to rGO, in order to avoid the simultaneous reduction of the as-prepared oxide, experimental conditions should be carefully chosen (strength and concentration of the agent, temperature, pressure, duration, etc.). The sol–gel method is a popular approach for preparation of metal oxide structures and film coatings. One of the greatest advantages of this process is the fact that surface hydroxyl groups of the GO/rGO sheets can act as nucleation sites for the hydrolysis step. Hence, the resulting metal oxide nanostructures are chemically bonded to the GO/rGO surfaces.25,243,245,252,268,288,318 The hydrothermal/solvothermal process is another effective method for the preparation of semiconductor composites with graphene. This route is a powerful tool for the synthesis of inorganic nanocrystals. The one-pot process can give rise to nanostructures with high crystallinity without post-synthetic annealing or calcination, and at the same time reduce GO to rGO.26,31,234,238–240,330,331,335,341
An interesting approach that has been mainly developed for thin film-based applications is electrochemical deposition. This allows the decoration of inorganic crystals on graphene-based substrates without the requirement for post-synthetic transfer of the composite materials.253–254,319,342 Microwave irradiation has also been used to prepare metal oxide–rGO composites, such as rGO–MnO2282 and rGO–Co3O4.310 In spite of the ease of process and scalable production, microwave-assisted synthesis does not display a fine control over the size uniformity and surface distribution of NPs on rGO surfaces.
Another preparation method consists in the addition of pre-synthesized nanoparticles to the GO suspension, followed by chemical and/or thermal reduction to yield the final composite. This ex situ synthesis allows a more precise control over the particle size and surface properties of nanoparticles as there is no interference from the GO/rGO and respective reducing agents, as observed in the in situ case. However, the synthesis process involves a chemical/thermal reduction to obtain the NPs/rGO composite which may change the NPs surface properties and damage graphene lattice. Table 4 shows several preparation methods commonly used to obtain different graphene–metal oxide composites.
| Composite | Metal precursor | Preparation method | Amount of metal oxide/wt% | Particle size/nm | Reference |
|---|---|---|---|---|---|
| G-TiO2 nanorod | Titanium isopropoxide | Ex situ synthesis | 55 | 2–4 diameter 20–30 length | 24 |
| G-TiO2 | Titanium trichloride | Direct growth | 85 | <15 | 235 |
| G-TiO2 anatase | Titanium trichloride | Sol–gel | 97.5 | 5 | 243 |
| G-TiO2 rutile | Titanium trichloride | Sol–gel | 90–99.5 | 6 | 243 |
| G-TiO2 | Tetrabutyl titanate | Hydrothermal synthesis | 67 | 9 | 238 |
| G-ZnO | Zinc acetate | Ex situ synthesis | Not specified | 4.5 | 261 |
| G-SnO2 | Tin(II) chloride dihydrate | Direct growth | 85 | ∼5–10 | 235 |
| G-MnO2 | Potassium permanganate | Microwave irradiation | 78 | 5–10 | 282 |
| G-Mn3O4 | Manganese(II) acetate | Direct growth and hydrothermal | 90 | 10–20 | 347 |
| G-Fe3O4 | Iron(III) chloride | Direct growth | 5.3–57.9 | 1.2–6.3 | 298 |
| G-Fe2O3 | Iron(III) chloride | Direct growth | 80 | 60 | 304 |
| G-Co3O4 | Cobalt nitrate | Direct growth | 75.4 | 10–30 | 307 |
| G-RuO2 | Ruthenium(III) chloride | Sol–gel | 38.3 | 5–20 | 132 |
| G-Al2O3 | α-Aluminium oxide | Mechanical mixture | 85–98 vol. % | 2.5–20 | 325 |
4. Application of graphene-based nanomaterials in catalysis
Ideally, graphene is a single-layer material, but samples with few layers are also subject to increasing interest. This is mainly due to the fact that single crystals can be obtained on top of non-crystalline substrates, in liquid stabilized suspensions or as suspended membranes.49 When a dispersion of isolated graphene sheets is dried, graphene sheets tend to couple with one another to stabilize into thicker layers, due to their thermodynamical instability,348 forming aggregates with very small interlayer spacing. This makes the application of atomic monolayers in catalysis challenging, for which reason, most catalytic applications have used few-layered graphene instead. This limitation can be partially overcome by intercalating nanoparticles within the graphene layers. This decreases the chances of formation of a stacked graphitic structure, by working as a “spacer” (the nanoparticles increase the distance between the graphene sheets to several nanometres).174 Additionally, incorporation of a second component onto an individual graphene or reduced graphene oxide sheet with good distribution aims to achieve unique properties from their interaction, targeting at catalytic, electrocatalytic and photocatalytic applications.Recent progresses have shown that graphene can have a deep impact on electronic and optoelectronic devices, chemical sensors, nanocomposites, energy conversion and storage, and catalysis. In the following sections, we will only focus on the properties of graphene and graphene-based composites for energy conversion and catalysis. For a detailed analysis regarding other applications of graphene-based materials, the reader can refer to several review articles recently published.9,50,54,56,57,65,102,106,118,349–354
4.1 Metal-free graphene-based materials as catalysts
The use of metal-free carbons instead of metal-supported catalysts in synthetic chemistry has largely progressed over the last decade. This results mainly from diminishing supplies of metals used in common industrial processes, and the discovery and development of novel carbon forms as fullerenes, nanotubes, nanofibers, graphene, among others.The performance of a catalyst is influenced by the nature, concentration and accessibility of the active sites that are capable of chemisorbing the reactants and form surface intermediates. It has been readily observed that functionalized carbons (containing oxygen, nitrogen, or other surface groups) are more efficient materials for catalysis than unfunctionalized ones. While functional group-rich materials, such as GO, exhibit high reactivity under mild conditions (their structure is not fully understood rendering mechanistic elucidation challenging), unfunctionalized graphene may not have a sufficient number of reactive sites to be a viable catalyst for many reactions. In the absence of defects, the basal planes are not very reactive, with the only active sites being present at the edges of the graphene layers as unsaturated carbon atoms. Nevertheless, pristine graphene can still find catalytic applications that make use of its delocalized π-electron system, such as complexation reactions.355 On the other hand, cracking and dehydrogenation of hydrocarbons have been reported using unfunctionalized carbon nanomaterials like nanotubes and fullerenes,2,356 which could be advantageously replaced by graphene in the future.
In chemistry, graphitic forms of carbon have an intriguing potential for catalysis. Early studies focused on simple redox processes, but the field has progressed to demonstrate that carbons enable reactions that are more sophisticated. These include complex functional group transformations and carbon–carbon or carbon–heteroatom bond formations. For example, C60 has been reported to catalyze the hydrogenation of nitrobenzene to aniline at room temperature under UV irradiation.357Carbon nanotubes have been used as catalysts for methane decomposition,358oxidation of p-toluidine,359 and conversion of aniline to azobenzene.360 However, the most prominent example of heterogeneous gas-phase catalysis by oxidized carbon materials is the selective oxidative dehydrogenation of hydrocarbons (ODH).361–363Ethylbenzene is predominantly used as the substrate, as the stable conjugated system of styrene allows for high yields. Earlier studies performed over activated carbons found that quinone surface groups were the active sites for this reaction.364 Since then, carbon nanostructures have also been reported to be active in the ODH of ethylbenzene to styrene.361,365–367 Furthermore, due to their remarkable stability and coke-resistance for this reaction, low-dimensional nanocarbons with well-defined microstructures often present higher yields when compared to other carbon forms. In contrast, light alkanes are much less reactive than the corresponding alkenes.363ODH is thought to proceed by nucleophilic oxygen atoms located at the prismatic edges of stacked graphene sheets or at surface defects in the (0001) graphitic surface.368 Unfortunately, ODH of light alkanes suffers from lower selectivities because the C–H bond in the product molecule is weaker than in the substrate.
Trunschke et al.368 recently demonstrated that graphene could also be a selective catalyst for model chemical reactions involving the insertion of oxygen into organic molecules (Fig. 8). They observed a 26.5 mmol g−1 h−1 productivity for the selective oxidation of acrolein to acrylic acid, almost half as high as that obtained with the industrial doped MoV mixed oxide (ca. 60 mmol g−1 h−1). Liu et al.19 studied the catalytic oxidation of SO2 gas to SO3 over porous graphene oxide foams. According to the authors, GO not only acted as catalyst to promote the reaction of SO2 and O2 to form SO3, but also as oxidant in the reaction. Upon prolonged exposure, the GO foams color gradually changed from brown to black. This implied that the GO was partially reduced and some of the oxygen-containing groups were lost (the GO turned from hydrophilic to hydrophobic and precipitated). Since the reaction takes place at room temperature and does not need noble metal catalysts, it could be a green and inexpensive method for the treatment of SO2 gas.
 | ||
| Fig. 8 Suggested reaction pathway for the oxidation of acrolein to acrylic acid at the graphitic carbon surface. Reprinted with permission from ref. 368. Copyright 2011 Wiley-VCH. | ||
Given its structural features, GO also finds application as a photocatalyst for H2 generation from water,369 providing a suitable alternative to metal-containing photocatalysts. An application that has received growing attention is the oxygen reduction reaction (ORR) in fuel cells. This reaction is commonly carried out using metal nanoparticles (particularly Pt). The major drawback of metal-based cathode materials is that they tend to be deactivated by CO poisoning and by sintering. Hence, preparation of metal-free catalysts is one of the most effective approaches for overcoming these problems with supported metal catalysts. Recently, nitrogen-containing graphene (N-graphene) has emerged as a promising candidate for the cathode catalyst due to the excellent oxygen reduction reaction activity without using any metal and the simple preparation procedure.370–374 Although a full understanding of the active sites in the catalysts is not yet completely understood, nitrogen atoms present in graphitic carbon are considered to play an essential role in high activity.
The use of metal-free carbon materials has been recently reviewed by Bielawski et al., who investigated the catalytic activity of various synthetic reactions under mild conditions in the liquid-phase.18,21,375–379 Their results highlight the unique role that large-area, functionalized carbon materials (GO) may find in the activation of small molecules, such as O2, for catalysis. Exploiting the reactivity intrinsic to graphite oxide (GO tends to be highly acidic and strongly oxidizing), Bielawski et al.375 have identified this material as a powerful catalyst to be used in the generation of aldehydes or ketones from various alcohols, alkenes and alkynes (Table 5). The authors demonstrated the efficient oxidation of benzyl alcohol to benzaldehyde (conversion >98%) in the presence of GO as a heterogeneous catalyst. Further oxidation to benzoic acid was observed in only minimal amounts and only under certain conditions (e.g. at elevated temperatures). Interestingly, this and other oxidation reactions of alcohols were performed under ambient conditions and did not proceed under a nitrogen atmosphere, suggesting that oxygen could be functioning as the terminal oxidant. In the same study, they also demonstrated that the scope of GO catalysis extends beyond simple oxidation reactions of alcohols. The successful oxidation of cis-stilbene to benzyl and hydration of various alkynes indicate that the scope of the reactivity of GO may be quite broad. The ability of GO to function as a carbocatalyst (metal-free carbon material as catalyst) was further confirmed in another work by Bielawski et al.376 where they produced chalcones in a single reaction vessel (>60% isolated yields). GO was found to function as an auto-tandem oxidation–hydration–aldol coupling catalyst, as various alkynes were hydrated in situ to their corresponding methyl ketones or alcohols were oxidized in situ to their corresponding aldehydes. The condensation of the methyl ketones with the aldehydes was believed to proceed via a Claisen–Schmidt-type process, where the GO acted as an acid catalyst. When an alkyne was substituted for the methyl ketone, however, the condensation was likely preceded by hydration of the alkyne. Similarly, when an alcohol was substituted for the aldehyde, the condensation was preceded by oxidation of the alcohol.


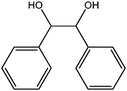


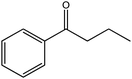
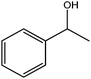


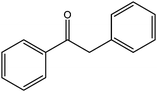
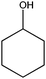






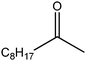
In yet another work by the same authors, GO was found to be an effective oxidant for use in a broad range of reactions, including the oxidation of olefins to their respective diones, methylbenzenes to their respective aldehydes, diarylmethanes to their respective ketones, and dehydrogenation of various hydrocarbons.21 In this case, GO was found to be capable of oxidizing cis-stilbene to benzyl, optimizing the yield with regard to GO loading and reaction temperature. The authors then explored the ability of GO to oxidize more challenging substrates, including hydrocarbons possessing sp3-hybridized C–H bonds. Various substrates with benzylic methylene groups were successfully converted to their corresponding ketone and unsaturated products.
Since no reactivity was observed when hydrazine-reduced graphene oxide or natural flake graphite were used, as an explanation for the results obtained with GO, the authors suggested that the presence of the surface bound oxygen-containing functionalities played an important role in the observed reactivities and product formation.
The majority of the reactions discussed in this section were performed under relatively mild conditions and produced the desired product (aldehyde, acid or ketone) in high yields, without the need for additional oxidants or metals as co-catalysts. However, reactivity results should be carefully evaluated, as to exclude the possibility of metal-mediated catalysis, due to potential contaminations (even in trace levels). Several advantages can be pointed out in these reactions using GO: use of a simple and inexpensive catalyst, metal-free reactivity, and facile recovery of the GO from the reaction media by simple filtration.
4.2 Metal supported graphene-based materials as catalysts
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1, accompanied by very low palladium leaching (<1 ppm) in the Suzuki–Miyaura coupling reaction. Reuse of the catalysts could be achieved despite some activity loss, depending on the recycling procedure. In another work, a Pd/G catalyst demonstrated excellent catalytic activity for both the Suzuki and Heck carbon–carbon cross-coupling reactions.200 The Pd/G catalyst was recycled eight times with a quantitative reaction yield. The remarkable reactivity of Pd/G toward Suzuki cross-coupling reactions (TOF = 108000 h−1) was attributed to the high degree of the dispersion and loading of Pd(0) supported on graphene sheets (particle size of 7–9 nm). Li et al.198 prepared a Pd/G catalyst using sodium dodecyl sulfate as both surfactant and reducing agent. They obtained Pd nanoparticles with an average particle size of 4 nm, which demonstrated a good efficiency for the Suzuki reaction under aqueous and aerobic conditions.
000 h−1, accompanied by very low palladium leaching (<1 ppm) in the Suzuki–Miyaura coupling reaction. Reuse of the catalysts could be achieved despite some activity loss, depending on the recycling procedure. In another work, a Pd/G catalyst demonstrated excellent catalytic activity for both the Suzuki and Heck carbon–carbon cross-coupling reactions.200 The Pd/G catalyst was recycled eight times with a quantitative reaction yield. The remarkable reactivity of Pd/G toward Suzuki cross-coupling reactions (TOF = 108000 h−1) was attributed to the high degree of the dispersion and loading of Pd(0) supported on graphene sheets (particle size of 7–9 nm). Li et al.198 prepared a Pd/G catalyst using sodium dodecyl sulfate as both surfactant and reducing agent. They obtained Pd nanoparticles with an average particle size of 4 nm, which demonstrated a good efficiency for the Suzuki reaction under aqueous and aerobic conditions.
Zhang et al.14 reported the use of Au/GO nanocomposites, which showed an unusually high activity for the Suzuki–Miyaura coupling reaction of chlorobenzene with arylboronic acid (yield as high as 98%). Graphene modified with Au nanoparticles was also used as an efficient catalyst for the Suzuki reaction in water under aerobic conditions.16 The catalytic activity of Au/G hybrids was related to the Au loading and particle size.
Metal-decorated graphene has provided enhanced electrocatalytic activity in alcohol oxidation reactions (energy production) more efficiently than any other commercially available material. Li et al.175 prepared a graphene-supported Pt catalyst (dPt = 5–6 nm) with higher electrochemically active surface area (ECSA) and electrocatalytic activity for methanol oxidation than a commercial Pt/C catalyst. The lower oxidation potential and higher current density over the Pt/G indicated a higher catalytic activity for the methanol oxidation, which the authors attributed to the higher ECSA and good Pt dispersion. In addition, the stability of Pt/G was also found to be better than that of Pt/C catalysts. Yoo et al.191 also reported an enhanced electrocatalytic activity for the same reaction using a Pt/G with metal particles smaller than 0.5 nm. The presence of extremely small Pt clusters suggests a strong interaction between graphene and Pt atoms. This interaction between Pt and graphene was thought to induce some modulation in the electronic structure of the Pt clusters. The Pt/G electrocatalyst revealed an unusually high activity for methanol oxidation reaction, and also exhibited quite a different behavior for CO oxidation compared to a Pt/carbon-black catalyst. In a work by Wang et al.173 the performance of Pt/G catalysts was directly compared to that of Pt/CNTs (dPt = ca. 3 nm for both supports, Fig. 9). Measurements showed that Pt/G catalyzed the methanol oxidation more efficiently (higher oxidation current density), which the authors attributed to a larger ECSA.
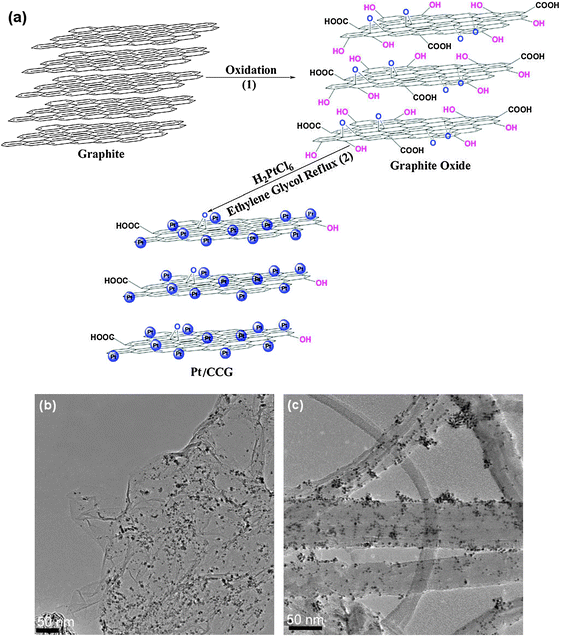 | ||
| Fig. 9 Formation route to anchor platinum nanoparticles onto chemically converted graphene (a); TEM images of (b) Pt/G and (c) Pt/CNTs hybrids. Reprinted with permission from ref. 173. Copyright 2009 Elsevier Ltd. | ||
Kundu et al.179 used ethylene glycol and microwave irradiation conditions to prepare the graphene-supported Pt catalyst (dPt = 2–3 nm) by the co-reduction of graphene oxide and Pt salt. The catalyst exhibited an excellent catalytic activity coupled with long-term stability for methanol oxidation. Similarly to other works, Pt/G was found to be a better catalyst than Pt/C in terms of both current density and CO tolerance, although morphologically similar.
On the other hand, Pt/G has also been used to catalyze the oxygen reduction reaction for use in fuel cells. Similar to the catalyzed alcohol oxidation reactions, Pt/G with large ECSA also promotes an efficient ORR catalysis. Xinet al.380 have recently demonstrated the use of a Pt/G catalyst for high catalytic activity of both methanol oxidation and oxygen reduction, when compared to Pt supported on carbon-black. The performance of Pt/G was further improved (ca. 3.5 times higher than Pt/C) after heat treatment in a N2 atmosphere at 300 °C. Shao et al.176 reported that the ORR performance of Pt/G was comparable to that of Pt/CNT catalysts. However, Pt/G showed an enhanced stability compared to Pt/CNTs and commercially available Pt/C materials. Kou et al.126 observed similar electrochemical results using Pt nanoparticles with an average diameter of 2 nm dispersed over GO. Pt/G showed not only larger specific surface area and higher ORR activity, but also excellent stability after 5000 cyclic voltammetry cycles. These improved properties were attributed to the smaller aggregation of Pt particles immobilized on graphene. In an attempt to further improve the ORR activity, carbon nitride was also incorporated into graphene to produce an electrocatalyst composite.382,383 In a work published by Shi et al.,382 this composite exhibited an electrocatalytic activity for ORR comparable to that of a rGO composite with ca. 23 wt% Pt nanoparticles. Metal-free graphene–carbon nitride composites also showed high visible-light photocatalytic activity (making them promising nanomaterials for applications in water treatment and dye-sensitized solar cells),384 and the ability to activate O2 for the selective oxidation of secondary C–H bonds of cyclohexane (good conversion and high selectivity to the corresponding ketones).385
A common problem when using Pt catalysts in fuel cells is its poisoning by carbon monoxide. One possible solution to this problem is the use of Pt based alloys as catalysts. Dong et al.172 studied the electrocatalytic activity of graphene supported Pt–Ru nanoparticles for methanol and ethanol oxidation. Compared to the widely used Vulcan XC-72R carbon-black, graphene strongly enhanced the oxidation efficiencies of both methanol and ethanol. Furthermore, the introduction of Ru greatly reduced the adsorption of CO-like carbonaceous species on the surfaces of Pt particles. Another similar work has demonstrated that using a graphene-supported Pt–Ru nanocomposite, an improvement in catalytic activity is achievable towards the oxidation of methanol when contrasted to Pt–Ru/C.180 Still in the field of bimetallic catalysts, Zhang et al.122 prepared a Pt–Au/G catalyst (Pt:Au = 1:1) via a polyelectrolyte-assisted process. The electrocatalytic activity of this bimetallic catalyst was compared against that obtained with Pt–Au/carbon-black in the formic acid oxidation. The Pt–Au/G catalyst displayed a 37% higher electrocatalytic activity toward formic acid oxidation than Pt–Au/carbon-black (Fig. 10). The higher electrocatalytic activity of Pt–Au/G was attributed to a strong electronic interaction between graphene and Pt–Au alloy nanoparticles, which suppressed the CO poisoning and facilitated the direct oxidation process of formic acid on the Pt–Au surface.
 | ||
| Fig. 10 TEM images of (a) Pt–Au/G and (b) Pt–Au/carbon-black; (c) Pt–Au/G show the highest electrocatalytic activity and stability toward formic acid oxidation compared to Pt–Au/carbon-black and E-TEK Pt/C. Reprinted with permission from ref. 122. Copyright 2011 American Chemical Society. | ||
A composite containing Pt-on-Pd bimetallic nanodendrites and graphene was synthesized and its catalytic activity investigated using the oxidation of methanol.170 The electrochemical data indicated that the as-prepared Pt–Pd/G composite exhibited much higher electrocatalytic activity toward methanol oxidation reaction than the platinum black and commercial E-TEK Pt/C catalysts. The current density of methanol oxidation catalyzed by this composite was about 3.0 and 9.5 times higher than those of E-TEK and platinum black catalysts, respectively.
Nitrogen doped carbon materials are recognized as good supports for Pt catalysts. The nitrogen atoms not only provide anchoring sites for the metal particles, but also act as chemically active sites for some catalytic reactions. Wu et al.227 prepared N-doped graphene by heating graphene oxide in an ammonia flow at different temperatures. The methanol oxidation current for Pt/N–graphene heated at 800 °C was found to be 3 times higher than that of those treated at lower temperatures (300, 500 or 700 °C). Moreover, it was much higher than that of the Pt/C commercial catalyst. This behavior was attributed to a higher conductivity and more uniformly dispersed Pt nanoparticles over the surface of the composite treated at 800 °C. Ramaprabhu et al.381 used graphene and nitrogen doped-graphene as catalytic support of Pt nanoparticles for ORR. The authors attributed the enhanced performance to an improved metal–carbon interaction and increased electrical conductivity induced by nitrogen doping.
One other possible application of graphene-based composites consists of the conversion of solar energy to electrical power, i.e. solar cells. These cells with transparent and conductive graphene film as window electrode have exhibited considerable power conversion efficiency. Among the most commonly studied are dye-sensitized and heterojunction solar cells.57,353,386–390Graphene films with excellent conductance, good transparency in both the visible and near-infrared regions, ultrasmooth surface with tunable wettability, high chemical and thermal stabilities and flexibility for transfer between alternative substrates, can be used not only in solar cells as electrode but also in many other optoelectronic devices.353
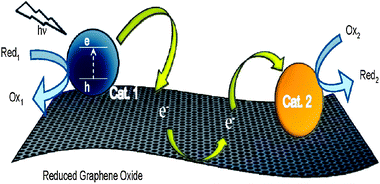 | ||
| Fig. 11 Illustration of the three-step electron transfer process involved in making a two-dimensional conducting support. Reprinted with permission from ref. 225. Copyright 2010 American Chemical Society. | ||
Possible applications of graphene-based materials in photocatalysis involve mainly the degradation of pollutants,24–26,234,249,266,391,392 and water splitting for hydrogen generation.27,369
A graphene–P25 TiO2 composite was applied to the photocatalytic degradation of organic compounds and compared against bare P25 TiO2 and a CNT–P25 TiO2 composite (Fig. 12).26 In their work, the graphene composite was found to have high dye absorptivity, extended light absorption range, and enhanced charge separation and transportation properties. The authors attributed the enhanced photocatalytic activity in the degradation of methylene blue dye under both UV and visible lights to the two-dimensional conjugated structure of graphene, which facilitated a better platform for dye adsorption and charge transportation. Liu et al.24 reported the application of a TiO2 nanorod–graphene composite in the degradation of methylene blue under UV light irradiation. They observed an effective reduction in charge recombination because of improved contact between graphene and TiO2 nanorods, increasing the photocatalytic activity. This result could open important perspectives for improving the photocatalytic activity of graphene–TiO2 composites by optimizing the morphology and distribution of TiO2 nanoparticles on graphene sheets. In an attempt to improve the visible-light response of graphene–TiO2 photocatalysts, Chen et al.231 reported a GO–TiO2 composite with p/n heterojunction in the degradation of methyl orange. In addition to graphene–TiO2 composites, other materials have also been used as efficient photocatalysts for decomposition of different pollutants in water, namely graphene–SnO2,235 and graphene–ZnO.265
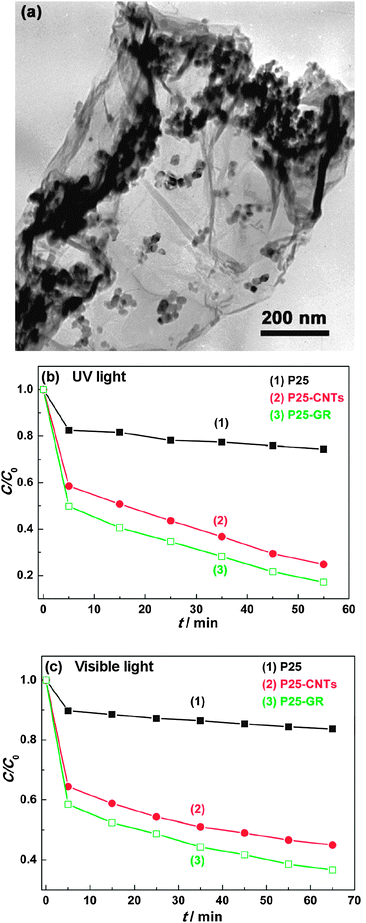 | ||
| Fig. 12 (a) TEM image of P25–graphene composite, and photocatalytic degradation of methylene blue under (b) UV and (c) visible light (λ > 400 nm) with P25 TiO2, CNT–P25 TiO2 and graphene–P25 TiO2. Reprinted with permission from ref. 26. Copyright 2010 American Chemical Society. | ||
Photocatalytic water splitting into hydrogen and oxygen using semiconductor photocatalysts has been considered as a promising and attractive approach to produce hydrogen energy. Unfortunately, due to the rapid recombination of photogenerated electrons and holes practical applications are quite limited. A possible way of improving photocatalytic hydrogen production using graphene-based materials requires the presence of a sacrificial agent. Using graphene–TiO2 composites with different graphene loadings (prepared by a sol–gel method), Zhang et al.27 studied the H2 evolution from aqueous solution containing Na2S and Na2SO3 as sacrificial agents under UV-Vis irradiation. The optimal graphene content was found to be 5 wt%, yielding a H2 production rate which exceeded that of pure P25 TiO2 over 2 times. In a similar work, Fan et al.393 prepared a graphene–P25 TiO2 (0.2:1 optimum ratio) composite that improved the H2 production rate by more than 10 times, when compared to that of pure P25 TiO2.
Despite some very promising results, the mechanism of photocatalytic enhancement by graphene-based composites is relatively uncertain. Some questions have raised the discussion on whether or not the graphene composites are truly different from other carbonaceous (activated carbon, fullerenes or carbon nanotubes) composite materials. In a work by Zhang et al.,234 a graphene–TiO2 composite was observed to be essentially the same as other carbon–TiO2 (activated carbon, fullerenes and carbon nanotubes) composite materials, regarding the enhancement of photocatalytic activity of TiO2.
5. Summary and outlook
Graphene has come a long way since it was first reported in 2004 by Novoselov and co-workers. The rise of graphene nanosheets has opened a new route for the use of two-dimensional carbon materials as catalytic supports, due to their high electrical and thermal conductivities, great mechanical strength and huge specific surface area and adsorption capacities. This has allowed researchers to design and develop countless combinations of graphene-based materials, some rather simple while others considerably more sophisticated. Graphene and its composites have been used as catalysts for chemical, electrochemical and photochemical reactions, showing promising results, especially when compared to conventional catalysts. One of the most attractive areas in catalysis involves the use of metal-free graphene. As we described in this review, this field has grown in complexity over the last few years and we expect it to know further developments, as theoretical calculations can be very useful to fine-tune the surface properties of graphene.Unfortunately, despite all of the promising results obtained so far, the generalization of graphene-based materials is dependent on one very important premise: the large-scale availability of high-quality graphene with controllable layer thickness, at relatively low cost. In addition to this issue, physical properties such as low bulk density can severely hinder the catalytic application of graphene at the industrial scale. Thus, there is still a long road ahead before graphene-based catalysts can effectively find a commercial application.
Acknowledgements
The authors gratefully acknowledge Dr Revathi Bacsa for the selected area diffraction pattern, TEM and HRTEM micrographs used in this work. B.F.M. acknowledges Fundação para a Ciência e a Tecnologia for the grant SFRH/BPD/70299/2010.References
- P. Serp and J. L. Figueiredo, Carbon Materials for Catalysis, John Wiley & Sons, 2009 Search PubMed.
- P. Serp, M. Corrias and P. Kalck, Appl. Catal., A, 2003, 253, 337 CrossRef CAS.
- H. W. Kroto, J. R. Heath, S. C. Obrien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162 CrossRef CAS.
- S. Iijima, Nature, 1991, 354, 56 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS.
- J. Y. Son, Y.-H. Shin, H. Kim and H. M. Jang, ACS Nano, 2010, 4, 2655 CrossRef CAS.
- X.-D. Zhuang, Y. Chen, G. Liu, P.-P. Li, C.-X. Zhu, E.-T. Kang, K.-G. Noeh, B. Zhang, J.-H. Zhu and Y.-X. Li, Adv. Mater., 2010, 22, 1731 CrossRef CAS.
- J. Liu, Z. Lin, T. Liu, Z. Yin, X. Zhou, S. Chen, L. Xie, F. Boey, H. Zhang and W. Huang, Small, 2010, 6, 1536 CrossRef CAS.
- L. L. Zhang, R. Zhou and X. S. Zhao, J. Mater. Chem., 2010, 20, 5983 RSC.
- A. K. Mishra and S. Ramaprabhu, J. Phys. Chem. C, 2011, 115, 14006 CrossRef CAS.
- S. Vivekchand, C. Rout, K. Subrahmanyam, A. Govindaraj and C. Rao, J. Chem. Sci., 2008, 120, 9 CrossRef CAS.
- G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Mülhaupt, J. Am. Chem. Soc., 2009, 131, 8262 CrossRef CAS.
- R. N. Singh and R. Awasthi, Catal.: Sci. Technol., 2011, 1, 778 RSC.
- N. Zhang, H. Qiu, Y. Liu, W. Wang, Y. Li, X. Wang and J. Gao, J. Mater. Chem., 2011, 21, 11080 RSC.
- H. He and C. Gao, Sci. China: Chem., 2011, 54, 397 Search PubMed.
- Y. Li, X. Fan, J. Qi, J. Ji, S. Wang, G. Zhang and F. Zhang, Mater. Res. Bull., 2010, 45, 1413 CrossRef CAS.
- Á. Mastalir, Z. Király, Á. Patzkó, I. Dékány and P. L'Argentiere, Carbon, 2008, 46, 1631 CrossRef CAS.
- D. R. Dreyer and C. W. Bielawski, Chem. Sci., 2011, 2, 1233 RSC.
- Y. Long, C. Zhang, X. Wang, J. Gao, W. Wang and Y. Liu, J. Mater. Chem., 2011, 21, 13934 RSC.
- J. Pyun, Angew. Chem., Int. Ed., 2011, 50, 46 CrossRef CAS.
- H.-P. Jia, D. R. Dreyer and C. W. Bielawski, Tetrahedron, 2011, 67, 4431 CrossRef CAS.
- Y. Zhang and C. Pan, J. Mater. Sci., 2011, 46, 2622 CrossRef CAS.
- H. Liu, S. Ryu, Z. Chen, M. L. Steigerwald, C. Nuckolls and L. E. Brus, J. Am. Chem. Soc., 2009, 131, 17099 CrossRef CAS.
- J. Liu, H. Bai, Y. Wang, Z. Liu, X. Zhang and D. D. Sun, Adv. Funct. Mater., 2010, 20, 4175 CrossRef CAS.
- J. Du, X. Lai, N. Yang, J. Zhai, D. Kisailus, F. Su, D. Wang and L. Jiang, ACS Nano, 2010, 5, 590.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2009, 4, 380.
- X.-Y. Zhang, H.-P. Li, X.-L. Cui and Y. Lin, J. Mater. Chem., 2010, 20, 2801 RSC.
- X. Wang, L. Zhi and K. Mullen, Nano Lett., 2007, 8, 323.
- C. X. Guo, H. B. Yang, Z. M. Sheng, Z. S. Lu, Q. L. Song and C. M. Li, Angew. Chem., Int. Ed., 2010, 49, 3014 CAS.
- H. Chang, X. Lv, H. Zhang and J. Li, Electrochem. Commun., 2010, 12, 483 CrossRef CAS.
- P. Wang, T. Jiang, C. Zhu, Y. Zhai, D. Wang and S. Dong, Nano Res., 2010, 3, 794 CrossRef CAS.
- Z. Liu, D. He, Y. Wang, H. Wu and J. Wang, Sol. Energy Mater. Sol. Cells, 2010, 94, 1196 CrossRef CAS.
- S. Park, N. Mohanty, J. W. Suk, A. Nagaraja, J. An, R. D. Piner, W. Cai, D. R. Dreyer, V. Berry and R. S. Ruoff, Adv. Mater., 2010, 22, 1736 CrossRef CAS.
- H. Chang, L. Tang, Y. Wang, J. Jiang and J. Li, Anal. Chem., 2010, 82, 2341 CrossRef CAS.
- Z. Wang, J. Zhang, P. Chen, X. Zhou, Y. Yang, S. Wu, L. Niu, Y. Han, L. Wang, F. Boey, Q. Zhang, B. Liedberg and H. Zhang, Biosens. Bioelectron., 2011, 26, 3881 CrossRef CAS.
- F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Nat. Mater., 2007, 6, 652 CrossRef CAS.
- L. Xie, X. Ling, Y. Fang, J. Zhang and Z. Liu, J. Am. Chem. Soc., 2009, 131, 9890 CrossRef CAS.
- X. Ling, L. Xie, Y. Fang, H. Xu, H. Zhang, J. Kong, M. S. Dresselhaus, J. Zhang and Z. Liu, Nano Lett., 2009, 10, 553.
- X. Ling and J. Zhang, Small, 2010, 6, 2020 CrossRef CAS.
- W. Cai and X. Chen, Small, 2007, 3, 1840 CrossRef CAS.
- C. Peng, W. Hu, Y. Zhou, C. Fan and Q. Huang, Small, 2010, 6, 1686 CrossRef CAS.
- O. Akhavan, E. Ghaderi and A. Esfandiar, J. Phys. Chem. B, 2011, 115, 6279 CrossRef CAS.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752 CrossRef CAS.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2009, 110, 132.
- C. N. R. Rao, K. Biswas, K. S. Subrahmanyam and A. Govindaraj, J. Mater. Chem., 2009, 19, 2457 RSC.
- M. Terrones, A. R. Botello-Méndez, J. Campos-Delgado, F. López-Urías, Y. I. Vega-Cantú, F. J. Rodríguez-Macías, A. L. Elías, E. Muñoz-Sandoval, A. G. Cano-Márquez, J.-C. Charlier and H. Terrones, Nano Today, 2010, 5, 351 CrossRef.
- C. N. R. Rao, A. K. Sood, R. Voggu and K. S. Subrahmanyam, J. Phys. Chem. Lett., 2010, 1, 572 Search PubMed.
- F. Molitor, et al. , J. Phys.: Condens. Matter, 2011, 23, 243201 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS.
- X. Huang, Z. Yin, S. Wu, X. Qi, Q. He, Q. Zhang, Q. Yan, F. Boey and H. Zhang, Small, 2011, 7, 1876 CrossRef CAS.
- V. Singh, D. Joung, L. Zhai, S. Das, S. I. Khondaker and S. Seal, Prog. Mater. Sci., 2011, 56, 1178 CrossRef CAS.
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109 CrossRef CAS.
- C. N. R. Rao, et al. , Sci. Technol. Adv. Mater., 2010, 11, 054502 CrossRef.
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718 CrossRef CAS.
- D. R. Dreyer, R. S. Ruoff and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 9336 CrossRef CAS.
- H. Bai, C. Li and G. Shi, Adv. Mater., 2011, 23, 1089 CrossRef CAS.
- Y. Sun, Q. Wu and G. Shi, Energy Environ. Sci., 2011, 4, 1113 RSC.
- P. V. Kamat, J. Phys. Chem. Lett., 2009, 1, 520 Search PubMed.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451 CrossRef CAS.
- J. Hass, et al. , J. Phys.: Condens. Matter, 2008, 20, 323202 CrossRef.
- C. Berger, Z. Song, T. Li, X. Li, A. Y. Ogbazghi, R. Feng, Z. Dai, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, J. Phys. Chem. B, 2004, 108, 19912 CrossRef CAS.
- C. Berger, Z. Song, X. Li, X. Wu, N. Brown, C. Naud, D. Mayou, T. Li, J. Hass, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, Science, 2006, 312, 1191 CrossRef CAS.
- W. A. de Heer, C. Berger, X. Wu, P. N. First, E. H. Conrad, X. Li, T. Li, M. Sprinkle, J. Hass, M. L. Sadowski, M. Potemski and G. Martinez, Solid State Commun., 2007, 143, 92 CrossRef CAS.
- W. Choi, I. Lahiri, R. Seelaboyina and Y. S. Kang, Crit. Rev. Solid State Mater. Sci., 2010, 35, 52 Search PubMed.
- S. Guo and S. Dong, Chem. Soc. Rev., 2011, 40, 2644 RSC.
- A. Srivastava, C. Galande, L. Ci, L. Song, C. Rai, D. Jariwala, K. F. Kelly and P. M. Ajayan, Chem. Mater., 2010, 22, 3457 CrossRef CAS.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and R. S. Ruoff, Science, 2009, 324, 1312 CrossRef CAS.
- P. W. Sutter, J.-I. Flege and E. A. Sutter, Nat. Mater., 2008, 7, 406 CrossRef CAS.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J.-H. Ahn, P. Kim, J.-Y. Choi and B. H. Hong, Nature, 2009, 457, 706 CrossRef CAS.
- A. Reina, X. Jia, J. Ho, D. Nezich, H. Son, V. Bulovic, M. S. Dresselhaus and J. Kong, Nano Lett., 2008, 9, 30.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS.
- M. J. McAllister, J.-L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396 CrossRef CAS.
- H. C. Schniepp, J.-L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228 RSC.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477 CrossRef CAS.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217 CrossRef CAS.
- A. K. Geim, Science, 2009, 324, 1530 CrossRef CAS.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463 CrossRef CAS.
- R. R. Nair, P. Blake, A. N. Grigorenko, K. S. Novoselov, T. J. Booth, T. Stauber, N. M. R. Peres and A. K. Geim, Science, 2008, 320, 1308 CrossRef CAS.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385 CrossRef CAS.
- L. Hu, X. Hu, X. Wu, C. Du, Y. Dai and J. Deng, Physica B (Amsterdam), 2010, 405, 3337 CrossRef CAS.
- Y. Tang, Z. Yang and X. Dai, J. Magn. Magn. Mater., 2011, 323, 2441 CrossRef CAS.
- K. Nakada and A. Ishii, Solid State Commun., 2011, 151, 13 CrossRef CAS.
- O. Leenaerts, B. Partoens and F. M. Peeters, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 125416 CrossRef.
- F. Ma, Z. Zhang, H. Jia, X. Liu, Y. Hao and B. Xu, THEOCHEM, 2010, 955, 134 CrossRef CAS.
- C. Rajesh, C. Majumder, H. Mizuseki and Y. Kawazoe, J. Chem. Phys., 2009, 130, 124911 CrossRef.
- M. Chi and Y.-P. Zhao, Comput. Mater. Sci., 2009, 46, 1085 CrossRef CAS.
- Y.-H. Lu, M. Zhou, C. Zhang and Y.-P. Feng, J. Phys. Chem. C, 2009, 113, 20156 CrossRef CAS.
- Y. Li, Z. Zhou, G. Yu, W. Chen and Z. Chen, J. Phys. Chem. C, 2010, 114, 6250 CrossRef CAS.
- Z. M. Ao, J. Yang, S. Li and Q. Jiang, Chem. Phys. Lett., 2008, 461, 276 CrossRef CAS.
- J. Dai and J. Yuan, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 165414 CrossRef.
- X. Qin, Q. Meng and W. Zhao, Surf. Sci., 2011, 605, 930 CrossRef CAS.
- E. N. Voloshina, D. Mollenhauer, L. Chiappisi and B. Paulus, Chem. Phys. Lett., 2011, 510, 220 CrossRef CAS.
- N. Ding, X. Lu and C.-M. L. Wu, Comput. Mater. Sci., 2012, 51, 141 CrossRef CAS.
- M. Zhou, A. Zhang, Z. Dai, C. Zhang and Y. P. Feng, J. Chem. Phys., 2010, 132, 194704 CrossRef.
- G. Lee, B. Lee, J. Kim and K. Cho, J. Phys. Chem. C, 2009, 113, 14225 CrossRef CAS.
- P. A. Denis, J. Phys. Chem. C, 2009, 113, 5612 CrossRef CAS.
- J. T. Paci, T. Belytschko and G. C. Schatz, J. Phys. Chem. C, 2007, 111, 18099 CrossRef CAS.
- S. Tang and Z. Cao, J. Chem. Phys., 2011, 134, 044710 CrossRef.
- J. D. Fowler, M. J. Allen, V. C. Tung, Y. Yang, R. B. Kaner and B. H. Weiller, ACS Nano, 2009, 3, 301 CrossRef CAS.
- T. Kuila, S. Bose, P. Khanra, A. K. Mishra, N. H. Kim and J. H. Lee, Biosens. Bioelectron., 2011, 26, 4637 CrossRef CAS.
- Y. Shao, J. Wang, H. Wu, J. Liu, I. A. Aksay and Y. Lin, Electroanalysis, 2010, 22, 1027 CAS.
- A. Ghosh, K. S. Subrahmanyam, K. S. Krishna, S. Datta, A. Govindaraj, S. K. Pati and C. N. R. Rao, J. Phys. Chem. C, 2008, 112, 15704 CrossRef CAS.
- G. Srinivas, Y. Zhu, R. Piner, N. Skipper, M. Ellerby and R. Ruoff, Carbon, 2010, 48, 630 CrossRef CAS.
- Hyperion Catalysis International, Inc, http://hyperioncatalysis.com/.
- D. Chen, L. Tang and J. Li, Chem. Soc. Rev., 2010, 39, 3157 RSC.
- J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth and S. Roth, Nature, 2007, 446, 60 CrossRef CAS.
- A. Fasolino, J. H. Los and M. I. Katsnelson, Nat. Mater., 2007, 6, 858 CrossRef CAS.
- G. Wang, J. Yang, J. Park, X. Gou, B. Wang, H. Liu and J. Yao, J. Phys. Chem. C, 2008, 112, 8192 CrossRef CAS.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51 CrossRef CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS.
- M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. G. Cancado, A. Jorio and R. Saito, Phys. Chem. Chem. Phys., 2007, 9, 1276 RSC.
- H.-L. Guo, X.-F. Wang, Q.-Y. Qian, F.-B. Wang and X.-H. Xia, ACS Nano, 2009, 3, 2653 CrossRef CAS.
- H. C. Schniepp, K. N. Kudin, J.-L. Li, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, ACS Nano, 2008, 2, 2577 CrossRef CAS.
- E. Stolyarova, K. T. Rim, S. Ryu, J. Maultzsch, P. Kim, L. E. Brus, T. F. Heinz, M. S. Hybertsen and G. W. Flynn, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9209 CrossRef CAS.
- A. Peigney, C. Laurent, E. Flahaut, R. R. Bacsa and A. Rousset, Carbon, 2001, 39, 507 CrossRef CAS.
- Q. Du, M. Zheng, L. Zhang, Y. Wang, J. Chen, L. Xue, W. Dai, G. Ji and J. Cao, Electrochim. Acta, 2010, 55, 3897 CrossRef CAS.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498 CrossRef CAS.
- Y. Zhu, S. Murali, M. D. Stoller, A. Velamakanni, R. D. Piner and R. S. Ruoff, Carbon, 2010, 48, 2118 CrossRef CAS.
- M. Käärik, M. Arulepp, M. Karelson and J. Leis, Carbon, 2008, 46, 1579 CrossRef CAS.
- K.-J. Jeon and Z. Lee, Chem. Commun., 2011, 47, 3610 RSC.
- S. Zhang, Y. Shao, H.-g. Liao, J. Liu, I. A. Aksay, G. Yin and Y. Lin, Chem. Mater., 2011, 23, 1079 CrossRef CAS.
- R. Pasricha, S. Gupta and A. K. Srivastava, Small, 2009, 5, 2253 CrossRef CAS.
- J. Shen, M. Shi, N. Li, B. Yan, H. Ma, Y. Hu and M. Ye, Nano Res., 2010, 3, 339 CrossRef.
- C. Nethravathi, E. A. Anumol, M. Rajamathi and N. Ravishankar, Nanoscale, 2011, 3, 569 RSC.
- R. Kou, Y. Shao, D. Wang, M. H. Engelhard, J. H. Kwak, J. Wang, V. V. Viswanathan, C. Wang, Y. Lin, Y. Wang, I. A. Aksay and J. Liu, Electrochem. Commun., 2009, 11, 954 CrossRef CAS.
- X. Chen, G. Wu, J. Chen, X. Chen, Z. Xie and X. Wang, J. Am. Chem. Soc., 2011, 133, 3693 CrossRef CAS.
- G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487 CrossRef CAS.
- Y.-L. Chen, Z.-A. Hu, Y.-Q. Chang, H.-W. Wang, Z.-Y. Zhang, Y.-Y. Yang and H.-Y. Wu, J. Phys. Chem. C, 2011, 115, 2563 CrossRef CAS.
- S. Chen, J. Zhu, X. Wu, Q. Han and X. Wang, ACS Nano, 2010, 4, 2822 CrossRef CAS.
- K. Zhou, Y. Zhu, X. Yang and C. Li, New J. Chem., 2010, 34, 2950 RSC.
- Z.-S. Wu, D.-W. Wang, W. Ren, J. Zhao, G. Zhou, F. Li and H.-M. Cheng, Adv. Funct. Mater., 2010, 20, 3595 CrossRef CAS.
- Z. Jiang, J. Wang, L. Meng, Y. Huang and L. Liu, Chem. Commun., 2011, 47, 6350 RSC.
- S. Q. Chen and Y. Wang, J. Mater. Chem., 2010, 20, 9735 RSC.
- D. Cai and M. Song, J. Mater. Chem., 2010, 20, 7906 RSC.
- H. Kim, A. A. Abdala and C. W. Macosko, Macromolecules, 2010, 43, 6515 CrossRef CAS.
- T. Kuilla, S. Bhadra, D. Yao, N. H. Kim, S. Bose and J. H. Lee, Prog. Polym. Sci., 2010, 35, 1350 CrossRef CAS.
- S. Wang, B. M. Goh, K. K. Manga, Q. Bao, P. Yang and K. P. Loh, ACS Nano, 2010, 4, 6180 CrossRef CAS.
- T. H. Han, W. J. Lee, D. H. Lee, J. E. Kim, E.-Y. Choi and S. O. Kim, Adv. Mater., 2010, 22, 2060 CrossRef CAS.
- C.-H. Lu, H.-H. Yang, C.-L. Zhu, X. Chen and G.-N. Chen, Angew. Chem., 2009, 121, 4879 CrossRef.
- Y. Wang, Z. Li, D. Hu, C.-T. Lin, J. Li and Y. Lin, J. Am. Chem. Soc., 2010, 132, 9274 CrossRef CAS.
- M. Jahan, Q. Bao, J.-X. Yang and K. P. Loh, J. Am. Chem. Soc., 2010, 132, 14487 CrossRef CAS.
- C. Petit and T. J. Bandosz, Adv. Funct. Mater., 2010, 20, 111 CrossRef CAS.
- C. Petit, J. Burress and T. J. Bandosz, Carbon, 2011, 49, 563 CrossRef CAS.
- V. C. Tung, L.-M. Chen, M. J. Allen, J. K. Wassei, K. Nelson, R. B. Kaner and Y. Yang, Nano Lett., 2009, 9, 1949 CrossRef CAS.
- E. Yoo, J. Kim, E. Hosono, H.-s. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277 CrossRef CAS.
- D. Yu and L. Dai, J. Phys. Chem. Lett., 2009, 1, 467 Search PubMed.
- Z. Fan, J. Yan, L. Zhi, Q. Zhang, T. Wei, J. Feng, M. Zhang, W. Qian and F. Wei, Adv. Mater., 2010, 22, 3723 CrossRef CAS.
- Z.-B. Liu, Y.-F. Xu, X.-Y. Zhang, X.-L. Zhang, Y.-S. Chen and J.-G. Tian, J. Phys. Chem. B, 2009, 113, 9681 CrossRef CAS.
- J. Xiang and L. T. Drzal, ACS Appl. Mater. Interfaces, 2011, 3, 1325 CrossRef CAS.
- Z. Xiong, L. L. Zhang, J. Ma and X. S. Zhao, Chem. Commun., 2010, 46, 6099 RSC.
- G. Gonçalves, P. A. A. P. Marques, C. M. Granadeiro, H. I. S. Nogueira, M. K. Singh and J. Grácio, Chem. Mater., 2009, 21, 4796 CrossRef CAS.
- R. Muszynski, B. Seger and P. V. Kamat, J. Phys. Chem. C, 2008, 112, 5263 CrossRef CAS.
- Z. Zhang, H. Chen, C. Xing, M. Guo, F. Xu, X. Wang, H. Gruber, B. Zhang and J. Tang, Nano Res., 2011, 4, 599 CrossRef CAS.
- C. Xu, X. Wang and J. Zhu, J. Phys. Chem. C, 2008, 112, 19841 CrossRef CAS.
- F. Li, H. Yang, C. Shan, Q. Zhang, D. Han, A. Ivaska and L. Niu, J. Mater. Chem., 2009, 19, 4022 RSC.
- J. Liu, Y. Li, Y. Li, J. Li and Z. Deng, J. Mater. Chem., 2010, 20, 900 RSC.
- Y. Fang, S. Guo, C. Zhu, Y. Zhai and E. Wang, Langmuir, 2010, 26, 11277 CrossRef CAS.
- J. Gong, T. Zhou, D. Song and L. Zhang, Sens. Actuators, B, 2010, 150, 491 CrossRef.
- W. Hong, H. Bai, Y. Xu, Z. Yao, Z. Gu and G. Shi, J. Phys. Chem. C, 2010, 114, 1822 CrossRef CAS.
- Y.-K. Kim, H.-K. Na and D.-H. Min, Langmuir, 2010, 26, 13065 CrossRef CAS.
- X. Huang, X. Zhou, S. Wu, Y. Wei, X. Qi, J. Zhang, F. Boey and H. Zhang, Small, 2010, 6, 513 CrossRef CAS.
- K. Jasuja and V. Berry, ACS Nano, 2009, 3, 2358 CrossRef CAS.
- B.-S. Kong, J. Geng and H.-T. Jung, Chem. Commun., 2009, 2174 RSC.
- C. Fu, Y. Kuang, Z. Huang, X. Wang, N. Du, J. Chen and H. Zhou, Chem. Phys. Lett., 2010, 499, 250 CrossRef CAS.
- Y. Hu, H. Zhang, P. Wu, H. Zhang, B. Zhou and C. Cai, Phys. Chem. Chem. Phys., 2011, 13, 4083 RSC.
- Z. Tang, S. Shen, J. Zhuang and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 4603 CrossRef CAS.
- J. Liu, S. Fu, B. Yuan, Y. Li and Z. Deng, J. Am. Chem. Soc., 2010, 132, 7279 CrossRef CAS.
- G. Lu, H. Li, C. Liusman, Z. Yin, S. Wu and H. Zhang, Chem. Sci., 2011, 2, 1817 RSC.
- S. Guo, S. Dong and E. Wang, ACS Nano, 2009, 4, 547.
- S. Guo, D. Wen, Y. Zhai, S. Dong and E. Wang, ACS Nano, 2010, 4, 3959 CrossRef CAS.
- L. Dong, R. R. S. Gari, Z. Li, M. M. Craig and S. Hou, Carbon, 2010, 48, 781 CrossRef CAS.
- Y. Li, W. Gao, L. Ci, C. Wang and P. M. Ajayan, Carbon, 2010, 48, 1124 CrossRef CAS.
- Y. Si and E. T. Samulski, Chem. Mater., 2008, 20, 6792 CrossRef CAS.
- Y. Li, L. Tang and J. Li, Electrochem. Commun., 2009, 11, 846 CrossRef CAS.
- Y. Shao, S. Zhang, C. Wang, Z. Nie, J. Liu, Y. Wang and Y. Lin, J. Power Sources, 2010, 195, 4600 CrossRef CAS.
- S. M. Choi, M. H. Seo, H. J. Kim and W. B. Kim, Carbon, 2011, 49, 904 CrossRef CAS.
- Y.-G. Zhou, J.-J. Chen, F.-b. Wang, Z.-H. Sheng and X.-H. Xia, Chem. Commun., 2010, 46, 5951 RSC.
- P. Kundu, C. Nethravathi, P. A. Deshpande, M. Rajamathi, G. Madras and N. Ravishankar, Chem. Mater., 2011, 23, 2772 CrossRef CAS.
- S. Bong, Y.-R. Kim, I. Kim, S. Woo, S. Uhm, J. Lee and H. Kim, Electrochem. Commun., 2010, 12, 129 CrossRef CAS.
- M. H. Seo, S. M. Choi, H. J. Kim and W. B. Kim, Electrochem. Commun., 2011, 13, 182 CrossRef CAS.
- H. J. Kim, S. M. Choi, M. H. Seo, S. Green, G. W. Huber and W. B. Kim, Electrochem. Commun., 2011, 13, 890 CrossRef CAS.
- B. Seger and P. V. Kamat, J. Phys. Chem. C, 2009, 113, 7990 CrossRef CAS.
- G. Gupta, D. A. Slanac, P. Kumar, J. D. Wiggins-Camacho, J. Kim, R. Ryoo, K. J. Stevenson and K. P. Johnston, J. Phys. Chem. C, 2010, 114, 10796 CrossRef CAS.
- N. Shang, P. Papakonstantinou, P. Wang and S. R. P. Silva, J. Phys. Chem. C, 2010, 114, 15837 CrossRef CAS.
- M. Bayati, J. M. Abad, R. J. Nichols and D. J. Schiffrin, J. Phys. Chem. C, 2010, 114, 18439 CrossRef CAS.
- S. Sharma, A. Ganguly, P. Papakonstantinou, X. Miao, M. Li, J. L. Hutchison, M. Delichatsios and S. Ukleja, J. Phys. Chem. C, 2010, 114, 19459 CrossRef CAS.
- K. Zhang, Q. Yue, G. Chen, Y. Zhai, L. Wang, H. Wang, J. Zhao, J. Liu, J. Jia and H. Li, J. Phys. Chem. C, 2010, 115, 379.
- E. Yoo, T. Okada, T. Akita, M. Kohyama, I. Honma and J. Nakamura, J. Power Sources, 2011, 196, 110 CrossRef CAS.
- C. Zhu, S. Guo, Y. Zhai and S. Dong, Langmuir, 2010, 26, 7614 CrossRef CAS.
- E. Yoo, T. Okata, T. Akita, M. Kohyama, J. Nakamura and I. Honma, Nano Lett., 2009, 9, 2255 CrossRef CAS.
- R. S. Sundaram, C. Gómez-Navarro, K. Balasubramanian, M. Burghard and K. Kern, Adv. Mater., 2008, 20, 3050 CrossRef CAS.
- J. L. Johnson, A. Behnam, S. J. Pearton and A. Ural, Adv. Mater., 2010, 22, 4877 CrossRef CAS.
- Á. Mastalir, Z. Király, M. Benkő and I. Dékány, Catal. Lett., 2008, 124, 34 CrossRef CAS.
- H. Zhao, J. Yang, L. Wang, C. Tian, B. Jiang and H. Fu, Chem. Commun., 2011, 47, 2014 RSC.
- Z. Jin, D. Nackashi, W. Lu, C. Kittrell and J. M. Tour, Chem. Mater., 2010, 22, 5695 CrossRef CAS.
- J. Yang, C. Tian, L. Wang and H. Fu, J. Mater. Chem., 2011, 21, 3384 RSC.
- Y. Li, X. Fan, J. Qi, J. Ji, S. Wang, G. Zhang and F. Zhang, Nano Res., 2010, 3, 429 CrossRef CAS.
- Z. Wen, S. Yang, Y. Liang, W. He, H. Tong, L. Hao, X. Zhang and Q. Song, Electrochim. Acta, 2010, 56, 139 CrossRef CAS.
- A. R. Siamaki, A. E. R. S. Khder, V. Abdelsayed, M. S. El-Shall and B. F. Gupton, J. Catal., 2011, 279, 1 CrossRef CAS.
- H. M. A. Hassan, V. Abdelsayed, A. E. R. S. Khder, K. M. AbouZeid, J. Terner, M. S. El-Shall, S. I. Al-Resayes and A. A. El-Azhary, J. Mater. Chem., 2009, 19, 3832 RSC.
- Z.-L. Hu, M. Aizawa, Z.-M. Wang, N. Yoshizawa and H. Hatori, Langmuir, 2010, 26, 6681 CrossRef CAS.
- J. Lee, K. S. Novoselov and H. S. Shin, ACS Nano, 2010, 5, 608.
- X. Zhou, X. Huang, X. Qi, S. Wu, C. Xue, F. Y. C. Boey, Q. Yan, P. Chen and H. Zhang, J. Phys. Chem. C, 2009, 113, 10842 CrossRef CAS.
- G. Lu, S. Mao, S. Park, R. Ruoff and J. Chen, Nano Res., 2009, 2, 192 CrossRef CAS.
- C. Xu and X. Wang, Small, 2009, 5, 2212 CrossRef CAS.
- J. Shen, M. Shi, B. Yan, H. Ma, N. Li and M. Ye, J. Mater. Chem., 2011, 21, 7795 RSC.
- J. Li and C.-y. Liu, Eur. J. Inorg. Chem., 2010, 2010, 1244 CrossRef.
- Z. Zhang, F. Xu, W. Yang, M. Guo, X. Wang, B. Zhang and J. Tang, Chem. Commun., 2011, 47, 6440 RSC.
- Z. Xu, H. Gao and H. Guoxin, Carbon, 2011, 49, 4731 CrossRef CAS.
- T. T. Baby and S. Ramaprabhu, J. Mater. Chem., 2011, 21, 9702 RSC.
- J. Yang, C. Zang, L. Sun, N. Zhao and X. Cheng, Mater. Chem. Phys., 2011, 129, 270 CrossRef CAS.
- Y.-H. Zou, H.-B. Liu, L. Yang and Z.-Z. Chen, J. Magn. Magn. Mater., 2006, 302, 343 CrossRef CAS.
- M. Stein, J. Wieland, P. Steurer, F. Tölle, R. Mülhaupt and B. Breit, Adv. Synth. Catal., 2011, 353, 523 CrossRef CAS.
- X. Bin, J. Chen, H. Cao, L. Chen and J. Yuan, J. Phys. Chem. Solids, 2009, 70, 1 CrossRef CAS.
- S. Yang, G. Cui, S. Pang, Q. Cao, U. Kolb, X. Feng, J. Maier and K. Müllen, ChemSusChem, 2010, 3, 236 CrossRef CAS.
- H. Paul and D. Mohanta, Appl. Phys. A: Mater. Sci. Process., 2011, 103, 395 CrossRef CAS.
- G. Wang, B. Wang, X. Wang, J. Park, S. Dou, H. Ahn and K. Kim, J. Mater. Chem., 2009, 19, 8378 RSC.
- K. Zhou, Y. Zhu, X. Yang and C. Li, Electroanalysis, 2010, 22, 259 CrossRef CAS.
- Z.-J. Fan, J. Yan, T. Wei, G.-Q. Ning, L.-J. Zhi, J.-C. Liu, D.-X. Cao, G.-L. Wang and F. Wei, ACS Nano, 2011, 5, 2787 CrossRef CAS.
- Y. Lin, D. W. Baggett, J.-W. Kim, E. J. Siochi and J. W. Connell, ACS Appl. Mater. Interfaces, 2011, 3, 1652 CrossRef CAS.
- Y.-K. Kim, H.-K. Na, Y. W. Lee, H. Jang, S. W. Han and D.-H. Min, Chem. Commun., 2010, 46, 3185 RSC.
- R. Guoqiang and X. Yangchuan, Nanotechnology, 2006, 17, 5596 CrossRef CAS.
- H. Zhou, C. Qiu, Z. Liu, H. Yang, L. Hu, J. Liu, H. Yang, C. Gu and L. Sun, J. Am. Chem. Soc., 2009, 132, 944.
- I. V. Lightcap, T. H. Kosel and P. V. Kamat, Nano Lett., 2010, 10, 577 CrossRef CAS.
- Y. Lin, K. A. Watson, M. J. Fallbach, S. Ghose, J. G. Smith, D. M. Delozier, W. Cao, R. E. Crooks and J. W. Connell, ACS Nano, 2009, 3, 871 CrossRef CAS.
- L.-S. Zhang, X.-Q. Liang, W.-G. Song and Z.-Y. Wu, Phys. Chem. Chem. Phys., 2010, 12, 12055 RSC.
- K. K. Manga, Y. Zhou, Y. Yan and K. P. Loh, Adv. Funct. Mater., 2009, 19, 3638 CrossRef CAS.
- C. Zhu, S. Guo, P. Wang, L. Xing, Y. Fang, Y. Zhai and S. Dong, Chem. Commun., 2010, 46, 7148 RSC.
- B. Li, X. Zhang, X. Li, L. Wang, R. Han, B. Liu, W. Zheng, X. Li and Y. Liu, Chem. Commun., 2010, 46, 3499 RSC.
- C. Chen, W. Cai, M. Long, B. Zhou, Y. Wu, D. Wu and Y. Feng, ACS Nano, 2010, 4, 6425 CrossRef CAS.
- T. Kamegawa, D. Yamahana and H. Yamashita, J. Phys. Chem. C, 2010, 114, 15049 CAS.
- Y. Liang, H. Wang, H. Sanchez Casalongue, Z. Chen and H. Dai, Nano Res., 2010, 3, 701 CrossRef CAS.
- Y. Zhang, Z.-R. Tang, X. Fu and Y.-J. Xu, ACS Nano, 2010, 4, 7303 CrossRef CAS.
- J. Zhang, Z. Xiong and X. S. Zhao, J. Mater. Chem., 2011, 21, 3634 RSC.
- F. Zou, Y. Yu, N. Cao, L. Wu and J. Zhi, Scr. Mater., 2011, 64, 621 CrossRef CAS.
- Q. Wang, X. Guo, L. Cai, Y. Cao, L. Gan, S. Liu, Z. Wang, H. Zhang and L. Li, Chem. Sci., 2011, 2, 1860 RSC.
- J. Shen, B. Yan, M. Shi, H. Ma, N. Li and M. Ye, J. Mater. Chem., 2011, 21, 3415 RSC.
- S. Ding, J. S. Chen, D. Luan, F. Y. C. Boey, S. Madhavi and X. W. Lou, Chem. Commun., 2011, 47, 5780 RSC.
- P. Wang, Y. Zhai, D. Wang and S. Dong, Nanoscale, 2011, 3, 1640 RSC.
- R. Leary and A. Westwood, Carbon, 2011, 49, 741 CrossRef CAS.
- T. N. Lambert, C. A. Chavez, B. Hernandez-Sanchez, P. Lu, N. S. Bell, A. Ambrosini, T. Friedman, T. J. Boyle, D. R. Wheeler and D. L. Huber, J. Phys. Chem. C, 2009, 113, 19812 CrossRef CAS.
- D. Wang, D. Choi, J. Li, Z. Yang, Z. Nie, R. Kou, D. Hu, C. Wang, L. V. Saraf, J. Zhang, I. A. Aksay and J. Liu, ACS Nano, 2009, 3, 907 CrossRef CAS.
- O. Akhavan, M. Abdolahad, A. Esfandiar and M. Mohatashamifar, J. Phys. Chem. C, 2010, 114, 12955 CrossRef CAS.
- Y.-B. Tang, C.-S. Lee, J. Xu, Z.-T. Liu, Z.-H. Chen, Z. He, Y.-L. Cao, G. Yuan, H. Song, L. Chen, L. Luo, H.-M. Cheng, W.-J. Zhang, I. Bello and S.-T. Lee, ACS Nano, 2010, 4, 3482 CrossRef CAS.
- S. Sun, L. Gao and Y. Liu, Appl. Phys. Lett., 2010, 96, 083113 CrossRef.
- Y. Wang, R. Shi, J. Lin and Y. Zhu, Appl. Catal., B, 2010, 100, 179 CrossRef CAS.
- Y. Qiu, K. Yan, S. Yang, L. Jin, H. Deng and W. Li, ACS Nano, 2010, 4, 6515 CrossRef CAS.
- G. Jiang, Z. Lin, C. Chen, L. Zhu, Q. Chang, N. Wang, W. Wei and H. Tang, Carbon, 2011, 49, 2693 CrossRef CAS.
- L. Zhang, S. Diao, Y. Nie, K. Yan, N. Liu, B. Dai, Q. Xie, A. Reina, J. Kong and Z. Liu, J. Am. Chem. Soc., 2011, 133, 2706 CrossRef CAS.
- X. Zhang, Y. Sun, X. Cui and Z. Jiang, Int. J. Hydrogen Energy DOI:10.1016/j.ijhydene.2011.04.053.
- K. K. Manga, S. Wang, M. Jaiswal, Q. Bao and K. P. Loh, Adv. Mater., 2010, 22, 5265 CrossRef CAS.
- S. Wu, Z. Yin, Q. He, X. Huang, X. Zhou and H. Zhang, J. Phys. Chem. C, 2010, 114, 11816 CrossRef CAS.
- Z. Yin, S. Wu, X. Zhou, X. Huang, Q. Zhang, F. Boey and H. Zhang, Small, 2010, 6, 307 CrossRef CAS.
- J. M. Lee, Y. B. Pyun, J. Yi, J. W. Choung and W. I. Park, J. Phys. Chem. C, 2009, 113, 19134 CrossRef CAS.
- J. Lin, M. Penchev, G. Wang, R. K. Paul, J. Zhong, X. Jing, M. Ozkan and C. S. Ozkan, Small, 2010, 6, 2448 CrossRef CAS.
- Y. Zhang, H. Li, L. Pan, T. Lu and Z. Sun, J. Electroanal. Chem., 2009, 634, 68 CrossRef CAS.
- W. T. Zheng, Y. M. Ho, H. W. Tian, M. Wen, J. L. Qi and Y. A. Li, J. Phys. Chem. C, 2009, 113, 9164 CrossRef CAS.
- J. O. Hwang, D. H. Lee, J. Y. Kim, T. H. Han, B. H. Kim, M. Park, K. No and S. O. Kim, J. Mater. Chem., 2011, 21, 3432 RSC.
- T. V. Cuong, V. H. Pham, J. S. Chung, E. W. Shin, D. H. Yoo, S. H. Hahn, J. S. Huh, G. H. Rue, E. J. Kim, S. H. Hur and P. A. Kohl, Mater. Lett., 2010, 64, 2479 CrossRef CAS.
- K. Prashant, et al. , Nanotechnology, 2010, 21, 385701 CrossRef.
- X. Liu, L. Pan, T. Lv, T. Lu, G. Zhu, Z. Sun and C. Sun, Catal.: Sci. Technol., 2011, 1, 1189 RSC.
- G. Williams and P. V. Kamat, Langmuir, 2009, 25, 13869 CrossRef CAS.
- Y. Yang and T. Liu, Appl. Surf. Sci., 2011, 257, 8950 CrossRef CAS.
- B. Li and H. Cao, J. Mater. Chem., 2011, 21, 3346 RSC.
- T. Xu, L. Zhang, H. Cheng and Y. Zhu, Appl. Catal., B, 2011, 101, 382 CrossRef CAS.
- S.-M. Paek, E. Yoo and I. Honma, Nano Lett., 2008, 9, 72.
- D. Wang, R. Kou, D. Choi, Z. Yang, Z. Nie, J. Li, L. V. Saraf, D. Hu, J. Zhang, G. L. Graff, J. Liu, M. A. Pope and I. A. Aksay, ACS Nano, 2010, 4, 1587 CrossRef CAS.
- L.-S. Zhang, L.-Y. Jiang, H.-J. Yan, W. D. Wang, W. Wang, W.-G. Song, Y.-G. Guo and L.-J. Wan, J. Mater. Chem., 2010, 20, 5462 RSC.
- L. Fenghua, et al. , Nanotechnology, 2009, 20, 455602 CrossRef.
- H. Song, L. Zhang, C. He, Y. Qu, Y. Tian and Y. Lv, J. Mater. Chem., 2011, 21, 5972 RSC.
- Y. Li, X. Lv, J. Lu and J. Li, J. Phys. Chem. C, 2010, 114, 21770 CrossRef CAS.
- H. Kim, S.-W. Kim, Y.-U. Park, H. Gwon, D.-H. Seo, Y. Kim and K. Kang, Nano Res., 2010, 3, 813 CrossRef CAS.
- Z. Wang, H. Zhang, N. Li, Z. Shi, Z. Gu and G. Cao, Nano Res., 2010, 3, 748 CrossRef CAS.
- J. Yao, X. Shen, B. Wang, H. Liu and G. Wang, Electrochem. Commun., 2009, 11, 1849 CrossRef CAS.
- P. Lian, X. Zhu, S. Liang, Z. Li, W. Yang and H. Wang, Electrochim. Acta, 2011, 56, 4532 CrossRef CAS.
- X. Wang, X. Zhou, K. Yao, J. Zhang and Z. Liu, Carbon, 2011, 49, 133 CrossRef CAS.
- M. Zhang, D. Lei, Z. Du, X. Yin, L. Chen, Q. Li, Y. Wang and T. Wang, J. Mater. Chem., 2011, 21, 1673 RSC.
- S. Chen, J. Zhu and X. Wang, ACS Nano, 2010, 4, 6212 CrossRef CAS.
- L. Li, Z. Du, S. Liu, Q. Hao, Y. Wang, Q. Li and T. Wang, Talanta, 2010, 82, 1637 CrossRef CAS.
- Z. Fan, J. Yan, T. Wei, L. Zhi, G. Ning, T. Li and F. Wei, Adv. Funct. Mater., 2011, 21, 2366 CrossRef CAS.
- J. Yan, Z. Fan, T. Wei, W. Qian, M. Zhang and F. Wei, Carbon, 2010, 48, 3825 CrossRef CAS.
- C. Zhu, S. Guo, Y. Fang, L. Han, E. Wang and S. Dong, Nano Res., 2011, 4, 648 CrossRef CAS.
- Q. Cheng, J. Tang, J. Ma, H. Zhang, N. Shinya and L.-C. Qin, Carbon, 2011, 49, 2917 CrossRef CAS.
- Z. Li, J. Wang, S. Liu, X. Liu and S. Yang, J. Power Sources, 2011, 196, 8160 CrossRef CAS.
- G. Yu, L. Hu, M. Vosgueritchian, H. Wang, X. Xie, J. R. McDonough, X. Cui, Y. Cui and Z. Bao, Nano Lett., 2011, 11, 2905 CrossRef CAS.
- L. Xing, C. Cui, C. Ma and X. Xue, Mater. Lett., 2011, 65, 2104 CrossRef CAS.
- G. Zhou, D.-W. Wang, F. Li, L. Zhang, N. Li, Z.-S. Wu, L. Wen, G. Q. Lu and H.-M. Cheng, Chem. Mater., 2010, 22, 5306 CrossRef CAS.
- P. Lian, X. Zhu, H. Xiang, Z. Li, W. Yang and H. Wang, Electrochim. Acta, 2010, 56, 834 CrossRef CAS.
- F. He, J. Fan, D. Ma, L. Zhang, C. Leung and H. L. Chan, Carbon, 2010, 48, 3139 CrossRef CAS.
- K. Morishige and T. Hamada, Langmuir, 2005, 21, 6277 CrossRef CAS.
- X. Yang, X. Zhang, Y. Ma, Y. Huang, Y. Wang and Y. Chen, J. Mater. Chem., 2009, 19, 2710 RSC.
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I.-C. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979 CrossRef CAS.
- M. Zhang, D. Lei, X. Yin, L. Chen, Q. Li, Y. Wang and T. Wang, J. Mater. Chem., 2010, 20, 5538 RSC.
- J. Shen, Y. Hu, M. Shi, N. Li, H. Ma and M. Ye, J. Phys. Chem. C, 2010, 114, 1498 CrossRef CAS.
- J. Liang, Y. Xu, D. Sui, L. Zhang, Y. Huang, Y. Ma, F. Li and Y. Chen, J. Phys. Chem. C, 2010, 114, 17465 CrossRef CAS.
- H. Yin, Y. Zhou, Q. Ma, S. Ai, Q. Chen and L. Zhu, Talanta, 2010, 82, 1193 CrossRef CAS.
- H. He and C. Gao, ACS Appl. Mater. Interfaces, 2010, 2, 3201 CrossRef CAS.
- W. Shi, J. Zhu, D. H. Sim, Y. Y. Tay, Z. Lu, X. Zhang, Y. Sharma, M. Srinivasan, H. Zhang, H. H. Hng and Q. Yan, J. Mater. Chem., 2011, 21, 3422 RSC.
- B. Li, H. Cao, J. Shao, M. Qu and J. H. Warner, J. Mater. Chem., 2011, 21, 5069 RSC.
- G. Wang, T. Liu, X. Xie, Z. Ren, J. Bai and H. Wang, Mater. Chem. Phys., 2011, 128, 336 CrossRef CAS.
- J. Zhu, T. Zhu, X. Zhou, Y. Zhang, X. W. Lou, X. Chen, H. Zhang, H. H. Hng and Q. Yan, Nanoscale, 2011, 3, 1084 RSC.
- V. K. Singh, M. K. Patra, M. Manoth, G. S. Gowd, S. R. Vadera and N. Kumar, New Carbon Mater., 2009, 24, 147 Search PubMed.
- X. Zhu, Y. Zhu, S. Murali, M. D. Stoller and R. S. Ruoff, ACS Nano, 2011, 5, 3333 CrossRef CAS.
- S. Bashkova and T. J. Bandosz, Ind. Eng. Chem. Res., 2009, 48, 10884 CrossRef CAS.
- G. Wang, T. Liu, Y. Luo, Y. Zhao, Z. Ren, J. Bai and H. Wang, J. Alloys Compd., 2011, 509, L216 CrossRef CAS.
- Z.-S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H.-M. Cheng, ACS Nano, 2010, 4, 3187 CrossRef CAS.
- S. Yang, X. Feng, S. Ivanovici and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 8408 CrossRef CAS.
- H. Kim, D.-H. Seo, S.-W. Kim, J. Kim and K. Kang, Carbon, 2011, 49, 326 CrossRef CAS.
- J. Yan, T. Wei, W. Qiao, B. Shao, Q. Zhao, L. Zhang and Z. Fan, Electrochim. Acta, 2010, 55, 6973 CrossRef CAS.
- S. Yang, X. Feng, L. Wang, K. Tang, J. Maier and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 4795 CAS.
- B. Wang, Y. Wang, J. Park, H. Ahn and G. Wang, J. Alloys Compd., 2011, 509, 7778 CrossRef CAS.
- Y. Zou and Y. Wang, Nanoscale, 2011, 3, 2615 RSC.
- I. R. M. Kottegoda, N. H. Idris, L. Lu, J.-Z. Wang and H.-K. Liu, Electrochim. Acta, 2011, 56, 5815 CrossRef CAS.
- Z. Ji, J. Wu, X. Shen, H. Zhou and H. Xi, J. Mater. Sci., 2011, 46, 1190 CrossRef CAS.
- H. Yang, G. H. Guai, C. Guo, Q. Song, S. P. Jiang, Y. Wang, W. Zhang and C. M. Li, J. Phys. Chem. C, 2011, 115, 12209 CrossRef CAS.
- D. Du, J. Liu, X. Zhang, X. Cui and Y. Lin, J. Mater. Chem., 2011, 21, 8032 RSC.
- S. Watcharotone, D. A. Dikin, S. Stankovich, R. Piner, I. Jung, G. H. B. Dommett, G. Evmenenko, S.-E. Wu, S.-F. Chen, C.-P. Liu, S. T. Nguyen and R. S. Ruoff, Nano Lett., 2007, 7, 1888 CrossRef CAS.
- S. Wu, Z. Yin, Q. He, G. Lu, X. Zhou and H. Zhang, J. Mater. Chem., 2011, 21, 3467 RSC.
- C. Xu, X. Wang, L. Yang and Y. Wu, J. Solid State Chem., 2009, 182, 2486 CrossRef CAS.
- B. Li, H. Cao, G. Yin, Y. Lu and J. Yin, J. Mater. Chem., 2011, 21, 10645 RSC.
- F. Kim, J. Luo, R. Cruz-Silva, L. J. Cote, K. Sohn and J. Huang, Adv. Funct. Mater., 2010, 20, 2867 CrossRef CAS.
- H. Wang, Y. Liang, T. Mirfakhrai, Z. Chen, H. Casalongue and H. Dai, Nano Res., 2011, 4, 729 CrossRef CAS.
- X. Wang, S. M. Tabakman and H. Dai, J. Am. Chem. Soc., 2008, 130, 8152 CrossRef CAS.
- Y. Fan, L. Wang, J. Li, J. Li, S. Sun, F. Chen, L. Chen and W. Jiang, Carbon, 2010, 48, 1743 CrossRef CAS.
- K. Wang, Y. Wang, Z. Fan, J. Yan and T. Wei, Mater. Res. Bull., 2011, 46, 315 CrossRef CAS.
- J.-H. Park, S.-W. Seo, J.-H. Kim, C.-J. Choi, H. Kim, D. K. Lee, W.-S. Jung and K.-S. Ahn, Mol. Cryst. Liq. Cryst., 2011, 538, 285 CAS.
- X. Yang, C. Lu, J. Qin, R. Zhang, H. Tang and H. Song, Mater. Lett., 2011, 65, 2341 CrossRef CAS.
- L. Xie, X. Wang, H. Mao, R. Wang, M. Ding, Y. Wang, B. Özyilmaz, K. P. Loh, A. T. S. Wee, Ariando and W. Chen, Appl. Phys. Lett., 2011, 99, 012112 CrossRef.
- Y. Fu and X. Wang, Ind. Eng. Chem. Res., 2011, 50, 7210 CrossRef CAS.
- E. Gao, W. Wang, M. Shang and J. Xu, Phys. Chem. Chem. Phys., 2011, 13, 2887 RSC.
- Y. Ding, Y. Jiang, F. Xu, J. Yin, H. Ren, Q. Zhuo, Z. Long and P. Zhang, Electrochem. Commun., 2010, 12, 10 CrossRef CAS.
- L. Wang, H. Wang, Z. Liu, C. Xiao, S. Dong, P. Han, Z. Zhang, X. Zhang, C. Bi and G. Cui, Solid State Ionics, 2010, 181, 1685 CrossRef CAS.
- C. Nethravathi, T. Nisha, N. Ravishankar, C. Shivakumara and M. Rajamathi, Carbon, 2009, 47, 2054 CrossRef CAS.
- A. Cao, Z. Liu, S. Chu, M. Wu, Z. Ye, Z. Cai, Y. Chang, S. Wang, Q. Gong and Y. Liu, Adv. Mater., 2010, 22, 103 CrossRef CAS.
- J. Wu, S. Bai, X. Shen and L. Jiang, Appl. Surf. Sci., 2010, 257, 747 CrossRef CAS.
- T. Dufaux, J. Boettcher, M. Burghard and K. Kern, Small, 2010, 6, 1868 CrossRef CAS.
- F. Miao, et al , Nanotechnology, 2010, 21, 075601 CrossRef.
- K. Wang, Q. Liu, X.-Y. Wu, Q.-M. Guan and H.-N. Li, Talanta, 2010, 82, 372 CrossRef CAS.
- X. Geng, L. Niu, Z. Xing, R. Song, G. Liu, M. Sun, G. Cheng, H. Zhong, Z. Liu, Z. Zhang, L. Sun, H. Xu, L. Lu and L. Liu, Adv. Mater., 2010, 22, 638 CrossRef CAS.
- Y. Lin, K. Zhang, W. Chen, Y. Liu, Z. Geng, J. Zeng, N. Pan, L. Yan, X. Wang and J. G. Hou, ACS Nano, 2010, 4, 3033 CrossRef CAS.
- Y.-T. Kim, J. H. Han, B. H. Hong and Y.-U. Kwon, Adv. Mater., 2010, 22, 515 CrossRef CAS.
- A. F. Zedan, S. Sappal, S. Moussa and M. S. El-Shall, J. Phys. Chem. C, 2010, 114, 19920 CrossRef CAS.
- L.-L. Li, K.-P. Liu, G.-H. Yang, C.-M. Wang, J.-R. Zhang and J.-J. Zhu, Adv. Funct. Mater., 2011, 21, 869 CrossRef CAS.
- N. Li, G. Liu, C. Zhen, F. Li, L. Zhang and H.-M. Cheng, Adv. Funct. Mater., 2011, 21, 1717 CrossRef CAS.
- Y. Park, S.-H. Kang and W. Choi, Phys. Chem. Chem. Phys., 2011, 13, 9425 RSC.
- H. Wang, L.-F. Cui, Y. Yang, H. Sanchez Casalongue, J. T. Robinson, Y. Liang, Y. Cui and H. Dai, J. Am. Chem. Soc., 2010, 132, 13978 CrossRef CAS.
- N. D. Mermin, Phys. Rev., 1968, 176, 250 CrossRef.
- D. A. C. Brownson, D. K. Kampouris and C. E. Banks, J. Power Sources, 2011, 196, 4873 CAS.
- Y. Wang, Z. Shi, Y. Huang, Y. Ma, C. Wang, M. Chen and Y. Chen, J. Phys. Chem. C, 2009, 113, 13103 CrossRef CAS.
- M. Pumera, Chem. Rec., 2009, 9, 211 CrossRef CAS.
- P. V. Kamat, J. Phys. Chem. Lett., 2011, 2, 242 Search PubMed.
- Y. H. Hu, H. Wang and B. Hu, ChemSusChem, 2010, 3, 782 CrossRef CAS.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012 10.1039/C1CS15078B.
- S. Sarkar, S. Niyogi, E. Bekyarova and R. C. Haddon, Chem. Sci., 2011, 2, 1326 RSC.
- N. F. Goldshleger, Fullerene Sci. Technol., 2001, 9, 255 CrossRef CAS.
- B. Li and Z. Xu, J. Am. Chem. Soc., 2009, 131, 16380 CrossRef CAS.
- N. Muradov, Catal. Commun., 2001, 2, 89 CrossRef CAS.
- M. Croston, J. Langston, R. Sangoi and K. S. V. Santhanam, Int. J. Nanosci., 2002, 1, 277 CrossRef CAS.
- M. Croston, J. Langston, G. Takacs, T. C. Morrill, M. Miri, K. S. V. Santhanam and P. Ajayan, Int. J. Nanosci., 2002, 1, 285 CrossRef CAS.
- D. S. Su, N. Maksimova, J. J. Delgado, N. Keller, G. Mestl, M. J. Ledoux and R. Schlögl, Catal. Today, 2005, 102–103, 110 CrossRef CAS.
- J. Zhang, X. Liu, R. Blume, A. Zhang, R. Schlögl and D. S. Su, Science, 2008, 322, 73 CrossRef CAS.
- B. Frank, J. Zhang, R. Blume, R. Schlögl and D. S. Su, Angew. Chem., Int. Ed., 2009, 48, 6913 CrossRef CAS.
- M. F. R. Pereira, J. J. M. Orfão and J. L. Figueiredo, Appl. Catal., A, 2000, 196, 43 CrossRef CAS.
- G. Mestl, N. I. Maksimova, N. Keller, V. V. Roddatis and R. Schlögl, Angew. Chem., Int. Ed., 2001, 40, 2066 CrossRef CAS.
- N. Keller, N. I. Maksimova, V. V. Roddatis, M. Schur, G. Mestl, Y. V. Butenko, V. L. Kuznetsov and R. Schlögl, Angew. Chem., Int. Ed., 2002, 41, 1885 CrossRef CAS.
- M. F. R. Pereira, J. L. Figueiredo, J. J. M. Órfão, P. Serp, P. Kalck and Y. Kihn, Carbon, 2004, 42, 2807 CrossRef CAS.
- B. Frank, R. Blume, A. Rinaldi, A. Trunschke and R. Schlögl, Angew. Chem., Int. Ed., 2011, 50, 10226 CrossRef CAS.
- T.-F. Yeh, J.-M. Syu, C. Cheng, T.-H. Chang and H. Teng, Adv. Funct. Mater., 2010, 20, 2255 CrossRef CAS.
- D. Geng, Y. Chen, Y. Chen, Y. Li, R. Li, X. Sun, S. Ye and S. Knights, Energy Environ. Sci., 2011, 4, 760 RSC.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321 CrossRef CAS.
- Y. Shao, S. Zhang, M. H. Engelhard, G. Li, G. Shao, Y. Wang, J. Liu, I. A. Aksay and Y. Lin, J. Mater. Chem., 2010, 20, 7491 RSC.
- L. Tang, Y. Wang, Y. Li, H. Feng, J. Lu and J. Li, Adv. Funct. Mater., 2009, 19, 2782 CrossRef CAS.
- K. R. Lee, K. U. Lee, J. W. Lee, B. T. Ahn and S. I. Woo, Electrochem. Commun., 2010, 12, 1052 CrossRef CAS.
- D. R. Dreyer, H.-P. Jia and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 6813 CAS.
- H.-P. Jia, D. R. Dreyer and C. W. Bielawski, Adv. Synth. Catal., 2011, 353, 528 CrossRef CAS.
- D. R. Dreyer, S. Murali, Y. Zhu, R. S. Ruoff and C. W. Bielawski, J. Mater. Chem., 2011, 21, 3443 RSC.
- D. R. Dreyer, K. A. Jarvis, P. J. Ferreira and C. W. Bielawski, Macromolecules, 2011, 44, 7659 CrossRef CAS.
- D. R. Dreyer, H.-P. Jia, A. D. Todd, J. Geng and C. W. Bielawski, Org. Biomol. Chem., 2011, 9, 7292 RSC.
- Y. Xin, J.-g. Liu, Y. Zhou, W. Liu, J. Gao, Y. Xie, Y. Yin and Z. Zou, J. Power Sources, 2011, 196, 1012 CrossRef.
- R. Imran Jafri, N. Rajalakshmi and S. Ramaprabhu, J. Mater. Chem., 2010, 20, 7114 RSC.
- Y. Sun, C. Li, Y. Xu, H. Bai, Z. Yao and G. Shi, Chem. Commun., 2010, 46, 4740 RSC.
- S. Yang, X. Feng, X. Wang and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 5339 CrossRef CAS.
- Q. Xiang, J. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355 CrossRef CAS.
- X.-H. Li, J.-S. Chen, X. Wang, J. Sun and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 8074 CrossRef CAS.
- F. Bonaccorso, Z. Sun, T. Hasan and A. C. Ferrari, Nat. Photonics, 2010, 4, 611 CrossRef CAS.
- G. Eda and M. Chhowalla, Adv. Mater., 2010, 22, 2392 CrossRef CAS.
- M. Liang, B. Luo and L. Zhi, Int. J. Energy Res., 2009, 33, 1161 CrossRef CAS.
- J. K. Wassei and R. B. Kaner, Mater. Today, 2010, 13, 52 CrossRef CAS.
- H. Chang, Y. Liu, H. Zhang and J. Li, J. Electroanal. Chem., 2011, 656, 269 CrossRef CAS.
- K. Zhou, Y. Zhu, X. Yang, X. Jiang and C. Li, New J. Chem., 2011, 35, 353 RSC.
- D.-H. Yoo, T. V. Cuong, V. H. Pham, J. S. Chung, N. T. Khoa, E. J. Kim and S. H. Hahn, Curr. Appl. Phys., 2011, 11, 805 CrossRef.
- W. Fan, Q. Lai, Q. Zhang and Y. Wang, J. Phys. Chem. C, 2011, 115, 10694 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |