Gold-catalyzed oxidation in organic synthesis: a promise kept
Cristina
Della Pina
* and
Ermelinda
Falletta
Dipartimento di Chimica Inorganica, Metallorganica e Analitica “L. Malatesta”, Università degli Studi di Milano, CNR-ISTM, via Venezian, 21-20133 Milano, Italy. E-mail: cristina.dellapina@unimi.it; Fax: +39 02 50314405; Tel: +39 02 50314406
First published on 25th October 2011
Abstract
An overview of Prof. Michele Rossi's recent research and related scientific context is presented, dealing with liquid and gas phase oxidation of some organic compounds by gold catalysis. The application of mono- and bimetallic gold nanoparticles as an aid for organic synthesis has been mainly devoted to the aerobic oxidation of alcohols and aldehydes, as well as to the new synthesis of conducting polymers, thus demonstrating the superior performance of gold in terms of activity, selectivity and durability in comparison with traditional Pd and Pt catalysts. Starting from the observation that oxygen activation towards organic compounds occurs mainly in the presence of nanometric gold clusters, the outstanding properties of colloidal “naked” particles are also discussed. Thorough kinetic studies suggest models for interpreting the aerobic oxidation of glucose, thereby shedding light on the molecular mechanism, whilst the perspective for industrial applications of supported gold catalysts shows how the “yellow metal” is more than a promise.
 Cristina Della Pina | Cristina Della Pina received the Master degree and was awarded the PhD degree in Industrial Chemistry at the Università degli Studi di Milano under the supervision of Prof. Michele Rossi. She is presently Assistant Professor of General Chemistry and her research work focuses on the development of heterogeneous catalysts for the selective oxidation of organic compounds. |
 Ermelinda Falletta | Ermelinda Falletta got her master degree in Organic Chemistry (2004) at the University of Palermo. In 2005 she joined Prof. Rossi's research group by a scholarship of the Centre of Excellence CIMAINA at the University of Milan and then she was involved in the research of gold catalysis as a graduate technician at the department of Inorganic Chemistry. She is working on her PhD thesis concerning new environmentally friendly synthetic routes for conducting organic polymers and their technological applications. |
1. Introduction
Two decades have passed since the pioneering works by Haruta and Hutchings presented gold as a new catalyst for CO oxidation and ethyne hydrochlorination, respectively.1,2Such breakthroughs, together with the subsequent employment of gold catalysis in the liquid and gas phase oxidation of alcohols and aldehydes performed by Rossi et al., have contributed to open up a new exciting scenario in organic synthesis.3–18 Compared to other metals, gold was shown to be markedly chemoselective, thus favouring high yields into the desired product. Therefore, glycols could be converted into monocarboxylates,3–6,8–14 unsaturated alcohols into unsaturated aldehydes,15 unsaturated aldehydes and ketones to unsaturated alcohols with selectivity approaching 100%. Moreover, due to its biocompatibility, availability and easy recovering, gold turns out to be a “green” catalyst for sustainable processes using clean oxidants, like O2 and H2O2, often allowing mild conditions, aqueous solution or the absence of any solvent.16–18
On the occasion of this special issue dedicated to Prof. Rossi, the present mini review focuses on the principal findings by gold catalysis achieved at Milan University since our joining his laboratories. Other research groups' notable achievements on this topic are also mentioned.
2. Selective oxidation of glucose
Glucose is a cheap and renewable starting material whose selective oxidation represents a strategic target for chemical intermediates.Among them, gluconic acid and gluconates are particularly appealing because they are industrially employed in food chemistry, cleaning agents and surfactants. The present commercial production follows an enzymatic route based on Aspergillus niger mould leading to sodium gluconate or calcium gluconate, according to slightly different protocols. However, the low productivity and the troublesome separation of the enzyme at the end of the process require alternative methods for improving the performance, using different catalytic technologies. Rossi and coworkers accepted the challenge and in 2002 they presented the first work on the application of gold supported on carbon for D-glucose selective oxidation in liquid phase under mild conditions (pH = 7–9.5, T = 323–373 K, pO2 = 1–3 bar).7 A comparative study between gold and commercial palladium and platinum-derived catalysts, so far employed as first attempts to substitute the biological process, highlighted the unique properties of the gold catalyst prepared by colloidal (sol) immobilization on carbon: not only gold was still active at low pH but, at a buffered higher value of 9.5, it displayed a selectivity comparable to that of bismuth-doped platinum–palladium on carbon catalysts, though with a higher activity. Moreover, gold was found to be more stable on recycling.
Encouraged by these first promising results, a thorough optimization work was carried out with the ambitious aim to introduce gold as an alternative catalyst to the enzyme. Thereby, starting from TOF of a few hundred h−1 units, we could reach the impressive value close to 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1, a catalytic performance comparable to the enzymatic behaviour.19 In this work, the catalytic activity of unsupported gold nanoparticles was also discovered.
000 h−1, a catalytic performance comparable to the enzymatic behaviour.19 In this work, the catalytic activity of unsupported gold nanoparticles was also discovered.
One of the most intriguing questions in heterogeneous catalysis is the so-called metal–support interaction: taking the aerobic oxidation of glucose in liquid phase as a model reaction, the separate contribution of gold nanoparticles and supporting material was evaluated. By showing that this reaction could be easily carried out using either unsupported colloidal gold particles (“naked” particles) or supported gold particles under mild conditions in basic solution, the key role of gold was demonstrated. The strategy adopted for achieving active “naked” colloidal particles was to avoid the traditional colloid stabilizers by using glucose in slight excess, which was found to act both as a protecting agent and reagent.
