Clarification of photocatalysis induced by iron ion species naturally contained in a clay compound†
Seiji
Kakuta
*a,
Toru
Okayama
a,
Megumi
Kato
b,
Akihiro
Oda
b and
Toshiyuki
Abe
*c
aNew Energy Technology Section, Aomori Prefectural Industrial Technology Research Center, 221-10, Yamaguchi, Nogi, Aomori 030-0142, Japan. E-mail: seiji_kakuta@aomori-itc.or.jp
bNihon Kohken Inc., 9-2, Hyoganzawa Fukuro, Kuroishi 036-0412, Japan
cDepartment of Frontier Materials Chemistry, Graduate School of Science and Technology, Hirosaki University, 3 Bunkyo-cho, Hirosaki 036-8561, Japan. E-mail: tabe@cc.hirosaki-u.ac.jp
First published on 19th September 2011
Abstract
When clay undergoes isomorphic replacement of a cation (e.g. Al3+, Mg2+) by an Fe2+ ion, the Fe ion in the crystal structure of clay is called structural-Fe. The octahedral sites of a structural-Fe are occupied by O2− ions as in Fe(III) oxide (e.g. α-Fe2O3); however, the photocatalysis of structural-Fe has not yet been understood in detail. In this study, structural-Fe(II) in clay was oxidized to Fe(III), following which the photocatalytic activity of clay was investigated in terms of the degradation of acetic acid. After the oxidation of structural-Fe(II), hydrous ferric oxide (HFO, Fe2O3·nH2O) as well as structural-Fe(III) coexisted as the products. To clarify the intrinsic photocatalyses of both species, clay compounds with varying amounts of HFO were prepared and examined in terms of the photocatalytic degradation. Kinetic analysis revealed that the photocatalytic activity of HFO is 30 times higher than that of structural-Fe(III), indicating that HFO is the main species that causes active and efficient photocatalysis in clay.
Introduction
Photocatalysts activate various types of reactions (e.g. water splitting, degradation of organic pollutants, etc.) under irradiation.1 The increasing demand for active and inexpensive photocatalysts has led to studies focusing mainly on the fabrication of nanomaterials having a small particle size and a large specific surface area.2,3 The use of clay compounds as a photocatalyst supporter has been recognized as a promising method for obtaining nanosized photocatalysts with a large specific surface area.4–6 Natural montmorillonite (MMT), a typical compound of clay, is composed of two silica tetrahedral sheets and an alumina octahedral sheet (Fig. 1).7 The intersheet layers of MMT include exchangeable cations (e.g. Na+ and Ca2+). These cations neutralize the negative charges generated by the partial substitution of Al3+ with Mg2+ or Fe2+ at the octahedral sites. A photocatalyst loaded on clay can usually be fabricated by the calcination of a precursor (e.g. metal ion, metal complex, etc.) incorporated through cation exchange.4–6 Iron(III) oxide (Fe2O3) embedded into clay is a typical example of a photocatalyst; it has been applied to the degradation of organic pollutants.8,9 Fe3+ ions in Fe2O3 are similar to those naturally contained in clay, i.e. structural-Fe(Z) (where Z represents the oxidation numbers, II or III in this case), particularly in terms of crystal structure.10 That is, the O2− ions in Fe2O3 occupy the octahedral sites of an Fe3+ ion in the same manner as structural-Fe(III) in clay. Fe2O3 usually exhibits photocatalytic activity in two types of polymorphs—α11,12 and amorphous13,14 phases; however, the photocatalysis of structural-Fe(III) has not yet been clarified in detail. Thus, we are interested in determining whether structural-Fe(III) itself can exhibit photocatalytic activity, especially after the oxidation of structural-Fe(II), or whether the ions can be transformed into an alternate type of Fe(III) species that can be made capable of exhibiting photocatalysis. Several natural clay compounds possess structural-Fe. Therefore, this clarification may provide information that will help us fabricate a clay-based photocatalyst of Fe(III) species, thus paving the way for a novel and clean preparation procedure for an active photocatalyst. | ||
| Fig. 1 Schematic illustration of the crystal structure of MMT.7 | ||
In the present study, the structural-Fe(II) in MMT was oxidized under ambient conditions. As a result, hydrous ferric oxide (HFO, Fe2O3·nH2O) and structural-Fe(III) were simultaneously produced in the clay. To verify the intrinsic photocatalyses of both Fe(III) species, clay compounds containing various amounts of HFO were prepared and examined with the degradation of acetic acid. The correlation between both types of Fe(III) species and their specific photocatalytic activities was determined on the basis of the results of their kinetic analyses.
Experimental
Purification of MMT
10 g of bentonite (Nihon Kohken, Tsugaru-2) was introduced into 1.0 dm3 of deionized water. The resulting suspension was stirred for 5 h, after which MMT was isolated from bentonite by centrifugation and then dried at 100 °C. As a result, ca. 2.1 g of MMT was obtained.Preparation of photocatalysts
The photocatalysts employed in this study were prepared by three different methods: the oxidation of structural-Fe(II) (method A), treatment with a strong acid (method B), and treatment with dithionite–citrate–bicarbonate (DCB) (method C).| Component | wt% |
|---|---|
| Na2O | 2.095 |
| MgO | 5.144 |
| Al2O3 | 19.341 |
| SiO2 | 68.802 |
| K2O | 0.404 |
| CaO | 0.188 |
| Fe2O3 | 4.026 |
Characterization of photocatalysts
The oxidation state of structural-Fe was characterized by infrared (IR) spectroscopy by using a Perkin-Elmer Spectrum One FTIR spectrometer equipped with a universal attenuated total reflectance sampling accessory (ZnSe cell). The specific surface areas of the photocatalyst samples were determined using the Brunauer–Emmett–Teller method by using a surface area analyzer (Shimadzu Micromeritics FlowSorb II 2300). X-ray powder diffraction (XRD) patterns of the samples were obtained using a Shimadzu XD-610 diffractometer with CuKα radiation (0.154 nm). The diffuse reflectance spectrum was obtained using a UV-Vis spectrophotometer (JASCO, V-570) equipped with an integrating sphere (JASCO, ISN-470). For obtaining the diffuse reflectance spectrum of the photocatalyst samples, a 1 wt% sample suspension was prepared; 1 cm3 of the suspension was filtered through a membrane filter (Millipore, pore size: 0.45 μm), and the filtrate was then dried at 60 °C. α-Fe2O3 (Nilaco, 99.99%) and HFO without MMT were used as reference samples. HFO was prepared by hydrolysing Fe3+ ions.14The HFO content in the photocatalysts (i.e. Fe(III)MMT, B-MMT, or C-MMT) was colorimetrically determined by the 1,10-phenanthroline method.17 In each case, HFO was extracted from 20 mg of the photocatalyst by the DCB method (vide supra). The resulting supernatant of Fe (extracted component) was appropriately diluted with deionized water to obtain the specimen for the colorimetric measurement (0.1–5.0 mg dm−3). The specimen solution (0.5 cm3) was mixed with 0.25 cm3 of 2 wt% hydroxylamine hydrochloride, 0.25 cm3 of 2 mol dm−3 sodium acetate/acetic acid buffer (pH 5.1), and 0.25 cm3 of 0.4 wt% 1,10-phenanthroline; then, the mixture solution was left to stand for 30 min. Its absorbance at 510 nm was measured using a UV-Vis spectrophotometer (Shimadzu, UVmini-1240).
Photocatalytic experiments
The photocatalytic activity of each sample was investigated in terms of the degradation of acetic acid. The photocatalytic reaction was carried out in a 20 cm3 airtight glass vessel containing 10 cm3 of 1 mol dm−3 acetic acid solution (pH was adjusted to 5.0 by adding NaOH to the solution) and 20 mg of the photocatalyst sample (i.e. SPO, MMT, Fe(III)MMT, B-MMT, or C-MMT) under air. A 100 W mercury lamp (USHIO, USH-102D) was used as the light source (light intensity: 110 mW cm−2), where irradiation with λ > 300 nm was conducted. The light intensity was measured using a power meter (Gentec, PSV-3103V2). Gaseous products were analyzed using a thermal conductivity detector (TCD) gas chromatograph (Shimadzu, GC-14B) with a packed column (ShinCarbon ST) and Ar carrier gas. After 10 h of the photocatalytic reaction, the HFO content in the photocatalysts employed was measured by the DCB and the 1,10-phenanthroline method because there is a possibility that HFO can be regenerated from structural-Fe(III) under weakly acidic conditions.15Results and discussion
The structural and optical characteristics of the samples employed were identified from the IR spectrum, XRD pattern, specific surface area, and diffuse reflectance spectrum.To investigate the oxidation state of structural-Fe, the IR spectra were measured in the range of 700 to 1000 cm−1 (Fig. 2); structural-Fe(II) has been recognized as an IR-inert species.18,19 However, in the case of MMT (Fig. 2a), some small peaks were observed. These peaks indicate that the present MMT includes structural-Fe(III) (i.e. Fe(III)MgOH and AlFe(III)OH deformation18,19) in small amounts. In the case of Fe(III)MMT (Fig. 2b), the peak intensity at around 790 cm−1 increased, evidently indicating that the structural-Fe(II) was successfully oxidized to Fe(III) by the above-mentioned procedure (i.e. method A). In order to account for the presence or absence of the unreacted structural-Fe(II) in MMT, Fe(III)MMT was additionally treated with hydrogen peroxide (see ESI†). It has been reported that the entire amount of structural-Fe(II) in clay can be oxidized when brought in contact with hydrogen peroxide.20 The IR spectrum of the treated Fe(III)MMT was obtained (Fig. S1, ESI†). However, the intensity of the peaks from the Fe(III) species remained unchanged. Therefore, this result verifies that the structural-Fe(II) was entirely converted to Fe(III) by method A. Fig. 3 shows the XRD patterns of MMT and Fe(III)MMT, in which no other crystalline components (e.g. α-Fe2O3) were confirmed.21–23 The diffuse reflectance spectra of MMT and Fe(III)MMT are shown in Fig. 4. The oxidation of structural-Fe(II) resulted in an increase in the absorption from 300 to 600 nm. As reported earlier, the colour of MMT can be attributed to the oxidation state of structural-Fe.24 In our study, we confirmed that the colour of the MMT employed changes from green to brownish yellow after oxidation; however, considering the crystal structure (see Introduction), the change in colour may have occurred because of the formation of an Fe2O3 species from structural-Fe(III). Therefore, the diffuse reflectance spectra were measured with the two types of iron oxide (i.e. α-Fe2O3 and HFO). From Fig. 4, we observe that both compounds can also absorb light with a wavelength of ca. <600 nm. Although the generation of crystalline components cannot be confirmed from the XRD pattern (Fig. 3), HFO, corresponding to amorphous Fe2O3, in addition to structural-Fe(III), may be present in Fe(III)MMT. The specific surface areas of MMT, Fe(III)MMT, and SPO were 154 m2 g−1, 142 m2 g−1, and 192 m2 g−1, respectively. These results imply that the specific surface area of MMT remained the same even after oxidation.
 | ||
| Fig. 2 IR spectra of both (a) MMT and (b) Fe(III)MMT. The arrows indicate the peak positions of the species originating from the structural-Fe(III) species. | ||
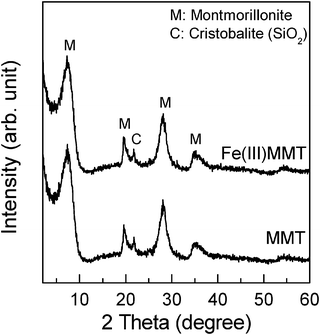 | ||
| Fig. 3 XRD patterns of MMT and Fe(III)MMT. | ||
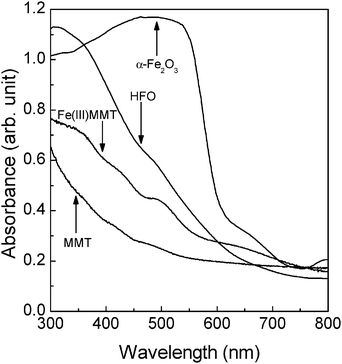 | ||
| Fig. 4 Diffuse reflectance spectra of MMT, Fe(III)MMT, HFO, and α-Fe2O3. | ||
The photocatalytic activities of SPO, MMT, and Fe(III)MMT were examined in terms of the degradation of acetic acid (CH3COOH + 2O2 → 2CO2 + 2H2O).25 During photocatalysis, both MMT and Fe(III)MMT oxidatively induced only the evolution of CO2. The evolution of CO2 was not observed in the dark, irrespective of the type of photocatalysts used. In addition, in the absence of photocatalysts under irradiation, CO2 was not generated. Hence, we verified that the acetic acid degrades through a photocatalytic process. Moreover, CO2 was not formed in the SPO system. Both SPO and MMT belong to the group of smectite clays, which are composed of aluminosilicate layers. However, the SPO employed does not contain any iron species, which distinguishes it from the present MMT. Therefore, the photocatalyses of MMT and Fe(III)MMT appear to originate from iron species naturally contained in the clay. Fig. 5 shows the time course of the amount of CO2 evolved under irradiation. In the MMT and Fe(III)MMT systems, the time course exhibited a linear dependence on irradiation time, indicating that both compounds can act as stable photocatalysts. The amount of CO2 was much higher in Fe(III)MMT than in MMT. The fact that the amount of structural-Fe(III) was higher in Fe(III)MMT than in MMT supports that the oxidation state of structural-Fe, i.e. the trivalent state, is responsible for the photocatalysis. In a separate experiment, Fe(III)MMT was used repeatedly as the photocatalyst for acetic acid degradation; the acetic acid solution was replaced through centrifugation after every irradiation for 10 h. In each cycle, the amount of CO2 evolved was kept constant (Table S2, ESI†). After three cycles, the cumulative amount of CO2 exceeded 40 μmol, which corresponds to a turnover number of 8 (cf. [the amount of CO2 molecules evolved]/[the total amount of Fe(III) species in Fe(III)MMT (ca. 5 μmol in the oxide (Fe2O3) content)]). This also proves that Fe(III) species in Fe(III)MMT can act as a stable photocatalyst.
 | ||
| Fig. 5 Time course of CO2 evolved in SPO, MMT, and Fe(III)MMT. Acetic acid concentration: 1 mol dm−3 (pH 5.0); light intensity: 110 mW cm−2. | ||
As mentioned earlier, it is inferred that two types of Fe(III) species, i.e. structural-Fe(III) and HFO, are formed in Fe(III)MMT. To evaluate the intrinsic activity of both species, the weight ratio of HFO to structural-Fe(III) in Fe(III)MMT was varied. In the following paragraph, we clarify the correlation between the amount of HFO in Fe(III)MMT and its photocatalytic activity.
First, the amount of HFO in Fe(III)MMT was varied according to method B (see Experimental), and the resulting B-MMT was used as the photocatalyst in the degradation of acetic acid (cf. the structural and optical characteristics of B-MMT are provided in Fig. S2–S4 in ESI†). Fig. 6 shows the linear dependence of the CO2 evolution rate on the HFO amount in B-MMT. The rate of evolution increased with the HFO amount, implying that the photocatalytic activity of HFO is superior to that of structural-Fe(III). Therefore, in order to investigate the differences in photocatalytic activities between structural-Fe(III) and HFO, the intrinsic photocatalytic activity of each Fe(III) species was estimated based on the plot shown in Fig. 6. The intrinsic activities were defined as kst (μmol h−1 μg−1) for structural-Fe(III) and kHFO (μmol h−1 μg−1) for HFO, respectively. The observed rate of CO2 evolution, VCO2 (μmol h−1), can be represented by the sum of the product of the amount of each Fe(III) species and its intrinsic photocatalytic activity, as follows:
| VCO2 = kst × Wst + kHFO × WHFO | (1) |
| VCO2 = (kHFO − kst) × WHFO + 800 × kst | (2) |
 | ||
| Fig. 6 Correlation between the rate of CO2 evolved and the amount of HFO in the photocatalysts employed. Total amount of photocatalyst: 20.0 mg; acetic acid concentration: 1 mol dm−3 (pH 5.0); light intensity: 110 mW cm−2. | ||
In a separate experiment, the HFO in Fe(III)MMT was removed by the DCB method (i.e. method C), and then, the photocatalytic activity of the resulting C-MMT was investigated. When 20 mg of C-MMT was employed for the degradation of acetic acid for 10 h under irradiation, the rate of CO2 evolution was ca. 0.73 μmol h−1, which was less than that in Fe(III)MMT (Fig. 5). In Fe(III)MMT, the amount of HFO removed was ca. 80 μg (vide supra) out of ca. 800 μg of Fe(III) species expressed as the oxide content. Therefore, the total amount of Fe(III) species as the oxide content in C-MMT was estimated to be ca. 720 μg. After the removal of HFO, it was swiftly regenerated from structural-Fe(III) under weakly acidic conditions (see experimental section), and the amount of HFO was estimated to be 28 μg. Incidentally, the HFO amount remained unchanged after long-term irradiation (5–20 h); this demonstrates a constant and stable photocatalysis of C-MMT for the evolution of CO2. Based on the intrinsic kinetic parameters as well as the amount of Fe(III) species (Wst: 692 μg and WHFO: 28 μg), the rate of CO2 evolution can be estimated to be ca. 0.71 μmol h−1 using eqn (1), which is consistent with the observed rate (vide supra). This also confirms that structural-Fe(III) is inferior to HFO in terms of photocatalysis.
To gain insight into the photocatalysis of Fe(III)MMT, its photocatalytic activity was examined with respect to the experimental conditions employed.
First, the photocatalytic activity of Fe(III)MMT was investigated in terms of irradiation. When the irradiation was conducted through a UV-pass filter for 300 < λ < 400 nm or a sharp-cut filter for λ > 420 nm, the rates of CO2 evolution from acetic acid were 0.57 μmol h−1 and 0.08 μmol h−1, respectively. Therefore, the photocatalytic degradation of acetic acid can occur under both UV-light and visible-light irradiation.
Two reference samples, commercially available Fe2O3 (Nilaco, 99.99%) and home-made HFO, were used as the photocatalysts for the degradation of acetic acid for comparison with Fe(III)MMT. The specific surface areas of the commercially available Fe2O3 and home-made HFO were 7 m2 g−1 and 241 m2 g−1, respectively. The amount of photocatalysts employed was ca. 80 μg in each system (i.e. the employed amount was the same as the amount of HFO in Fe(III)MMT). After 10 h of irradiation, the rates of CO2 evolution in commercial Fe2O3 and home-made HFO were 0.28 μmol h−1 and 0.72 μmol h−1, respectively. The resulting rates of evolution were lower than that in Fe(III)MMT (ca. 1.38 μmol h−1), most probably indicating that the specific surface area of HFO in Fe(III)MMT is greater than that in home-made HFO. It has been reported that HFO can be chemically synthesized in the interlayer space of clay,26 through which the aggregation of HFO can be suppressed to result in an increase in its specific surface area.4–6,14 Thus, a large specific surface area of HFO loaded in clay can lead to an efficient photocatalysis of Fe(III)MMT.
In a separate experiment, the photocatalytic degradation of acetic acid by Fe(III)MMT was performed particularly within an Ar atmosphere, where the photo-Kolbe reaction (i.e. CH3COOH → CH4 + CO2) occurred (cf. the detailed reaction scheme is shown in ESI†).27–29 After 10 h of irradiation, CH4 (4.8 μmol) and CO2 (4.7 μmol) were observed to be generated; that is, the ratio of the amount of CH4 to that of CO2 (CH4/CO2) was ca. 1, indicating that Fe(III)MMT is capable of photocatalysis for the stoichiometric degradation of acetic acid. By the photo-Kolbe reaction, acetic acid can be directly oxidized by a photogenerated hole at HFO (i.e. CH3COO− + h+ → CO2 + ˙CH3), and so, a similar formation of CO2 can also be expected to occur in the present system. By comparing the size of an acetic acid molecule (ca. 0.4 nm)30 to that of the HFO-loaded interlayer (0.58 nm)26 (cf. in the present study, the size of HFO in the Fe(III)MMT could not be determined, because the amount of HFO was much less than that of the parent clay), acetic acid can be intercalated into the interlayer space of clay, and thus, the above-mentioned photocatalytic oxidation is possible. In the photocatalytic reduction, particularly under air, molecular oxygen (O2) can work as an electron acceptor. Based on previous knowledge,31 the presence of O2 kinetically enhances the photocatalytic degradation of acetic acid. Moreover, the reduction of O2 leads to the generation of various types of reactive oxygen species,32 which may also participate in the degradation of acetic acid. In summary, the photocatalytic degradation of acetic acid by Fe(III)MMT involves the direct oxidation of acetic acid at HFO along with the reduction of O2.
Conclusion
The present study is the first to demonstrate that a natural clay, MMT, can exhibit active photocatalysis for the degradation of acetic acid on account of the presence of iron species in its crystal structure. After the oxidation of structural-Fe(II), two types of photocatalytically active species, structural-Fe(III) and HFO, were produced in MMT. Kinetic analysis revealed that the intrinsic activity of HFO is superior to that of structural-Fe(III), wherein the photocatalytic degradation of acetic acid involves its direct oxidation at HFO along with the reduction of O2. Thus, HFO preferentially functioned as the active photocatalyst in the clay even when the weight ratio of HFO to structural-Fe(III) was low. This paper presented a novel and simple preparation procedure for an active clay-based photocatalyst, especially without a precursor such as a metal complex; this can also open up avenues for the passive use of photocatalysts featuring natural materials.References
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
- C.-H. Kuo, C.-H. Chen and M. H. Hauang, Adv. Funct. Mater., 2007, 17, 3773–3780 CrossRef CAS.
- A. Kay, I. Cesar and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 15714–15721 CrossRef CAS.
- G. K. Zhang, X. M. Ding, F. S. He, X. Y. Yu, J. Zhou, Y. J. Hu and J. W. Xie, Langmuir, 2008, 24, 1026–1030 CrossRef CAS.
- S. Yamanaka, T. Doi, S. Sako and M. Hattori, Mater. Res. Bull., 1984, 19, 161–168 CrossRef CAS.
- H. Mori, H. Miyoshi, K. Takeda, H. Yoneyama and H. Fujita, J. Mater. Sci., 1992, 27, 3197–3199 CrossRef CAS.
- R. E. Grim, Clay Mineralogy, McGraw-Hill, New York, 2nd edn, 1968 Search PubMed.
- L. Yimin, L. Yueqing and Z. Xiaoliu, J. Hazard. Mater., 2006, 132, 196–201 CrossRef.
- J. Chen and L. Zhu, J. Photochem. Photobiol., A, 2007, 188, 56–64 CrossRef CAS.
- L. Vayssieres, C. Sathe, S. M. Butorin, J. Nordgren, D. K. Shuh and J. Guo, Adv. Mater., 2005, 17, 2320–2323 CrossRef CAS.
- S. U. M. Khan and J. Akikusa, J. Phys. Chem. B, 1999, 103, 7184–7189 CrossRef CAS.
- A. Duret and M. Grätzel, J. Phys. Chem. B, 2005, 109, 17184–17191 CrossRef CAS.
- N. Deng, F. Wu, S. Tian and T. Fang, Chemosphere, 1997, 4, 2725–2735 CrossRef.
- S. Kakuta and T. Abe, J. Mater. Sci., 2009, 44, 2890–2898 CrossRef CAS.
- H. Drame, Clays Clay Miner., 2005, 53, 335–347 CrossRef CAS.
- O. P. Mehra and M. L. Jackson, Clays Clay Miner., 1958, 7, 317–327 Search PubMed.
- D. V. D'Amore, S. R. Stewart and J. H. Huddleston, Soil Sci. Soc. Am. J., 2004, 68, 1012–1022 CrossRef CAS.
- C.-I. Fialips, D. Huo, L. Yan, J. Wu and J. W. Stucki, Clays Clay Miner., 2002, 50, 455–469 CrossRef CAS.
- S. Vingiani, D. Righi, S. Petit and F. Terribile, Clays Clay Miner., 2004, 52, 473–483 CrossRef CAS.
- I. Rozenson and L. Heller-Kallai, Clays Clay Miner., 1978, 26, 88–92 CAS.
- JCPDS card no. 29-1499.
- JCPDS card no. 33-0664.
- JCPDS card no. 11-0695.
- J. W. Stucki and P. R. Lear, ACS Symp. Ser., 1989, 415, 330–358 CrossRef.
- D. S. Muggli, S. A. Keyser and J. L. Falconer, Catal. Lett., 1998, 55, 129–132 CrossRef CAS.
- O. A. Ileperuma, W. C. B. Kiridena and W. D. D. Dissanayake, J. Photochem. Photobiol., A, 1991, 59, 191–197 CrossRef CAS.
- B. Kraeutler and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 2239–2240 CrossRef CAS.
- B. Kraeutler and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 5985–5992 CrossRef CAS.
- T. Sakata, T. Kawai and K. Hashimoto, J. Phys. Chem., 1984, 88, 2344–2350 CrossRef CAS.
- N. Alghezawi, O. anlı, L. Aras and G. Asman, Chem. Eng. Process., 2005, 44, 51–58 CrossRef CAS.
- B. Ohtani, Y. Nohara and R. Abe, Electrochemistry, 2008, 76, 147–149 CAS.
- T. E. Agustina, H. M. Ang and V. K. Vareek, J. Photochem. Photobiol., C, 2005, 6, 264–273 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The structural and optical characteristics of Fe(III)MMT after being treated with additional H2O2 are shown. The details of the structural and optical characteristics of B-MMT employed in the present study are provided. The amount of HFO in B-MMT is also shown as a function of the HCl concentration in acid treatment. The photocatalytic degradation data of the reused Fe(III)MMT system are shown. The equations in the photo-Kolbe reaction are represented. See DOI: 10.1039/c1cy00286d |
| This journal is © The Royal Society of Chemistry 2011 |