Starting from kinetic tests with a constant mass of metal of a given density, in the form of colloidal spherical particles having a monomodal distribution, it is possible to derive a correlation between activity and particle size, expressed by this simple model v = k/r, where “v” means the catalyst controlled rate and “r” is the nanoparticle radius.
In order to verify experimentally such a dependence, we prepared colloidal particles of different size (diameter range: 3–10 nm) by varying the chloroauric concentration from 50 to 600 mg l−1. Testing these particles at T = 303 K, pH = 9.5 and glucose:Au = 12000 molar ratio, using a glass reactor interfaced to an automatic titration device equipped with NaOH, we actually observed a catalytic activity inversely proportional to the diameter in the range of 2.5–4.5 nm. However, we also detected a deviation from linearity for particles larger than 6 nm, as well as the inactiveness for those larger than 10 nm (Fig. 1).19
![Correlation between activity and particle size of unsupported gold nanoparticles during the aerobic oxidation of glucose. Experimental conditions: [glucose] = 0.38 M; [Au] = 3.2 × 10−5 M; glucose : Au = 12 000, T = 303 K, pH = 9.5, pO2 = 1 bar.](/image/article/2011/CY/c1cy00283j/c1cy00283j-f1.gif) | ||
| Fig. 1 Correlation between activity and particle size of unsupported gold nanoparticles during the aerobic oxidation of glucose. Experimental conditions: [glucose] = 0.38 M; [Au] = 3.2 × 10−5 M; glucose:Au = 12000, T = 303 K, pH = 9.5, pO2 = 1 bar. | ||
The rapid decrease of activity for particles larger than 6 nm indicates a discontinuity in the catalytic behaviour which is not only due to geometrical factors but likely due to electronic factors, influencing the electrophilic properties of the particles. By the way, other properties show a sudden discontinuity in the region close to the metallic–non-metallic transition of gold, as the melting point collapses.20
Although the short lifetime of unsupported gold nanoparticles prevents them from being a practical catalyst, their activity is fundamental for evaluating metal–support interactions by determining the intrinsic role of gold in the oxidation reaction.
For this purpose, we synthesized 0.5% Au/C by contacting the gold sol with carbon X40S and we compared it to the corresponding unsupported gold with the same nanoparticles size of 3.6 nm, under identical experimental conditions. Considering the conversion–time plot of glucose oxidation during the first 200 s, the curve with supported particles was quite similar to that of unsupported gold (Fig. 2).19
![Comparison between supported (0.5% Au/C) and unsupported gold nanoparticles during the aerobic oxidation of glucose. Experimental conditions: [Au] = 10−4 M, [glucose] = 0.4 M, T = 303 K, pO2 = 1 bar.](/image/article/2011/CY/c1cy00283j/c1cy00283j-f2.gif) | ||
| Fig. 2 Comparison between supported (0.5% Au/C) and unsupported gold nanoparticles during the aerobic oxidation of glucose. Experimental conditions: [Au] = 10−4 M, [glucose] = 0.4 M, T = 303 K, pO2 = 1 bar. | ||
Such a trend means that metal–support interactions do not affect gold intrinsic activity, even though a stabilising effect of carbon on the original activity of the metal is clear, thus allowing to reach the total conversion of glucose.
These promising findings inspired us to carry out a systematic comparison between gold catalysis and enzymatic catalysis under strictly similar conditions.21
For these experiments, the following catalytic systems were employed:
(a) Hyderase (from Amano Enzyme Co., UK) as an enzymatic preparation containing glucose oxidase and catalase as active components and flavine–adenine dinucleotide (FAD) as the rate controlling factor (1.3 × 10−6 mol g−1).
(b) Gold catalyst, 0.5% w/w of Au on coconut derived carbon powder (X40S from Camel, As = 1200 m2 g−1), prepared by sol immobilization, containing metal particles of average diameter 3.6 nm, as determined by XRD analysis with Scherrer equation and confirmed by TEM, mainly at the surface (Au/C = 1.5 from XPS data). The catalyst was synthesized after optimizing the preparation method by reducing chloroauric acid (HAuCl4, Au = 10 mg ml−1) with NaBH4 (metal:NaBH4 = 1:1 w:w) in the presence of glucose as an innovative stabilizing agent (Au:glucose = 1:50 molar ratio) in substitution of the traditional protecting agents, such as polyvinylalcohol (PVA), polyvinylpyrrolidone (PVP) and tetrahydroxymethylphosphonium chloride (THMP).21,22
A proper tuning of different parameters such as pH, temperature, glucose concentration and stirring speed led to the results displayed in Table 1.
| Catalyst | [Glucose] (mol l−1) | Catalyst/glucose (g/Kg) | pH | Stirring speed/rpm | T/K | Specific activity/h−1 | Productivity/Kg m−3 h−1 |
|---|---|---|---|---|---|---|---|
| Au/C | 3 | 5 | 9.5 | 39000 |
323 | 217.7 | 514.3 |
| Hyderase | 1 | 6 | 5–7 | 900 | 303 | 144.6 | 121.7 |
The final superior productivity obtained with the inorganic catalyst can be explained taking into account the lower FAD concentration in the enzymatic extract and the possibility given by gold to start with a threefold higher glucose concentration. On the other hand, considering the molecular efficiency of the active FAD sites, a turnover frequency of 600000 h−1 was reached, a markedly better value than the efficiency of active external gold atoms in the inorganic catalyst, calculated as 90000 h−1.
Gold-catalyzed oxidation of glucose has recently drawn a general attention: in particular, Pruesse's research group has widely developed this topic, successfully extending the application of gold catalysis to a large number of carbohydrates,17,23,24 and Haruta et al. have shown how the solid grinding can offer an effective and easy protocol for preparing well dispersed catalysts suitable to improve glucose oxidation performance.25 The Japanese team has also re-investigated the selective oxidation of glucose using H2O2 as the oxidant, employing gold nanoparticles deposited directly from aqueous solution of diethylenediaminegold(III) complex onto commercially available polymer beads, such as poly(methyl methacrylate) (PMMA), polystyrene (PS) and polyaniline (PANI).26 They found the kinetics to be influenced by the kind of supports (PMMA > PS > PANI), rather than by the size of Au particles.
An effective method for directly achieving free gluconic acid is presently unknown. Generally, it is manufactured from calcium gluconate and sulfuric acid besides a large amount of CaSO4 as a by-product. This is due to the fact that at low pH values the enzymatic catalysis is inhibited, and also Pd, Pt, and Au catalysts are scarcely active. To attempt a successful direct synthesis of gluconic acid, we prepared and evaluated bimetallic catalysts, thereby discovering a strong synergistic effect between gold and platinum. The most promising Au + Pt combination was optimized leading to a quite active catalyst for alkali free oxidation of glucose containing gold and platinum in the ratio 2:1 (w/w), but critical experiments are still lacking concerning the catalyst lifetime.27
It is worth mentioning the innovative bulk nanoporous gold material, synthesized by selective leaching of Ag/Au alloy and tested by Yin et al. in the liquid phase oxidation of glucose to gluconic acid under mild conditions, thus finding that the activity strongly depends on pH, temperature and porous ligament size.28 In comparison with the traditional catalysts, such a catalytic system does not match the performances of Au nanoparticles but it is advantageous in terms of easy preparation, recycling and recovery.
2.1. Kinetic study and molecular mechanism
The exciting results so far reported were interpreted by a series of mechanistic studies on the selective liquid phase oxidation of glucose catalysed both by gold and enzyme.29–31The first fundamental contribution came out from Claus in Germany and our group in Italy.
These kinetic investigations were performed using carbon supported gold particles32 and unsupported colloidal gold particles30 as the catalysts, leading to two different models for explaining the mechanistic aspects of the reaction.
Our kinetic tests were carried out employing unsupported colloidal particles (“naked” gold particles) and led to the fundamental detection of hydrogen peroxide, instead of water, as the reduction product of dioxygen.30,31 In fact, during the conversion of glucose, we were able to quantify H2O2 besides gluconate with values close to the 1:1 stoichiometry.31
Moreover, the comparative kinetic study between the inorganic catalytic system and enzyme highlighted how gold catalysis and biological catalysis are able to promote the fast and selective oxidation of glucose under mild conditions according to the same stoichiometry, thus leading to the formation of hydrogen peroxide as an unstable by-product. On the other hand, the two catalytic systems follow up different reaction mechanisms: in the case of enzymatic catalysis, the rate determining step is the oxidation of the substrate by the enzyme, which is converted into the reduced form according to a faster step and showing a zero order with respect to dioxygen;29 in gold catalysis, the rate determining step is due to the adsorbed glucose which is oxidized by dioxygen dissolved in water, according to a first order dependence of the reaction rate on pO2.30
A molecular mechanism of glucose oxidation on a gold nanoparticle could be suggested on the basis of the promoting role of alkali and the detection of H2O2 as a product: the fundamental step is the formation of the electron-rich gold species derived by the hydrated glucose anion with gold. This species is supposed to activate oxygen by nucleophilic attack and the derived dioxogold intermediate can then behave as a bridge for the two electron-transfer from glucose to dioxygen (Scheme 1).31
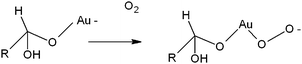 | ||
| Scheme 1 Electron-rich gold and dioxogold species suggested as key intermediates in the mechanism of glucose oxidation. | ||
A geometrical model has been then derived for describing the morphological properties of two catalysts made up of carbon supported gold particles, prepared according to colloidal gold deposition and having an average size of 3.30 nm and 7.89 nm, respectively.33 The tests were carried out by progressively poisoning these catalysts with thiocyanate, cyanide, cysteine and thiourea during the aerobic oxidation of glucose: the observed deactivation trend followed the order thiocyanate > cyanide ≈ cysteine > thiourea and each of them obeyed an exponential law. The kinetics of the catalyst deactivation led to suppose a long range poison-catalyst interaction influencing the entire metal particle, as a contribution of electronic factors which overlap the space shielding of active sites. The thorough evaluation of the kind of molecules causing a detectable poisoning effect, as well as the promoting effect of OH−, allowed us to conclude that the dioxygen reduction step is differently influenced by soft and hard-nucleophiles: whereas a hard nucleophile prevents from a back-donation to the Lewis base from the metal, thus leaving in the reacting solution the original or a higher catalytic effect as in the case of OH−, a soft nucleophile, showing π back-bonding ability, can withdraw the electron density from the metal, thus inhibiting dioxygen reduction and decreasing the catalytic property of the entire gold particle (Scheme 2).33
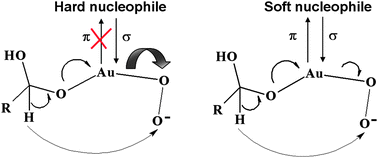 | ||
| Scheme 2 Effect of softness and hardness nucleophiles on gold nanoparticles. | ||
3. Selective oxidation of benzyl alcohol
The worldwide interest in benzyl alcohol oxidation is related to the high added value of the corresponding products, benzaldehyde in primis being the artificial bitter-almond aroma so widely employed in food and cosmetic industry. Moreover, this selective oxidation is often taken as a model reaction for gold catalysis, both in liquid and gas phases mainly depending on the thermal stability and volatility of compounds, studying also the effect of the ring substitution on the reactivity. Gold nanoparticles have been shown to be very active in aqueous base, but under these conditions the product is the corresponding mono-acid and not the aldehyde (Scheme 3).4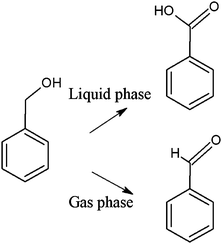 | ||
| Scheme 3 Possible products of benzyl alcohol oxidation. | ||
Even though the role of the supporting material in favouring many gold-catalyzed reactions is a well known leit motiv, this kind of reaction displays good catalytic performances independently of the support. Therefore, carbon nanotubes and nanofibers have been used as an alternative to activated carbon and, after a careful dispersion of Au and Pd nanoparticles on the surface, 96% selectivity towards aldehyde could be reached.34
The prolific application of polymers as the supporting material for gold includes also the use of a porous polyimide membrane. Mertens et al.35 stabilized gold–palladium clusters with polyvinylpyrrolidone without any loss of nanodispersion. The resulting catalytic membrane was highly active in the solvent free oxidation of benzyl alcohol (TOF 22500 h−1) with total selectivity to the carbonyl compound.
Among many papers dealing with this vast topic, the one which reports on ceria prepared under supercritical conditions for supporting gold–palladium nanoparticles is quite innovative.36 Hutchings et al. synthesized a very effective catalyst for the aerobic oxidation of benzyl alcohol under solvent-free conditions: the monometallic Au and Pd and bimetallic Au–Pd catalysts, prepared from the scCeO2 support, were evaluated at 413 K with O2 in the absence of solvent resulting to be highly active and selective for benzaldehyde (ca. 91% selectivity). The other products were mainly benzyl benzoate (3%), benzoic acid (2%) and toluene (3%). The Au–Pd supported on a supercritical CeO2 catalyst was approximately 4 times more active than Au–Pd supported on conventional CeO2.
Our research group has also dealt with the selective oxidation of benzyl alcohol. Following Rossi's first study on the catalytic conversion of benzyl alcohol in gas phase,15 we have extended this research by testing a series of mono- and bimetallic catalysts, made up of gold and copper supported on silica, in order to improve the previous performances (>99.5% selectivity to benzaldehyde at 50% conversion and T = 523 K; 98% selectivity to benzaldehyde at 75% conversion and T = 553 K). The catalysts were synthesized using an incipient wetness technique as reported elsewhere,15,37 thus obtaining a series of 1% Au/SiO2 and 1% Au–Cu/SiO2 of different Au/Cu w/w. The aerobic oxidation of benzyl alcohol was carried out in a fixed bed vertical glass reactor, fitted with a glass frit carrying the catalyst and provided with an electronically controlled furnace. The thermal interval was chosen on the basis of the reaction feasibility: at T < 523 K no benzyl alcohol oxidation occurred and at T > 623 K the product underwent degradation with coking. The results (99% selectivity to benzaldehyde at 98% conversion) underline the key role of gold in the reaction, along with a synergistic effect between gold and copper: whereas gold alone led to interesting values of selectivity and conversion, the most promising achievements have been obtained with the ratio Au/Cu = 4 w/w in the catalyst 1% Au–Cu/SiO2, resulting in over 99% selectivity to benzaldehyde at 98% conversion, at a relatively low temperature of 533 K. These findings could favour the way to potential applications in perfumery, where high purity grade of benzaldehyde (>99%) is always requested.37
4. Selective oxidation of allyl alcohol
One of the most strategic building blocks in organic synthesis is 3-hydroxypropionic acid (3-HP), which is involved in the manufacture of high performance polymers. Many efforts have been addressed to find effective biological and chemical routes, but no large scale process for synthesizing 3-HP is now industrially applied. One potentially useful starting point could be represented by allyl alcohol, in principle coming from the dehydration of glycerol thereof introducing a bio-renewable green pathway to this molecule. Using iron oxide as a catalyst, in fact, Schüth et al. were able to convert glycerol to allyl alcohol through dehydration and consecutive hydrogen transfer.38 Although the details of how the hydrogen transfer takes place are still under investigation, iron oxide appears to be a promising catalyst due to its high activity and good stability, as well as its low price and easily tuneable structures and morphologies. Further improvements are expected thus opening the route to practical applications.We recently found a new route to 3-hydroxypropionic acid by oxidizing allyl alcohol in the presence of a gold catalyst.39
Reacting allyl alcohol in aqueous alkali solution with O2, a slow oxidation took place thus leading to the expected product acrylate, besides small amounts of glycerate and, surprisingly, 3-hydroxypropionate. Nanometric gold particles dispersed on activated carbon were shown to be the catalyst of choice, whereas the selectivity to 3-HP could be tuned by acting on the amount of alkali and temperature. Thus, a high selectivity (79% at full conversion after 24 h) to the desired 3-hydroxypropionate was obtained with NaOH in excess (3:1) and a mild temperature (323 K).
We also suggested a possible mechanism, where acrolein is the intermediate undergoing a Michael-type addition of water to produce 3-HP (Scheme 4).
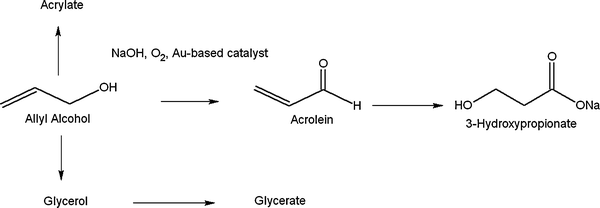 | ||
| Scheme 4 Suggested pathway to 3-HP from allyl alcohol oxidation by gold catalysis. | ||
Despite the great performance, a weak point related to the catalyst recycling came out: Au/C catalyst, in fact, was not stable as the selectivity to 3-HP decreased after the second reaction cycle, therefore questioning the viability of this new process. In order to overcome the problem of selectivity loss, we then prepared and tested a series of bimetallic Au–M nanoparticles (M = Pt, Pd, Cu or Ag) supported on carbon, as alloying gold with a second metal has been shown to be often effective in enhancing gold performances.40
The conversion, selectivity and lifetime of the catalysts could be tuned and markedly improved by optimizing the experimental conditions, as well as the preparation protocols, thus leading to notable performances also after 4 reaction cycles, especially for the 0.3% Au99Cu1/C bimetallic system: the selectivity to 3-HP decreased from 95.1% (1st cycle) to 78.3% (4th cycle), retaining the same conversion (around 98%). The stabilization of the catalytic activity due to the presence of the dilute second metal could be a consequence of electronic modifications in the metal nanoparticle. It is well known that metal particle size is often fundamental in affecting the catalytic performance. However, in this case, it seems not to be so important, as STEM analyses showed the definite signs of metal particles sintering. Most likely, the low redox potential of copper could be implicated in its sustained high performance, thus outweighing any limitations imposed by the particle growth during recycling.
5. Selective oxidation of amines and aminoalcohols
The catalytic oxidation of aminoalcohols is far from being a general method for the synthesis of aminoacids, owing to the doping effect of the amino-group on traditional metals.17 Rossi et al. demonstrated that gold catalysis partly overcomes such a limit, by comparing the performance of gold with palladium and platinum under similar conditions where the presence of alkali promoted the oxidation rate (T = 343 K, pO2 = 3 bar and substrate/NaOH = 1 molar ratio).41 We then extended the aerobic oxidation to tertiary amines, thus discovering the ability of supported mono- and bi-metallic catalysts (Au/C, Pt/C, Rh/C, Au–Pt/C, Au–Rh/C, Au/Al2O3 and Au/TiO2) to transform these amines into the corresponding N-oxides under “green” conditions, that is working in aqueous solution with molecular oxygen under mild conditions (T = 363 K, pO2 = 2 bar).42 Accordingly, 100% yield was achieved with triethylamine, pyridine and 3-dimethylaminopropan-1-ol using Au/C. In the case of N-substituted aminoalcohols, the oxidation took place exclusively at the nitrogen atom. Thereby, the reaction of 3-dimethylamino-1-propanol led to the corresponding N-oxide with 100% regioselectivity. In principle, the oxidation of the amino group is possible both in the absence and in the presence of alkali: without any alkali, 100% selectivity was observed with different metal catalysts, but only gold containing catalysts allowed 100% conversion, while Pt/C resulted to be inert and Rh/C allowed only 20% conversion towards unidentified compounds.42Over the last three years many papers on this topic have exponentially flourished.18
Angelici's group is particularly active in this field, thus discovering the catalytic activity of the large gold particles (around 1000 nm) in the reactions of carbon monoxide, or isocyanides, with primary amines and molecular oxygen under mild conditions to achieve, respectively, ureas or carbodiimides.43–45 Furthermore, they showed that bulk gold is also effective in the oxidative dehydrogenation of secondary amines to imines.
Also Mullins et al. have demonstrated that bulk gold can be an active catalyst as in the case of the selective oxidation of propylamine with oxygen.46
Anyway, the two decades of outstanding results in organic synthesis have underpinned the superiority of nanometric gold, with respect to the bulk status, in several oxidation reactions and Corma's group has confirmed it successfully performing the aerobic oxidation of various ranges of amines.47 Gold supported on titania was shown to be particularly effective.
The catalytic effectiveness of nanogold on titania in the aerobic oxidation of amines was supported also by Christensen and co-workers.48 They reported on gold ability in promoting the oxidation of n-hexyl amine and 1,6-hexanediamine with high selectivity into the corresponding amides, N-hexyl hexanoic amide and caprolactam, respectively. Such achievements sound particularly strategic as they open up for new and green routes to caprolactam and cyclohexanone oxime, basic precursors for nylon-6.
Finally, it is worth noting Baiker and Mallat's recent works describing a novel generation of gold catalysts for amines oxidation, showing how it is possible to synthesize a valuable catalyst without using any dedicated step for the creation of supported gold nanoparticles.49,50 Among the examined substrates, benzylamine, dibenzylamine and indoline were converted to the corresponding imines in 89–100% yield. By comparison, bulk gold powder did not show any detectable catalytic activity under similar conditions.
6. Gold catalysis in materials science: oxidative polymerization of aniline and pyrrole
Conducting polymers are appealing materials due to their eclectic peculiarities: they display electronic properties of both metals and semiconductors, associated with the mechanic properties of organic macromolecules. Since the end of the 1970s, when the first highly conducting polyacetylene was synthesized, a progressive interest has emerged in the synthesis of other organic conducting polymers mainly polyaniline (PANI) and polypyrrole (PPy). These efforts have led to effective applications of such materials in many devices combining optical, electrochemical and conducting properties, and polyaniline as well as polypyrrole (Scheme 5) are particularly strategic owing to their stability in air and tuneable conductivity.51–54 PANI conductivity is related to the degree of acid-doping (pH) and oxidation state of the material: equal numbers of oxidized and reduced units (emeraldine form), with one proton doping every two units, guarantee optimum conductivity of the polymer.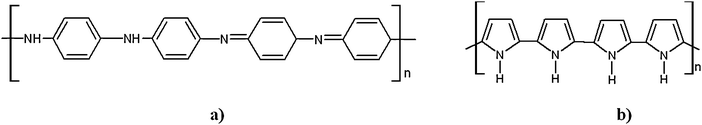 | ||
| Scheme 5 (a) Polyaniline structure, (b) polypyrrole structure. | ||
Another fundamental factor influencing characteristics and performance of the material is the morphology of the conducting polymers. Various geometries such as films, hollow spheres, and nanoparticles allow these materials to be widely employed as OLED and materials for absorption and separation and in drug delivery. Catalytic processes employing transition metals as the catalysts and green oxidizing reagents, such as dioxygen and hydrogen peroxide, represent a strategic tool.
6.1. Polyaniline
Aniline polymerization is traditionally performed through oxidative coupling of aniline and its dimer, N-(4-aminophenyl)aniline, using oxidants such as ammonium peroxydisulfate (APS), K2Cr2O7, KIO3. When aniline is oxidized in an acidic aqueous medium with ammonium peroxydisulfate, a PANI precipitate is produced besides sulfuric acid as a by-product, this latter representing a drawback owing to environmental restrictions. On the other hand, employing metals in a high oxidation state does not offer the best solution.51The new usage of gold and copper as the catalysts for polyaniline synthesis has opened up a novel green route to this conducting material.52,53
We have recently showed that the addition of a gold catalyst (aniline:Au = 100–1000, molar ratio) allowed us to achieve the insoluble green polymer “emeraldine”, then purified by extraction with 1-methyl-2-pirrolidone and evaporation of the solvent under vacuum.51,52
Different gold-based catalytic systems were tested (colloidal gold, gold supported on carbon and gold supported on titania), but Au/TiO2 displayed the best performance, especially in terms of yield to PANI. Regarding oxygen as the oxidizing reagent, actually no oxidative polymerisation of aniline was observed even using different catalytic systems under a slight oxygen pressure (3 bar) at room temperature. Only a scarce catalytic effect (typically 4–5% yield) was detected when a small amount of colloidal gold (Au:aniline = 0.001, molar ratio) was employed as a catalyst in the presence of H2O2. No product was isolated without any catalyst and no benefit was observed from using H2O2 in excess, but the PANI yield could be improved by increasing the gold amount in the range Au:aniline = 0.001–0.004 (molar ratio), thus reaching the asymptotic value of 27% after 1 day reaction.
The well known short lifetime of gold “naked” particles in oxidation reactions19 could explain the modest conversion of aniline to PANI. In order to support such hypothesis, the oxidative polymerisation of aniline was carried out also in the presence of supported gold catalysts (0.5% Au/C, 1% Au/TiO2), under the same experimental conditions ([aniline] = 0.05 M, [HCl] = 0.025 M, T = 293 K, t = 1 day, H2O2/aniline = 1 molar ratio), actually allowing improved performances.
In particular, the superior performance of Au/TiO2 (70% yield to PANI) with respect to Au/C (12% yield to PANI) can be ascribed to a powerful synergistic effect of the supporting material titania with gold. In fact, differently from unloaded carbon which was inert in aniline polymerisation by H2O2, P25 titania itself catalyzed the partial oxidation of aniline to soluble dark oligomers. However, without any gold loading, no long-chain polymer was then formed.
The products obtained in all the preparations were identified as ES (emeraldine salt) according to the FT-IR, UV–vis and XRD spectra, while the morphology of the products was pictured by transmission electron microscopy (TEM) and scanning electron microscopy (SEM), thus showing emeraldine mainly in the form of nanospheres of 44–160 nm obtained both with Au/C and Au/TiO2 catalysts. The conductivity of the polymer obtained in the high-yield conversion of aniline with Au/TiO2 catalyst reached the value of 1.5 × 10−1 S cm−1, as determined with a standard conductivity cell (CON-H Material Mates), and this value is similar to those obtained through other polymerisation methods.51,52
6.2. Polypyrrole
The metal-assisted polymerisation of pyrrole has drawn minor attention when compared to aniline polymerisation, but our interesting findings demonstrate the effectiveness of such a route, by presenting the first example of the facile polymerization of pyrrole using O2 and H2O2 as the oxidants, in aqueous solution and under mild conditions catalysed by colloidal gold.54Before experimenting with the catalyst, blank tests were conducted both with air or oxygen and hydrogen peroxide as the oxidizing reagents: by stirring the aqueous solution of pyrrole hydrochloride at room temperature for several days, under air and in the absence of gold nanoparticles, no insoluble product was collected from the dark solution, meaning that only short-chain oligomers came out from a spontaneous auto-oxidation. The use of pure dioxygen under pressure (3 bar) led to a modest yield of insoluble polymer (12%) after 3 days. On the other hand, with hydrogen peroxide as the oxidant, a notable pyrrole polymerization was achieved even in the absence of any catalyst, thus producing 57% of the polymeric material after 1 day.
The addition of gold, in the form of colloidal nanoparticles, to pyrrole (Py) in acidic (HCl) aqueous solution, under oxygen (pO2 = 3 bar) at room temperature, allowed a slow oxidative polymerization. The observed yields of polypyrrole, up to 75% after 1 day and almost 100% after 3 days, depended on the total gold amount ranging in the interval Py:Au = 1000–10000 molar ratio.
A great improvement in pyrrole polymerisation performance could be obtained employing H2O2, instead of O2, as the oxidizing reagent.51,54
We have already underlined that this oxidant favours the polymerization of pyrrole, stirred in HCl aqueous solution under nitrogen atmosphere and room temperature, even without any catalyst thus leading to a good 57% yield after 1 day.
However, the addition of colloidal gold markedly implemented the polymerisation reaching 99% yield of the product after 24 h (Fig. 3).
![Oxidative polymerization of pyrrole with H2O2, with and without gold. Reaction conditions: [Py] = 0.15 M, [HCl] = 0.15 M, Py/H2O2 = 1, water, T = 20 °C, t = 1 day under an N2 atmosphere.](/image/article/2011/CY/c1cy00283j/c1cy00283j-f3.gif) | ||
| Fig. 3 Oxidative polymerization of pyrrole with H2O2, with and without gold. Reaction conditions: [Py] = 0.15 M, [HCl] = 0.15 M, Py/H2O2 = 1, water, T = 20 °C, t = 1 day under an N2 atmosphere. | ||
As far as the morphological aspects are concerned, a concert of different analytical techniques allowed the characterization of the obtained polypyrrole structures. Independently of the synthetic method, all the prepared polymers displayed a similar IR spectrum.
Also X-ray diffraction patterns of PPy, synthesized with or without gold catalysis, confirmed the typical peaks of such material.
TEM analyses pictured various structures as a consequence of different experimental protocols. The low-yield, non-catalyzed polymerization with gaseous oxygen produced more ordered structures, made up of 40–80 nm spheres, whereas a large amount of peculiar thin squares (20–60 nm) were observed in the high yield polymerization catalysed by gold.
By using H2O2, the quick polymerization without gold catalysis led to a partly reticulated structure, whereas an amorphous material could be achieved in the gold-catalyzed polymerization. This latter structure is unusual, also in the context of conventional polymerizations of pyrrole, and could be of interest for tailor-made composite applications.
The conductivity correlated with the bulk resistance of the synthesized polypyrrole materials ranged from 2.7 × 10−4 to 5 × 10−3 S cm−1. As already observed in the case of polyaniline, also these values are in line with the typical conductivity registered using conventional preparation methods with stoichiometric reagents, but much lower than the values generally found in electrochemical film deposition.
7. Conclusions
The discovery of the eclectic catalytic power of gold has signed a breakthrough in the selective oxidation of organic compounds, thus favouring novel “green” ways towards strategic feedstock testified by the increasing number of papers and patents over the last two decades. In this context where great scientists have started and developed a new chapter of chemistry, Michele Rossi at University of Milano has undoubtedly played an important role. Although methods and techniques need further refinement, the great versatility and effectiveness of gold catalysis are evident in a plenty of different applications ranging from the selective oxidation of alcohols and amines to the novel polymerization of aniline and pyrrole via eco friendly protocols.Acknowledgements
We heartily thank our Mentor Michele Rossi, for his high value as a scientist and as a man. We deeply acknowledge Mario Pagliaro, Graham Hutchings and RSC for the realization of this themed issue dedicated to Michele Rossi.References
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405 CAS.
- B. Nkosi, M. D. Adams, N. J. Coville and G. J. Hutchings, J. Catal., 1991, 128, 378 CrossRef CAS.
- L. Prati and M. Rossi, Stud. Surf. Sci. Catal., 1997, 110, 509 CrossRef CAS.
- L. Prati and M. Rossi, J. Catal., 1998, 176, 552 CrossRef CAS.
- C. Bianchi, F. Porta, L. Prati and M. Rossi, Top. Catal., 2000, 13, 231 CrossRef CAS.
- F. Porta, L. Prati, M. Rossi, S. Coluccia and G. Martra, Catal. Today, 2000, 61, 165 CrossRef CAS.
- S. Biella, L. Prati and M. Rossi, J. Catal., 2002, 206, 242 CrossRef CAS.
- F. Porta, L. Prati, M. Rossi and G. Scari, J. Catal., 2002, 211, 464 CAS.
- F. Porta, L. Prati, M. Rossi and G. Scari, Colloids Surf., A, 2002, 211, 43 CrossRef CAS.
- S. Biella, G. L. Castiglioni, C. Fumagalli, L. Prati and M. Rossi, J. Catal., 2002, 72, 43 CAS.
- S. Biella, L. Prati and M. Rossi, Inorg. Chim. Acta, 2003, 349, 253 CrossRef CAS.
- S. Biella, L. Prati and M. Rossi, J. Mol. Catal. A: Chem., 2003, 197, 207 CrossRef CAS.
- C. L. Bianchi, S. Biella, A. Gervasini, L. Prati and M. Rossi, Catal. Lett., 2003, 85, 91 CrossRef CAS.
- S. Biella, F. Porta, L. Prati and M. Rossi, Catal. Lett., 2003, 90, 23 CrossRef CAS.
- S. Biella and M. Rossi, Chem. Commun., 2003, 378 RSC.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef.
- C. Della Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077 RSC.
- C. Della Pina, E. Falletta and M. Rossi, Chem. Soc. Rev., 2011 10.1039/C1CS15089H.
- M. Comotti, C. Della Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812 CrossRef CAS.
- G. C. Bond and D. Thompson, Catal. Rev. Sci. Eng., 1999, 41, 319 CrossRef CAS.
- M. Comotti, C. Della Pina, E. Falletta and M. Rossi, J. Catal., 2006, 244, 122 CrossRef CAS.
- M. Comotti, C. Della Pina, R. Matarrese, M. Rossi and A. Siani, Appl. Catal., A, 2005, 291, 204 CrossRef CAS.
- C. Baatz, N. Decker and U. Pruesse, J. Catal., 2008, 258, 165 CrossRef CAS.
- C. Baatz and U. Pruesse, J. Catal., 2007, 249, 34 CrossRef CAS.
- T. Ishida, N. Kinoshita, H. Okatsu, T. Akita, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2008, 47, 9265 CrossRef CAS.
- T. Ishida, K. Kuroda, N. Kinoshita, W. Minagawa and M. Haruta, J. Colloid Interface Sci., 2008, 323, 105 CrossRef CAS.
- M. Comotti, C. Della Pina and M. Rossi, J. Mol. Catal. A: Chem., 2006, 251, 92.
- H. Yin, C. Zhou, C. Xu, P. Liu, X. Xu and Y. Ding, J. Phys. Chem. C, 2008, 112, 9673 CrossRef CAS.
- P. Beltrame, M. Comotti, C. Della Pina and M. Rossi, J. Catal., 2004, 228, 282 CrossRef CAS.
- P. Beltrame, M. Comotti, C. Della Pina and M. Rossi, Appl. Catal., A, 2006, 297, 1 CrossRef CAS.
- M. Comotti, C. Della Pina, E. Falletta and M. Rossi, Adv. Synth. Catal., 2006, 348, 313 CrossRef CAS.
- Y. Őnal, S. Schimpf and P. Claus, J. Catal., 2004, 223, 122 CrossRef CAS.
- C. Della Pina, E. Falletta, M. Rossi and A. Sacco, J. Catal., 2009, 263, 92 CrossRef CAS.
- A. Villa, D. Wang, P. Spontoni, R. Arrigo, D. Sub and L. Prati, Catal. Today, 2010, 157, 89 CrossRef CAS.
- P. G. N. Mertens, P. Vendezande, X. Ye, H. Poelman, D. E. De Vos and I. F. J. Vankelecom, Adv. Synth. Catal., 2008, 350, 1241 CrossRef CAS.
- P. J. Miedziak, Z. Tang, T. E. Davies, D. I. Enache, J. K. Bartley, A. F. Carley, A. A. Herzing, C. J. Kiely, S. H. Taylor and G. J. Hutchings, J. Mater. Chem., 2009, 19, 8619 RSC.
- C. Della Pina, E. Falletta and M. Rossi, J. Catal., 2008, 260, 384 CrossRef CAS.
- Y. Liu, H. Tüysüz, C.-J. Jia, M. Schwickardi, R. Rinaldi, A.-H. Lu, W. Schmidt and F. Schüth, Chem. Commun., 2010, 46, 1238 RSC.
- C. Della Pina, E. Falletta and M. Rossi, ChemSusChem, 2009, 2, 57 CrossRef.
- E. Falletta, C. Della Pina, M. Rossi, Q. He, C. J. Kiely and G. J. Hutchings, Faraday Discuss., 2011, 152, 367 RSC.
- S. Biella, G. Castiglioni, C. Fumagalli, L. Prati and M. Rossi, Catal. Today, 2002, 72, 42.
- C. Della Pina, E. Falletta and M. Rossi, Top. Catal., 2007, 44, 325 CrossRef CAS.
- M. Lazar and R. J. Angelici, J. Am. Chem. Soc., 2006, 128, 10613 CrossRef CAS.
- B. Zhu and R. J. Angelici, Chem. Commun., 2007, 2157 RSC.
- B. Zhu, M. Lazar, B. G. Trewyn and R. J. Angelici, J. Catal., 2008, 260, 1 CrossRef CAS.
- J. Gong, T. Yan and C. B. Mullins, Chem. Commun., 2009, 761 RSC.
- A. Grirrane, A. Corma and H. Garcia, J. Catal., 2009, 264, 138 CrossRef CAS.
- S. Kegnaes, J. Mielby, U. V. Mentzel, C. H. Christensen and A. Riisager, Green Chem., 2010, 12, 1437 RSC.
- L. Aschwanden, T. Mallat, F. Krumeich and A. Baiker, J. Mol. Catal. A: Chem., 2009, 309, 57 CrossRef CAS.
- L. Aschwanden, T. Mallat, M. Maciejewski, F. Krumeich and A. Baiker, ChemCatChem, 2010, 2, 666 Search PubMed.
- C. Della Pina, E. Falletta and M. Rossi, Catal. Today, 2011, 160, 11 CrossRef CAS.
- Z. Chen, C. Della Pina, E. Falletta, M. Lo Faro, M. Pasta, M. Rossi and N. Santo, J. Catal., 2008, 259, 1 CrossRef CAS.
- Z. Chen, C. Della Pina, E. Falletta and M. Rossi, J. Catal., 2009, 267, 93 CrossRef CAS.
- C. Della Pina, E. Falletta, M. Lo Faro, M. Pasta and M. Rossi, Gold Bull. (London, U. K.), 2009, 42, 27 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |