Reactions in “sacrificial” solvents
Tamas
Mallat
* and
Alfons
Baiker
*
Institute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, Hönggerberg, HCI, CH-8093 Zurich, Switzerland. E-mail: baiker@chem.ethz.ch; mallat@chem.ethz.ch
First published on 16th August 2011
Abstract
The involvement of organic solvents in redox reactions and their influence on the efficiency and “greenness” of the desired chemical transformation are critically reviewed. Some representative examples are presented on the co-oxidation and co-reduction of widely used solvents (alcohols, ethers, nitriles, amides, sulfoxides, hydrocarbons, dense CO2), and on the impact of these frequently hidden reactions. The transformation of the solvent may be limited to its unintended consumption leading to an increase in the E-factor, but it may also play a critical role in the reaction investigated and lead to false interpretation of reaction mechanisms. In fact, the approach of using a “sacrificial” solvent as a part of the oxidizing system may be an attractive—but surely not a green—alternative in synthetic chemistry. Finally, the crucial but often unrecognized role of the solvent in stabilizing or destabilizing catalytic metal nanoparticles is discussed. Surface redox reactions involving the solvent can be decisive for switching between heterogeneous and homogeneous catalytic pathways.
 Tamas Mallat | Tamas Mallat received his PhD in chemical engineering in 1976 at the Technical University of Budapest, Hungary. He worked as a postdoctoral associate at Brunel University, London and then as a scientist at the Organic Chemical Technology Research Group of the Hungarian Academy of Sciences. In 1990 he moved to ETH Zurich, where he is a senior scientist. His main scientific interest is the application of heterogeneous catalysts in the synthesis of fine chemicals. |
 Alfons Baiker | Alfons Baiker studied chemical engineering at ETH Zurich and earned his Ph.D. degree in 1974. After several postdoctoral stays at various universities abroad he returned to ETH where he moved up to the ranks to become Full Professor in 1990. His research interests, documented in over 850 articles in refereed journals and numerous patents, are centered on catalyst design and novel catalytic materials, mechanisms and kinetics of catalytic surface processes, asymmetric hydrogenation, selective oxidation, environmental catalysis, chiral surfaces, in situ spectroscopy, and the application of supercritical fluids and ionic liquids in catalysis. |
1. Introduction
A basic requirement that a chemical reaction occurs efficiently is that the reactants are optimally mixed and brought into contact with each other on a molecular scale. This is relatively easy to achieve in gas-phase reactions. However, if the reactants possess labile covalent bonds and are relatively involatile, or even solids at the suitable reaction temperature, the use of a solvent becomes inevitable and reactions have to be performed in the liquid phase. This is often the case in organic synthesis. The fact that the interaction forces in solution are much stronger and vary much more than in the gas-phase results in a number of complicating phenomena which are comprised under the term “solvent effect”.The solvent effect is one of the fundamental phenomena in general chemistry, in kinetics and catalysis. Numerous reviews and books addressed this point and suggested the use of various solvent parameters (e.g. polarity, H-bond donor and acceptor properties, acid/base character, state of aggregation) for rationalizing the influence of solvents and understanding the mechanisms of the reactions.1–3 The solvent may play a passive or an active role, as it may be involved in the elementary reaction steps.4,5 Solubility of the reactant, for example oxygen or hydrogen, can be a limiting parameter in oxidation and hydrogenation reactions, respectively. In heterogeneous catalysis the solvent can modify the chemisorption of substrates and products, and thus influence the product distribution.6 Protic solvents may increase the acidity of metal-exchanged zeolites and control the selectivity in epoxidation reactions.5
The solvents are commonly used in a considerable excess relative to the substrate, hence their environmental impact and safety and health issues are also critical in practical applications.7–9 Another important aspect in the selection of the reaction medium is the stability or reactivity. An organic solvent may be transformed to a significant extent under the reaction conditions. In particular, it is rarely considered that the reaction medium itself can be oxidized or reduced and the products may take part in the target reaction by, for example, taking over the role of the actual oxidizing agent. The majority of the common organic solvents are resistant to oxidation in ambient air,3 but under more severe conditions and in the presence of a good catalyst their transformation is not negligible. Undetected involvement of the solvent may lead to a false mechanistic interpretation of the results. This involvement may have an inhibiting or an accelerating effect on the target reaction. Even in the case of a minor conversion of the solvent, the amount of by-products related to the substrate is significant and leads to a decrease of atom economy10 or an increase of the E factor (mass ratio of waste to product).11,12 Considering all these aspects makes the selection of the appropriate solvent a really demanding task.
The topic of this brief review is the frequently overlooked involvement of common solvents in redox reactions during oxidation and hydrogenation type transformations. The collection is not comprehensive; the focus will be on two practically important fields. At first we discuss the so-called “green” oxidation in an organic solvent, which is in many instances only a dream. In this respect, dense CO2 has also its limitation. Another important and new topic is the stability of metal nanoparticles and the role of organic solvents in controlling metal leaching via surface redox reactions.
2. Catalytic oxidation and hydrogenation in organic solvents
The use of molecular oxygen as the source of oxidant without any additive (co-reductant) is commonly considered as an environment friendly oxidation technology, particularly in the presence of a solid, easily recyclable catalyst. Hydrogen peroxide may be an industrially attractive alternative to oxygen,13 since the only co-product is water and handling of the mass transport is easier. In this chapter we will focus on the application of these two oxidants and analyze the stability or reactivity of some common solvents in redox reactions.The reactivity of organic compounds under hydrogenation conditions is well established and the necessary information for their use as solvents can easily be collected from the pertinent literature.14–16 Accordingly, only some special cases will be mentioned here.
2.1 Are alcohols really green solvents?
There is a trend in fine chemical and pharmaceutical industries to replace hydrocarbon and chlorinated hydrocarbon solvents by lower alcohols, esters and ethers.12 In the Pfizer solvent selection guide for medicinal chemistry, and in a recent review on green solvents,17 several short chain aliphatic alcohols appear as preferred solvents in green chemistry.18 The high boiling point alcohols, glycerol,19,20 poly(ethylene glycol)7,9 and poly(propylene glycol),7 are also considered green solvents for various reactions.Admittedly, alcohols are excellent solvents for many purposes but their application in oxidation reactions needs caution. Alcohols are proven reducing agents and commonly used in transfer hydrogenation reactions under mild conditions, in the presence of supported Pd or other noble metal catalysts.21–23 Aerobic oxidation of alcohols and polyols to carbonyl compounds and carboxylic acids are thoroughly investigated transformations, which are accelerated by a broad range of homogeneous24–26 and heterogeneous27–30 catalysts.
Experimental observations also support the limited usefulness of alcohols as inert solvents in oxidation reactions. In the CoCl2 catalyzed oxidative cleavage of α-methylstyrene (Table 1) no conversion of the substrate was detected in 2-propanol, rather the solvent was oxidized extensively to acetone.31 Similar difficulties were encountered with methanol as a solvent. In 1,2-dimethoxyethane the major products were polymers. Oxidation of this solvent produced peroxy radicals that initiated the polymerization of the substrate. The highest yield of 74% was achieved in tert-butanol, which solvent was relatively resistant to self-oxidation, i.e. less peroxides were formed under the reaction conditions than in the case of 1,2-dimethoxyethane. It is also probable that the actual reaction mechanism was not Co-catalyzed aerobic oxidation of α-methylstyrene, but rather the in situ formed tert-butylhydroperoxide was the actual oxidant.
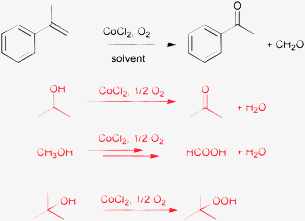
Note that the less reactive tertiary alcohol might be used as a solvent even in the oxidation of primary or secondary alcohols.32 For example, the highly reactive benzylic alcohols can safely be oxidized (dehydrogenated) on Pt-group metal catalysts with molecular oxygen under mild conditions in tert-butanol, but more active catalysts, such as CoCl2 in the previous example, convert also tert-butanol to the corresponding hydroperoxide.
Oxidation of p-cresol (and its aryl-substituted derivatives) with oxygen and various homogeneous and heterogeneous cobalt, copper, manganese, and cerium catalysts provides up to 95% yield to p-hydroxybenzaldehyde (Scheme 1).33–39 Typically, the reaction is carried out in a basic methanolic solution. Peeters and coworkers recognized at first that a considerable fraction of oxygen was consumed by the oxidation of the solvent methanol to formic acid, which was neutralized by the large amount of NaOH.40 The fraction of oxygen used for p-cresol oxidation varied in the range of 9–85%, depending on the catalyst composition and reaction conditions.40,41 Interestingly, most of the reports published after Peeters' criticism do not mention this drawback of the method.
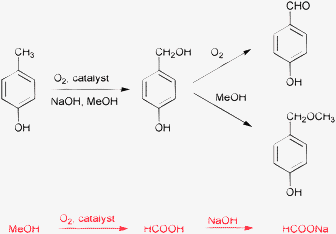 | ||
| Scheme 1 Catalytic oxidation of p-cresol to p-hydroxybenzaldehyde in methanol and the accompanying oxidation of the solvent (40–120 °C and 1–10 bar).40 | ||
A well studied cascade reaction for the production of propene oxide is the use of in situ generated hydrogen peroxide for the epoxidation of propene (Scheme 2).42–44 The catalyst contains Pd and Pt, deposited onto the surface of titanium-substituted molecular sieve TS-1. In this approach the actual oxidant is formed from oxygen and hydrogen on the metal surface, the peroxide migrates to the pores of TS-1 and reacts there with propene. Methanol is the favored solvent, since its interaction with the zeolite surface improves the epoxide selectivity.45 Continuous epoxidation in a fixed bed reactor revealed a serious catalyst deactivation with time on stream and a dramatic shift in the selectivity pattern.46 At the early stage of the reaction the epoxide selectivity was over 90% (at only 3.5% conversion). The rate of epoxide formation decreased with time-on-stream and more and more by-products were formed (acetone, acrolein, acrylic acid, monomethylated glycols, oligomers). After 35 h, propene oxide was barely detectable and methyl formate became the main product. Methyl formate was produced from methanol by oxidation—esterification (Scheme 2). The formic acid intermediate was probably responsible for the acid-catalyzed side reactions and the high molecular weight byproducts blocked the narrow pores of TS-1 and hindered epoxidation. It was found later that dense CO2 was a more appropriate solvent for this process.47,48
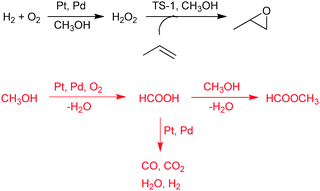 | ||
| Scheme 2 Direct epoxidation of propene with a mixture of H2 and O2 in the presence of a Pt–Pd/TS-1 catalyst, and the competing transformation of the solvent methanol.46 | ||
2.2 Ethers
Ethers are very useful and popular solvents in a variety of organic transformations, although they are known to form peroxides with oxygen under mild conditions, even in the absence of a catalyst.49 The reactivity of THF is similar to that of linear ethers and detectable amounts of hydroperoxide are formed at ambient temperature already in a few hours after distillation (A, Scheme 3).50 The oxidation at the α-carbon of ethers has been thoroughly investigated.51–54 The transformation with molecular oxygen is catalyzed by numerous salts or complexes of Fe,50,55 Co,52,56,57 Cu,58,59 Ni,59 Ag,59 Pt,60 Ru,61,62 Pd,63 Ir and Rh,64 and Sr and Ba.65 The heterogeneous catalysts Pd/C,64 Rh/C, 64 and Pt53,60,62 have also been used to oxidize THF. In the specific case of THF, further transformation of the hydroperoxide (A, Scheme 3) usually gives tetrahydrofuran-2-ol (B), γ-butyrolactone (C), and 4-hydroxybutanal (D, the tautomer of tetrahydrofuran-2-ol), but products of more complex transformations (E,52 F,61 G,61 and H66) have also been described.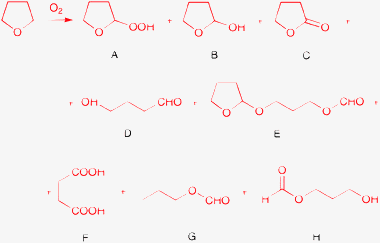
Moreira et al. found a unique solvent effect in the aerobic oxidation of triarylphosphines to oxides (Ar3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in the presence of diiron complexes.66 Under ambient conditions in THF the transformation was complete within a few minutes. In diethyl ether and methyl tert-butyl ether the conversion was ca. 50% after 12 h. The slower reaction may be due to the poor solubility of the catalyst. Replacement of the ethers with toluene, acetone, acetonitrile, chloroform, or dichloromethane suppressed completely the oxidation reaction. A detailed mechanistic study confirmed the involvement of THF in the catalytic cycle. Major products of THF oxidation were A, B, and C (Scheme 3); these species formed even in the absence of the Ar3P substrate. In addition, a minor species H (Scheme 3) was produced in a complex reaction network via C–C bond cleavage, where the substrate acted as a sacrificial reductant.
O) in the presence of diiron complexes.66 Under ambient conditions in THF the transformation was complete within a few minutes. In diethyl ether and methyl tert-butyl ether the conversion was ca. 50% after 12 h. The slower reaction may be due to the poor solubility of the catalyst. Replacement of the ethers with toluene, acetone, acetonitrile, chloroform, or dichloromethane suppressed completely the oxidation reaction. A detailed mechanistic study confirmed the involvement of THF in the catalytic cycle. Major products of THF oxidation were A, B, and C (Scheme 3); these species formed even in the absence of the Ar3P substrate. In addition, a minor species H (Scheme 3) was produced in a complex reaction network via C–C bond cleavage, where the substrate acted as a sacrificial reductant.
It has been recently forwarded that diethoxymethane would be a green industrial solvent, when ethers are the preferred medium for a certain reaction.67 This diether (formaldehyde acetal) has several advantages, including its resistance to peroxide formation upon exposure to air. It was, however, slowly oxidized with Cu- and Co-based catalysts with oxygen,68,69 and the reaction rate was multiplied in the presence of TBHP.69
As mentioned in the previous chapter, in the CoCl2 catalyzed oxidative cleavage of α-methylstyrene (Scheme 1) 1,2-dimethoxyethane was the worst solvent. A peroxide test revealed that oxidation of the diether produced 2.5 times more peroxy radicals than that of tert-butanol and the fast transformation of the solvent was responsible for the shift in the major reaction route from oxidative cleavage to polymerization.31
2.3 Dimethyl sulfoxide, the “sacrificial” solvent
Sulfoxides play an important role in chemistry as solvents, ligands and reagents. The redox chemistry of sulfoxides has been thoroughly investigated. The reactions include the deoxygenation to sulfides (i.e. oxidation with DMSO)70 and the reverse process, the oxidation of sulfides to sulfoxides and further to sulfones (Scheme 4).71–73 Sulfoxides are only weak oxidizing agents, based on the redox potential of the R2SO/R2S system, but their reactivity changes considerably by coordination via the S or O atom. The scope of synthetic methods involving DMSO as a mild and selective oxidant is very broad.74–80 Various transition metal complexes (Ti, Mo, V, Fe, Co, W, Re, Ru, Rh, Pt), and metal ions (Ti, V) incorporated into zeolites, have been found active in oxidations with DMSO. DMSO can be used as an oxygen donor even in the absence of activating reagents.81 On the other hand, sulfoxides and sulfones are oxidized rapidly with peroxides and the acidic products can decompose further peroxide molecules.82–85 | ||
| Scheme 4 Reductive and oxidative transformation of sulfoxides.71–73 | ||
In the light of the redox activity of sulfoxides, inertness is not expected to be a general feature of DMSO in reduction and oxidation reactions.
In the oxidation of internal epoxides with molecular oxygen, Bi(OTf)3 afforded 30–77% yield to α-diketones.86 The reaction proceeded only in DMSO; no diketone was formed in other solvents, such as acetonitrile or DMF. A similar striking solvent effect was described in the Bi-catalyzed aerobic oxidation of styrene oxide to benzoic acid: the reaction was facile in DMSO, but the unchanged epoxide was recovered quantitatively in DMF.87 Some years later a detailed mechanistic study of the oxidation of cyclohexene oxide revealed that the first, critical step is a DMSO-based ring opening to an oxysulfonium intermediate (Scheme 5).88 Oxidation of the intermediate leads to α-ketol and the quantitative formation of dimethyl sulfide. In this transformation DMSO acts as the only oxygen donor. On the contrary, DMSO is not involved in the subsequent step resulting in the α-diketone, which may proceed also in DMF or acetonitrile.
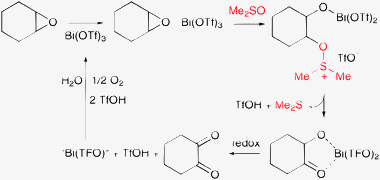 | ||
| Scheme 5 Bismuth-catalyzed oxidation of internal epoxides to α-diketones in DMSO (Me2SO) with molecular oxygen, at 100 °C and 1 bar.88 | ||
Oxidation of 1,2-diarylalkynes to benzyl derivatives with oxygen and a heterogeneous Pd/C catalyst ran smoothly in DMSO, but barely any product formed in toluene or N,N-dimethylacetamide.89 On the other hand, the reaction was extremely slow under Ar. The authors concluded that DMSO was the stoichiometric oxidant and oxygen was required only to oxidize Pd(0) to Pd(II) that was the actual catalyst of the complex transformation (Scheme 6). Interestingly, the reaction was still regarded as an environmentally benign process, despite the consumption of the solvent and the co-formation of two equivalents of dimethyl sulfide. As mentioned previously, we consider only molecular oxygen an environment friendly oxidant—and maybe hydrogen peroxide, if the environmental impact of its synthesis is neglected. Concerning atom economy,10 the above two methods are not more attractive than oxidation with a stoichiometric oxidant, or using oxygen together with a sacrificial aldehyde.90–93
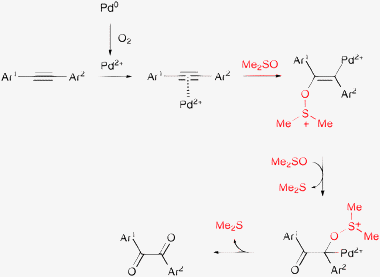 | ||
| Scheme 6 Pd-catalyzed oxidation of 1,2-diarylalkynes to benzyl derivatives with oxygen in DMSO as a solvent and a co-oxidant (120 °C, 1 bar).89 | ||
In the aerobic oxidation of benzyl alcohol, polyoxovanadomolybdate catalysts afforded benzaldehyde in DMSO, but the primary product was dibenzyl ether in other solvents, such as acetonitrile and nitromethane. The unique solvent effect was clarified by a detailed mechanistic study, which confirmed the role of DMSO as a “sacrificial” solvent: it was the actual oxygen donor, not oxygen.74,75
2.4 Further examples of the use of “sacrificial” solvents
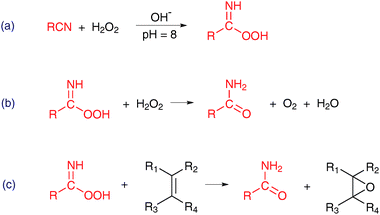 | ||
| Scheme 7 Alkali-catalyzed epoxidation using a nitrile as a co-reductant.95–97 | ||
Nitriles can decompose also organic hydroperoxides, such as TBHP.98 The structure of the intermediate formed from peroxides and nitriles was confirmed by Raman and IR spectroscopy.99,100 It was shown that acetonitrile is more reactive than benzonitrile and the deprotonated form of H2O2 is more reactive than the deprotonated form of TBHP.
Solid base catalysts, such as hydrotalcites, can take over the role of base promotion in the formation of peroxycarboximidic acid and afford excellent yields to epoxides.101–106 Hydrotalcite-based catalysts can promote also the Bayer–Villiger oxidation of ketones using the nitrile–H2O2 system.107 Here again, no reaction occurred without nitrile.108
In the uncatalyzed oxidation of tertiary amines to amine-oxides with H2O2 in neutral solution, addition of acetonitrile induced a rate acceleration by a factor of 200.109
In the oxidation of tetralin in chlorobenzene with oxygen and 2,2′-azobisisobutyronitrile initiator, addition of acetonitrile increased the reaction rate.110 The rate enhancement was proportional to the amount of acetonitrile and it was attributed to a nitrile–tetralin hydroperoxide interaction, analogous to Scheme 7.
In the oxidation of benzene to phenol with H2O2 a Cu(II)-substituted molecular sieve afforded close to 100% selectivity but only in acetonitrile.111
An obvious conclusion from this short collection is that nitrile solvents cannot be considered inert in oxidation reactions with hydroperoxides or when hydroperoxides are formed as reaction intermediates. Curiously, there are still numerous reports where H2O2 is considered as a green, environmentally friendly oxidant and the outstanding rates in the presence of acetonitrile are attributed to its excellent solvent properties.112,113
| Solvent | Conversion (%) | Selectivity (%) | Yield (%) |
|---|---|---|---|
| DMF | 92 | 89 | 82 |
| tert-BuOH | 0 | 0 | 0 |
| Chlorobenzene | 8 | 0 | 0 |
| DMSO | 78 | 6 | 5 |
A common interpretation of the outstanding epoxide selectivity is the aprotic nature and high dielectric constant of DMF.123,125,126 This explanation is, however, not satisfactory. In the epoxidation of styrene a hydroxyapatite-supported Co-oxide catalyst (CoHAp)127,128 gave more than 10% epoxide yield only in dialkylamide type solvents and no reaction was detectable in hexane, ethyl acetate, 1,2-dichloroethane, and tert-BuOH.129 No correlation could be established between the epoxide yield and the typical solvent characteristics such as polarity, acidity, or basicity.
In the case of Co-substituted zeolites, a DMF–Co3+OO˙ superoxo complex was assumed to play the key role in the oxygen transfer to the CC bond (Scheme 8).130–132 A critical question is how the bulky DMF–Co–O–substrate adduct can be accommodated in the small cages of the X-zeolite.130,131
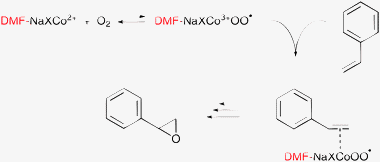
A more probable interpretation of the role of N,N-dialkylamides is related to their reactivity towards oxygen. The basic chemistry and mechanism of the autoxidation of amides was established already in the 1960s.133–135 Oxidation in the absence of added initiator is initially autocatalytic and the abstraction of a H from the C adjacent to N leads to the formation of hydroperoxide. This 1-amidoalkyl hydroperoxide may be the real oxidant of olefins and other substrates in amide solvents, as illustrated in a simplified route in Scheme 9 for olefin epoxidation in DMF. The autoxidation of amides is accelerated by transition metal ions.136–138
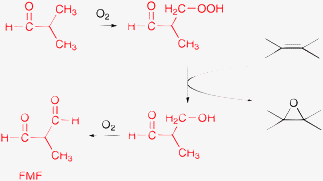 | ||
| Scheme 9 Simplified route to illustrate the role of DMF in the cobalt-catalyzed olefin epoxidation with oxygen and the co-production of N-formyl-N-methylformamide (FMF).133–135 | ||
Decomposition of the 1-amidoalkyl hydroperoxide gives various products, depending on the conditions and the catalyst. In the epoxidation of styrene with oxygen and CoHAp catalyst, N-formyl-N-methylformamide (FMF, Scheme 9) was produced in a considerable amount.129 Formation of FMF was detected only in the presence of styrene and its amount was related to the amount of epoxide. For example, at 110 °C and 1 bar around 10% of DMF was converted to FMF within 2 h and this was not the only product of solvent conversion. These observations support the involvement of DMF solvent as a co-reductant of molecular oxygen in the Co-catalyzed epoxidation reactions.
The cobalt-catalyzed oxidation of the cyclic amide N-methylpyrrolidinone to 5-hydroperoxy-1-methylpyrrolidin-2-one with oxygen and the application of this intermediate in oxidation reactions are also well documented.139–142 In addition, epoxidation of olefins with hydrogen peroxide and an amide is a known, efficient synthetic method.143,144 A key step in this complex transformation is the formation of the corresponding peracid, which is the actual oxidant of the olefin (Scheme 10).
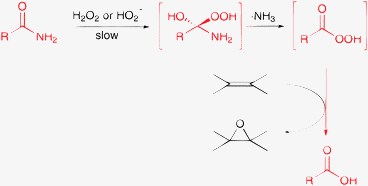 | ||
| Scheme 10 Epoxidation of olefins with H2O2 and amides.143,144 | ||
Summing up, nitriles and amides are excellent solvents in many organic transformations but their role in oxidation reactions with oxygen or H2O2 goes well beyond that of a good solvent. Considering atom economy10 or the E factor,11,12 oxidation in these solvents is not more attractive than the use of a sacrificial aldehyde as a co-reductant.
2.5 Stability of hydrocarbon solvents against oxidation
Hydrocarbons are popular solvents that are considered relatively inert compared to the substrate to be oxidized. For example, toluene has been widely used (also in our laboratory) as a solvent in the aerobic oxidation of alcohols.28,145–149 Recently, in the aerobic oxidation of benzyl alcohol on Pt/ZrO2 the mass balance indicated that a part of the product benzoic acid originated from the solvent toluene (Table 3).150 Note that the excess benzoic acid corresponds to only 0.14% conversion of toluene, which is negligible when the mass balance is related to the solvent.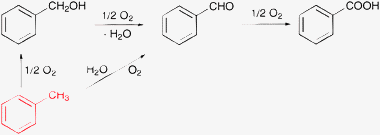
| Solvent | Conversion (%) | Benzaldehyde (%) | Benzoic acid (%) |
|---|---|---|---|
| n-Heptane | 100 | 100 | 0 |
| n-Octane | 43 | 43 | 0 |
| Benzene | 17.3 | 17.3 | 0 |
| Toluene | 15.1 | 0 | 21.6 |
Industrially, the oxidation of toluene to benzoic acid with molecular oxygen is carried out at elevated temperatures, but there are efficient catalysts that activate the C–H bond already at 60–100 °C.151–153 It has also been demonstrated that co-oxidation of benzyl alcohol and benzaldehyde has a strong influence on the autoxidation of toluene.154 In the presence of hydroperoxides numerous catalysts are active in the oxidation of toluene under mild conditions.155–159
Toluene is by far not a unique hydrocarbon based on its reactivity. There is an immense literature on the catalytic oxidation of hydrocarbons with molecular oxygen and hydroperoxides.160–166 Clearly, it is a demanding task to find a chemically suitable and also inert solvent for an oxidation reaction in the presence of an efficient catalyst or a strong oxidant. For example, during epoxidation of cyclohexanol with TBHP at 90 °C even nonane—the solvent of the peroxide—was oxifunctionalized.167 After 4 h the conversion of the uncatalyzed reaction exceeded 1%.
2.6 The green solution: ionic liquid, water, dense CO2, or no solvent?
The previous chapters deal with some well-known and also some less frequently recognized involvement of organic solvents in oxidation reactions. These data show that even under mild conditions and using only molecular oxygen as the oxidant, several widely used solvents are not inert in the presence of an active catalyst. These frequently overlooked or neglected reactions may diminish the ecologic and economic attractivity of the process or lead to a false mechanistic interpretation. A logical conclusion is to avoid the use of organic solvents and apply water or dense CO2, when a solvent is necessary. These media are relatively resistant to redox reactions and commonly considered as green solvents that should be used preferentially in organic transformations.12,168–170 Unfortunately, for many organic compounds they are poor solvents and work best as biphasic systems in the presence of an organic solvent (aqueous biphasic system)7 or an ionic liquid (together with CO2).171,172For polar substrates water is frequently the best medium. Examples include the aerobic oxidation of primary alcohols173–175 and polyols176,177 over Pt-group metal catalysts and the hydroxylation of phenol.178 The latter reaction was carried out with a solid catalyst Cu2(OH)PO4 and hydrogen peroxide, and replacement of water by acetonitrile diminished the conversion to one-third. In acetone and 1,2-dichloroethane the catalyst was inactive.
Dense carbon dioxide is an attractive medium for various reactions168,179–182 including hydrogenation169,183 and aerobic oxidation.170,184,185 Although CO2 is not oxidizable, its reduction is feasible under the usual reaction conditions and this transformation may be at the origin of some unexpected catalytic observations. An illustrating example is the Pt-catalyzed enantioselective hydrogenation of α-ketoesters. Hydrogenation of ethyl pyruvate (Scheme 11, R = H) is more than three times faster in dense ethane than in toluene, under otherwise identical conditions.186 The higher rate was attributed to the higher solubility of hydrogen in ethane. On the contrary, a serious catalyst deactivation was observed in dense CO2. Even in the presence of an organic solvent, addition of CO2 deactivated the catalyst. A similar deactivation during hydrogenation of ethyl benzoylformate (Scheme 11, R = Ph) proved that the catalyst poisoning was not due to the reactivity of the α-H atom in ethyl pyruvate, resulting in site blocking by the high molecular weight byproducts.187,188 ATR-IR spectroscopy of the catalytic solid–liquid interface revealed the hydrogenation of CO2 at the boundary sites of Pt nanoparticles and the Al2O3 support.189 ATR-IR measurements under high pressure conditions proved that the reverse water–gas shift reaction in dense CO2 occurs on all Pt-group metal catalysts.190 In addition to the formation of the strongly adsorbing (poisoning) species CO, its further transformation to methanol and formate on the Pt surface was also detected (Scheme 11). The dramatic poisoning effect is related to the structure sensitivity of the enantioselective hydrogenation reaction. Interaction of the chiral modifier cinchonidine with the substrate forms a bulky adduct that occupies about 20–25 surface Pt atoms.191,192 Simpler reactions require much smaller active site ensembles and are less prone to poisoning by CO.
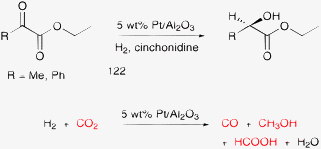 | ||
| Scheme 11 Enantioselective hydrogenation of ethyl pyruvate and ethyl benzoylformate, and that of dense CO2 used as solvent (35 °C, 80 bar CO2, 70 bar H2).187,188 | ||
Since CO adsorbs preferentially on certain surface metal sites,194 formation of CO during reactions in dense CO2 may lead to higher selectivities in some complex reactions due to site blocking. In the partial hydrogenation of 2-chloronitrobenzene in dense CO2, Pt/C allowed almost quantitative yield to 2-chloroaniline (Table 4).193 The solvent-free reaction was slower and less selective. It was shown that the second step, the dehalogenation of 2-chloroaniline was retarded by CO. In a control experiment, injection of a small amount of CO into the reactor in the absence of solvent did not affect the hydrogenation of the nitro group but suppressed dehalogenation.
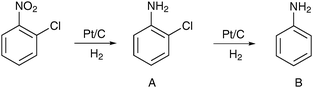
Carbon dioxide may act as a mild oxidant. A well-known example is the oxidative dehydrogenation of ethyl benzene to styrene over V- and Fe-containing catalysts.195,196 Transient response studies in the presence and absence of CO2 showed that lattice oxygen of vanadium oxide and iron oxide was consumed by ethyl benzene and the catalyst was reoxidized by carbon dioxide.197 The necessary reaction temperature is, however, very high, around 550–600 °C.
In the study of Rh-catalyzed hydrogenation of carbon dioxide to formic acid, a significant amount of formic acid was observed already in the absence of hydrogen at close to ambient temperature, when the THF solution of the catalyst [Rh(2,5-norbornene)(PMe2Ph)3]BF4 was pressurized with CO2.198 The dominant reaction is shown in Scheme 12. Addition of oxygen and working at lower pressure (1–5 bar) accelerated the (slow) reaction. Isotope labeling experiments indicated a complex reaction network, where formic acid only partially originated from CO2 and partially from the oxidative degradation of THF. The method could be extended to various other ethers, and also N-methylpyrrole was converted to N-methlypyrrolidone at room temperature.
 | ||
| Scheme 12 Rh-catalyzed aerobic oxidation at the α-C atom of ethers, mediated by carbon dioxide (20–60 °C, 50 bar).198 | ||
Processes based on the use of ionic liquids are generally considered environmentally benign and eco-compatible,199,200 but recent research has raised questions concerning their eco-toxicity and degradability.171,201,202 Various ionic liquids, including some popular alkyl-imidazolium based salts, show acute toxicity for freshwater organisms (algae, fish)203 and some reactivity in hydrogenation and oxidation reactions.204 The catalyst system [nPr4N][RuO4]/O2/2-aminopyridine was used for the oxidation of 4-trifluoromethylbenzyl alcohol to the corresponding aldehyde in the ionic liquid 1-butyl-3-methylimidazolium trifluoromethanesulfonimide ([bmim][(CF3SO2)2N]).205 After reaction at 60 °C, the presence of 1-butyl-3-methylimidazolidone and 1-butyl-3-methylimidazolidine-2,4,5-trione was detected by GC-MS. It was speculated that decomposition of the solvent was due to deprotonation at C2 of the imidazolium ring, followed by the formation of azolone, dearomatization of the ring, and oxidation of the CC bond. Replacement of [bmim][(CF3SO2)2N] by 1-butyl-2,3-dimethylimidazolium trifluoromethanesulfonimide ([bmmim][(CF3SO2)2N]) ceased the oxidation of the ionic liquid and supported the mechanistic considerations of the authors.
Working under solvent-free conditions may offer a solution to these problems (“the best solvent is no solvent”).12 This is the obvious choice to avoid the oxidation of the solvent in reactions carried out under forcing conditions with an active catalyst.206 Unfortunately, the solventless technology is restricted to liquid substrates and products, which is a strong limitation in the synthesis of fine chemicals. Another shortcoming is that under solvent-free conditions the reaction sometimes stops at medium conversions. In the aerobic oxidation of monoterpenic alkenes only 30–40% conversions were obtained (Table 5).207 Addition of fresh catalyst did not help, although the catalyst could be reused several times with fresh reactants. It was speculated that the oxygenated products, accumulated at high concentrations, acted as radical scavengers and retarded the further oxidation.
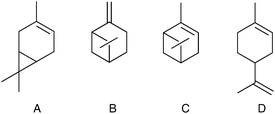
Oxidation of α-pinene208 and the oxidative double bond cleavage of styrenes209 ran smoothly under solvent-free conditions and addition of an organic solvent retarded or completely blocked the transformations. The probable explanation may be again the involvement of solvent as a radical scavenger. An indication to this assumption is that in the Co2+-catalyzed oxidation of α-pinene with molecular oxygen, addition of acetonitrile solvent increased the induction period from 1 h to 3 h, while using old, peroxidized α-pinene eliminated the induction period.208
3. Metal nanoparticles: homogeneous or heterogeneous catalysts depending on the solvent
Another often disregarded role of solvents is related to their influence on the stability of metal nanoparticles. (Note that the following discussion is valid to nanoparticles stabilized by either an organic polymer or a solid surface.)Application of metal nanoparticles in various chemical transformations has been a topic of great academic and industrial importance.210–213 A major application of Pt-group metal nanoparticles is catalytic hydrogenation. In these reactions the metal particles possess high stability and during reaction even soluble metal complexes often form nanoparticles as the actual catalyst under the governing reducing conditions.214 The reason for the high stability is that the metal/hydrogen system (M0/Had) can be considered as a hydrogen electrode,215 the potential of which is determined by the hydrogen concentration at the surface and the pH of the medium (Had ↔ H+ + e−). Under hydrogen “starvation” conditions, i.e. when the slow hydrogen mass transport is the rate limiting step, this protection against oxidation and leaching may not be sufficient. Further risk for metal dissolution is a strongly oxidizing substrate (e.g. a nitro compound), that shifts the mixed potential of the system.216,217 In addition, the presence of a good ligand for the metal (typically N-, S-, and P-containing compounds and carboxylic acids) can stabilize the oxidized (ionic) state. Hydrogenolysis of a Cl-, Br- and I-containing solvent or substrate leads also to the stabilization of the metal ion in solution as halogen-complexes (in the order Cl < Br < I).218
In the presence of oxygen and an appropriate anion or ligand, oxidation of the metal and subsequent leaching become significant, although Pt-group metals may be kept in the reduced, metallic state by the substrate during oxidative dehydrogenation type reactions.32,219–221 In these reactions dehydrogenation of the substrate reduces and molecular oxygen oxidizes the catalyst, an inverted situation compared to hydrogenation reactions. The actual oxidation state of the metal is determined by the relative rates of the two processes.222
The oxidation state of the active metal is less predictable in those transformations, in which neither hydrogen nor oxygen is involved. A good representative with high practical importance is the Pd-catalyzed coupling reactions. There has been an extensive debate on whether the reactions are governed by surface or molecular catalysis, since identification of the active species as a surface site on a tiny nanoparticle or as a dissolved complex is a delicate task.223–226
In the first report on the enantioselective allylic alkylation of rac-(E)-1,3-diphenylallyl acetate, Pd nanoparticles stabilized by a chiral diphosphite were found to act as the actual, heterogeneous catalyst.227Although numerous classical heterogeneity tests supported this conclusion, a subsequent study in a membrane reactor uncovered the molecular nature of the reactive species.228 In the dichloromethane solution at ambient temperature Pd nanoparticles were only the precursors to the active, soluble Pd species.
On the contrary, in the same test reaction Pd/Al2O3 was considered a truly heterogeneous catalyst in the presence of BINAP (Scheme 13) and other diphosphines,229 and also ferrocenyl phosphines,230 as chiral surface modifiers. Major deviation from the original work227,228 was the application of THF instead of dichloromethane as solvent and the relatively high reaction temperature of 60–120 °C. At ambient temperature or in some other solvents the catalyst performance was very poor.
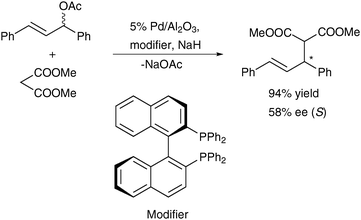 | ||
| Scheme 13 Enantioselective allylic substitution of (E)-1,3-diphenylallyl acetate with dimethyl malonate on Pd/Al2O3 modified by (R)-BINAP (60–120 °C, 6 h).229 | ||
The contradiction was clarified with a combined catalytic–in situ XANES study.231 Pd/Al2O3 stored in air is oxidized at the metal surface. The XANES measurement through the catalyst bed during allylic alkylation demonstrated that the supported Pd particles were rapidly reduced by the reaction mixture already at 60 °C (Fig. 1). The degree of reduction was close to that achieved with dilute hydrogen. No leached Pd was detectable by measurements through the supernatant solution. In a control experiment in neat THF the degree of Pd reduction after 40 min was even higher (Fig. 1), indicating the critical role of the solvent. The probable explanation for the rapid reduction of Pd by THF is the consumption of surface oxygen via oxidation of the ether solvent, as depicted in Scheme 3. This interpretation is supported by reactions in another cyclic ether, dioxane. Also dioxane reduced rapidly Pd under nitrogen and the catalyst was kept in a reduced state during reaction. The reaction was slower but the chemo- and enantioselectivities were similar to that achieved in THF. On the other hand, serious Pd leaching was observed in chloroform.231 Chloroform and dichloromethane, which are popular solvents in alkylation reactions, are easily dehalogenated by Pd.16,232 As discussed previously, these solvents favor leaching of Pd and homogeneous catalysis, a process analogous to the classical metal corrosion.215
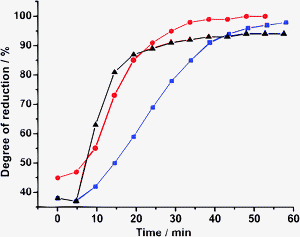 | ||
Fig. 1 Reduction of 5% Pd/Al2O3 at 10 bar N2, as monitored by the linear combination fit using PdO and hydrogen-reduced Pd/Al2O3 in THF as references;  —with 5% H2 in He, —with 5% H2 in He,  —in THF, ▲—in the reaction mixture. The reaction temperature (60 °C) was attained in 10 min.231 —in THF, ▲—in the reaction mixture. The reaction temperature (60 °C) was attained in 10 min.231 | ||
Note also that the alkylation reactions are rarely carried out under truly oxygen-free conditions. Hence, the solvent (and other reaction components that can reduce the metal) has a double role: it has to initially reduce the oxidized metal (Mn+) to the active, metallic state and has to maintain its reduced state during reaction by consuming (reducing) the oxygen diffused into the reactor. In the absence of such a good reducing solvent, homogeneous catalysis is the probable reaction pathway.
The critical role of substrates containing weakly bound halogen is illustrated by the Heck coupling of PhBr with styrene, using the same Pd/Al2O3 catalyst (Scheme 14).233 In the presence of the reactive aryl-bromide substrate, rapid Pd leaching was detected by an in situ quick scanning EXAFS investigation, as soon as the reaction temperature of 150 °C was reached. Changes in the size of the Pd particles during reaction supported the interpretation of the spectra. The solvent N-methylpyrrolidone was expected to promote the metal corrosion by stabilizing the Pd2+ species.
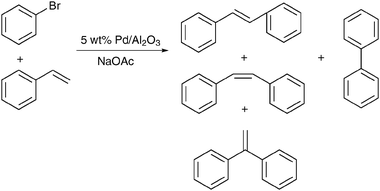 | ||
| Scheme 14 Heck coupling of PhBr with styrene in N-methylpyrrolidone, at 150 °C.233 | ||
4. Summary
A redox reaction in an organic solvent is probably rarely a green process. The only safe way to avoid the unintended involvement of the solvent in redox transformations is to carry out the reactions without solvent. Solvent-free conditions are, however, limited to liquid reactants and products and the yield may also be diminished in the absence of solvent. Water, dense CO2 and ionic liquids are good alternatives but in many cases organic solvents are difficult to substitute, and dense CO2 and ionic liquids are also not truly inert. When the organic reaction medium is inevitable, a careful test of the solvent stability should be included in the selection process. This step is particularly relevant to mechanistic studies and to the test of new, powerful catalysts. Unusually high solvent specificity of a reaction, the appearance of unexpected byproducts, and significant deviation in the mass balance related to the substrate are warning signs that the solvent is not inert in the transformation.On the other hand, involvement of the solvent in redox processes offers an attractive possibility for the synthetic chemist to control a catalytic reaction. As discussed in the last chapter, application of a solvent that acts as a mild reducing agent may be a practically useful approach to stabilize a metal nanoparticle and avoid its leaching (corrosion). In this case the solvent is the reducing agent that consumes the oxygen present in the reaction mixture, but the “price” for this approach is the partial degradation of the solvent. Consideration of the solvent as a potential reducing agent that stabilizes the metallic state, or on the contrary, a reaction component that accelerates metal corrosion and leaching by providing a halogen anion or a good ligand that stabilizes the oxidized metal as a soluble species, may help understanding the reaction mechanism.
References
- J.-L. M. Abboud and R. Notario, Pure Appl. Chem, 1999, 71, 645–718 CrossRef CAS.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, 2003 Search PubMed.
- E. Buncel, R. A. Stairs and H. Wilson, The Role of the Solvent in Chemical Reactions, Oxford Univ. Press, Oxford, 2003 Search PubMed.
- M. G. Clerici, Top. Catal., 2001, 15, 257–263 CrossRef CAS.
- M. Ziolek, Catal. Today, 2004, 90, 145–150 CrossRef CAS.
- L. Gilbert and C. Mercier, in Heterogeneous Catalysis and Fine Chemicals III, ed. M. Guisnet, J. Barbier, J. Barrault, C. Bouchoule, D. Duprez, G. Perot and C. Montassier, 1993, pp. 51–66 Search PubMed.
- R. A. Sheldon, Green Chem., 2005, 7, 267–278 RSC.
- C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–934 RSC.
- N. Yan, C. X. Xiao and Y. Kou, Coord. Chem. Rev., 2010, 254, 1179–1218 CrossRef CAS.
- B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS.
- R. A. Sheldon, Chem. Ind. (London), 1992, 70, 903–906.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- R. A. Sheldon, Top. Curr. Chem., 1993, 164, 21–43 CAS.
- M. Freifelder, Practical Catalytic Hydrogenation, Wiley, New York, 1971 Search PubMed.
- P. Rylander, Catalytic Hydrogenation in Organic Syntheses, Academic Press, New York, 1979 Search PubMed.
- S. Nishimura, Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, Wiley, New York, 2001 Search PubMed.
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- J. Chen, S. K. Spear, J. G. Huddleston and R. D. Rogers, Green Chem., 2005, 7, 64–82 RSC.
- Y. L. Gu and F. Jerome, Green Chem., 2010, 12, 1127–1138 RSC.
- A. Hartikka, S. A. Modin, P. G. Andersson and P. I. Arvidsson, Org. Biomol. Chem., 2003, 1, 2522–2526 RSC.
- J. X. Gao, X. D. Yi, P. P. Xu, C. L. Tang, H. L. Wan and T. Ikariya, J. Organomet. Chem., 1999, 592, 290–295 CrossRef CAS.
- M. L. Kantam, R. S. Reddy, U. Pal, B. Sreedhar and S. Bhargava, Adv. Synth. Catal., 2008, 350, 2231–2235 CrossRef CAS.
- T. Naota, H. Takaya and S.-I. Murahashi, Chem. Rev., 1998, 98, 2599–2660 CrossRef CAS.
- I. E. Marko, P. R. Giles, M. Tsukazaki, I. Chelle-Regnaut, A. Gautier, R. Dumeunier, F. Philippart, K. Doda, J. L. Mutonkole, S. M. Brown and C. J. Urch, Adv. Inorg. Chem., 2004, 56, 211–240 CAS.
- I. W. C. E. Arends, T. Kodama and R. A. Sheldon, Top. Organomet. Chem., 2004, 11, 277–320 CAS.
- M. Besson and P. Gallezot, in Fine Chemicals Through Heterogeneous Catalysis, ed. R. A. Sheldon and H. van Bekkum, Wiley-VCH, Weinheim, 2001, pp. 491–506 Search PubMed.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS.
- B. Z. Zhan and A. Thompson, Tetrahedron, 2004, 60, 2917–2935 CrossRef CAS.
- K. Kaneda, K. Ebitani, T. Mizugaki and K. Mori, Bull. Chem. Soc. Jpn., 2006, 79, 981–1016 CrossRef CAS.
- Y. H. Lin, I. D. Williams and P. Li, Appl. Catal., A, 1997, 150, 221–229 CrossRef CAS.
- T. Mallat and A. Baiker, Catal. Today, 1994, 19, 247–284 CrossRef CAS.
- S. N. Sharma and S. B. Chandalia, J. Chem. Technol. Biotechnol., 1990, 49, 141–153 CAS.
- T. Yoshikuni, J. Mol. Catal., 1992, 72, 29–36 CrossRef CAS.
- Y. M. Liu, S. T. Liu, K. Z. Zhu, X. K. Ye and Y. Wu, Appl. Catal., A, 1998, 169, 127–135 CrossRef CAS.
- T. Joseph, C. S. Sajanikumari, S. S. Deshpande and S. Gopinathan, Indian J. Chem., Sect. A: Inorg., Bio-Inorg., Phys., Theor. Anal. Chem., 1999, 38, 792–796.
- F. Wang, G. Y. Yang, W. Zhang, W. H. Wu and J. Xu, Chem. Commun., 2003, 1172–1173 RSC.
- F. Wang, G. Y. Yang, W. Zhang, W. H. Wu and J. Xu, Adv. Synth. Catal., 2004, 346, 633–638 CrossRef CAS.
- C. V. Rode, V. S. Kshirsagar, J. M. Nadgeri and K. R. Patil, Ind. Eng. Chem. Res., 2007, 46, 8413–8419 CrossRef CAS.
- M. P. J. Peeters, M. Busio and P. Leijten, Appl. Catal., A, 1994, 118, 51–62 CrossRef CAS.
- D. S. Kim, S. H. Chang and W. S. Ahn, J. Mol. Catal. A: Chem., 2002, 179, 175–183 CrossRef CAS.
- M. G. Clerici and P. Ingallina, Catal. Today, 1998, 41, 351–364 CrossRef CAS.
- R. Meiers, U. Dingerdissen and W. F. Hölderich, J. Catal., 1998, 176, 376–386 CrossRef CAS.
- R. Meiers and W. F. Hölderich, Catal. Lett., 1999, 59, 161–163 CrossRef CAS.
- M. G. Clerici and P. Ingallina, J. Catal., 1993, 140, 71–83 CrossRef CAS.
- G. Jenzer, T. Mallat, M. Maciejewski, F. Eigenmann and M. Baiker, Appl. Catal., A, 2001, 208, 125–133 CrossRef CAS.
- Q. L. Chen and E. J. Beckman, Green Chem., 2007, 9, 802–808 RSC.
- Q. L. Chen and E. J. Beckman, Green Chem., 2008, 10, 934–938 RSC.
- T. Uchida, M. Wakakura, A. Miyake and T. Ogawa, J. Therm. Anal. Calorim., 2008, 93, 247–251 CrossRef CAS.
- H. Rein, Angew. Chem., Int. Ed., 1950, 62, 120 Search PubMed.
- J. A. Howard and K. U. Ingold, Can. J. Chem., 1969, 47, 3809–3815 CAS.
- P. Li and H. Alper, J. Mol. Catal., 1992, 72, 143–152 CrossRef CAS.
- T. J. Wang, Z. H. Ma, M. Y. Huang and Y. Y. Jiang, Polym. Adv. Technol., 1996, 7, 88–91 CrossRef CAS.
- R. C. Luckay, X. Sheng, C. E. Strasser, H. G. Raubenheimer, D. A. Safin, M. G. Babashkina and A. Klein, New J. Chem., 2010, 34, 2835–2840 RSC.
- M. T. Hay, S. J. Geib and D. A. Pettner, Polyhedron, 2009, 28, 2183–2186 CrossRef CAS.
- E. Hata, T. Takai and T. Mukaiyama, Chem. Lett., 1993, 1513–1516 CAS.
- M. T. Reetz and K. Tollner, Tetrahedron Lett., 1995, 36, 9461–9464 CrossRef CAS.
- S. Minakata, E. Imai, Y. Ohshima, K. Inaki, I. Ryu, M. Komatsu and Y. Ohshiro, Chem. Lett., 1996, 19–20 CAS.
- A. M. Romano and M. Ricci, J. Mol. Catal. A: Chem., 1997, 120, 71–74 CrossRef CAS.
- A. Sen, M. R. Lin, L. C. Kao and A. C. Hutson, J. Am. Chem. Soc., 1992, 114, 6385–6392 CrossRef CAS.
- T. Straub and A. M. P. Koskinen, Inorg. Chem. Commun., 2002, 5, 1052–1055 CrossRef CAS.
- A. Datta, M. Agarwal, S. Dasgupta and R. Y. Kelkar, J. Mol. Catal. A: Chem., 2003, 198, 205–214 CrossRef CAS.
- M. Sommovigo and H. Alper, J. Mol. Catal., 1994, 88, 151–158 CrossRef CAS.
- M. Shi, J. Chem. Res., Synop., 1998, 592–593 RSC.
- J. Morales-Juarez, R. Cea-Olivares, M. M. Moya-Cabrera, V. Jancik, V. Garcia-Montalvo and R. A. Toscano, Inorg. Chem., 2005, 44, 6924–6926 CrossRef CAS.
- R. F. Moreira, E. Y. Tshuva and S. J. Lippard, Inorg. Chem., 2004, 43, 4427–4434 CrossRef CAS.
- M. T. Coleman, Chim. Oggi, 2009, 27, 43–45 CAS.
- T. Ikoma, M. Takemoto and A. Okamoto, Jpn. Kokai Tokkyo Koho, Mitsubishi Gas Chem. Co., JP 1998-285353, Japan, 2000 Search PubMed.
- A. Corma, M. E. Domine and M. Renz, Cons. Superior de Investig. Cientif., Univ. Politec. Valencia, WO 2005123654, Spain 2005 Search PubMed.
- V. Y. Kukushkin, Coord. Chem. Rev., 1995, 139, 375–407 CrossRef CAS.
- V. Hulea and P. Moreau, J. Mol. Catal. A: Chem., 1996, 113, 499–505 CrossRef CAS.
- P. Moreau, V. Hulea, S. Gomez, D. Brunel and F. DiRenzo, Appl. Catal. A: Gen., 1997, 155, 253–263 CrossRef CAS.
- A. L. Maciuca, E. Dumitriu, F. Fajula and V. Hulea, Chemosphere, 2007, 68, 227–233 CrossRef CAS.
- A. M. Khenkin and R. Neumann, J. Org. Chem., 2002, 67, 7075–7079 CrossRef CAS.
- R. Neumann and A. M. Khenkin, Chem. Commun., 2006, 2529–2538 RSC.
- T. T. Tidwell, Synthesis, 1990, 857–870 CrossRef CAS.
- W. Li, J. C. Li, M. Lin, S. Wacharasindhu, K. Tabei and T. S. Mansour, J. Org. Chem., 2007, 72, 6016–6021 CrossRef CAS.
- A. M. Khenkin and R. Neumann, J. Am. Chem. Soc., 2002, 124, 4198–4199 CrossRef CAS.
- W. Li, J. C. Li, Z. K. Wan, J. J. Wu and W. Massefski, Org. Lett., 2007, 9, 4607–4610 CrossRef CAS.
- K. Görmer, H. Waldmann and G. Triola, J. Org. Chem., 2010, 75, 1811–1813 CrossRef.
- W. Li, H. C. Li, D. DeVincentis and T. S. Mansour, Tetrahedron Lett., 2004, 45, 1071–1074 CrossRef CAS.
- K. U. Ingold, Chem. Rev., 1961, 61, 563–589 CrossRef CAS.
- A. M. Cojocariu, P. H. Mutin, E. Dumitriu, A. Vioux, F. Fajula and V. Hulea, Chemosphere, 2009, 77, 1065–1068 CrossRef CAS.
- J. J. Wu, M. Muruganandham, J. S. Yang and S. S. Lin, Catal. Commun., 2006, 7, 901–906 CrossRef CAS.
- R. Asatryan and J. W. Bozzelli, Phys. Chem. Chem. Phys., 2008, 10, 1769–1780 RSC.
- S. Antoniotti and E. Dunach, Chem. Commun., 2001, 2566–2567 RSC.
- C. Coin, V. Le Boisselier, I. Favier, M. Postel and E. Dunach, Eur. J. Org. Chem., 2001, 735–740 CrossRef CAS.
- S. Antoniotti and E. Dunach, Eur. J. Org. Chem., 2004, 3459–3464 CrossRef CAS.
- S. Mori, M. Takubo, T. Yanase, T. Maegawa, Y. Monguchi and H. Sajiki, Adv. Synth. Catal., 2010, 352, 1630–1634 CrossRef CAS.
- P. Mastrorilli, C. F. Nobile, G. P. Suranna and L. Lopez, Tetrahedron, 1995, 51, 7943–7950 CrossRef CAS.
- B. B. Wentzel, P. A. Gosling, M. C. Feiters and R. J. M. Nolte, J. Chem. Soc., Dalton Trans., 1998, 2241–2246 RSC.
- R. I. Kureshy, N. H. Khan, S. H. R. Abdi, P. Iyer and S. T. Patel, Polyhedron, 1999, 18, 1773–1777 CrossRef CAS.
- N. Theyssen, Z. S. Hou and W. Leitner, Chem.–Eur. J., 2006, 12, 3401–3409 CrossRef.
- K. B. Wiberg, J. Am. Chem. Soc., 1953, 75, 3961–3964 CrossRef CAS.
- G. B. Payne, P. H. Williams and P. H. Deming, J. Org. Chem., 1961, 26, 659–663 CrossRef CAS.
- G. B. Payne and P. H. Williams, J. Org. Chem., 1961, 26, 651–659 CrossRef CAS.
- G. B. Payne, Tetrahedron, 1962, 18, 763–765 CrossRef CAS.
- H. Berger, Trans. Faraday Soc., 1962, 58, 1137–1147 RSC.
- V. Vacque, N. Dupuy, B. Sombret, J. P. Huvenne and P. Legrand, J. Mol. Struct., 1996, 384, 165–174 CrossRef CAS.
- V. Vacque, N. Dupuy, B. Sombret, J. P. Huvenne and P. Legrand, J. Mol. Struct., 1997, 410, 555–558.
- S. Ueno, K. Yamaguchi, K. Yoshida, K. Ebitani and K. Kaneda, Chem. Commun., 1998, 295–296 RSC.
- M. L. A. von Holleben, P. R. Livotto and C. M. Schuch, J. Braz. Chem. Soc., 2001, 12, 42–46 CrossRef CAS.
- K. Kaneda, K. Yamaguchi, K. Mori, T. Mizugaki and K. Ebitani, Catal. Surv. Jpn., 2000, 4, 31–38 CrossRef CAS.
- M. A. Aramendia, V. Borau, C. Jimenez, J. M. Luque, J. M. Marinas, J. R. Ruiz and F. J. Urbano, Appl. Catal., A, 2001, 216, 257–265 CrossRef CAS.
- I. Kirm, F. Medina, X. Rodriguez, Y. Cesteros, P. Salagre and J. Sueiras, Appl. Catal., A, 2004, 272, 175–185 CrossRef CAS.
- R. Ionescu, O. D. Pavel, R. Birjega, R. Zavoianu and E. Angelescu, Catal. Lett., 2010, 134, 309–317 CrossRef CAS.
- U. R. Pillai and E. Sahle-Demessie, J. Mol. Catal. A: Chem., 2003, 191, 93–100 CrossRef CAS.
- J. R. Ruiz, C. Jimenez-Sanchidrian and R. Llamas, Tetrahedron, 2006, 62, 11697–11703 CrossRef CAS.
- G. Laus, J. Chem. Soc., Perkin Trans. 2, 2001, 2, 864–868 Search PubMed.
- S. N. Chaudhuri and U. S. Nandi, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 1976, 14, 759–762 CAS.
- B. Y. Chou, J. L. Tsai and S. F. Cheng, Microporous Mesoporous Mater., 2001, 48, 309–317 CrossRef CAS.
- R. K. Jha, S. Shylesh, S. S. Bhoware and A. P. Singh, Microporous Mesoporous Mater., 2006, 95, 154–163 CrossRef CAS.
- Y. Y. Karabach, A. M. Kirillov, M. Haukka, M. N. Kopylovich and A. J. L. Pombeiro, J. Inorg. Biochem., 2008, 102, 1190–1194 CrossRef CAS.
- S. R. Gun and U. S. Nandi, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 1977, 15, 483–485.
- F. Mizukami, Y. Horiguchi, M. Tajima and J. Imamura, Bull. Chem. Soc. Jpn., 1979, 52, 2689–2695 CrossRef CAS.
- M. Gupta, S. Paul, R. Gupta and A. Loupy, Tetrahedron Lett., 2005, 46, 4957–4960 CrossRef CAS.
- Y. K. Liu, D. Q. Xu, Z. Y. Xu and Y. M. Zhang, Synlett, 2007, 1671–1674.
- J. Liang, Q. H. Tang, G. Y. Meng, H. L. Wu, Q. H. Zhang and Y. Wang, Chem. Lett., 2004, 33, 1140–1141 CrossRef CAS.
- Q. H. Tang, Q. H. Zhang, H. L. Wu and Y. Wang, J. Catal., 2005, 230, 384–397 CrossRef.
- G. Xu, Q. H. Xia, X. H. Lu, Q. Zhang and H. J. Zhan, J. Mol. Catal. A: Chem., 2007, 266, 180–187 CrossRef CAS.
- J. Liang, Q. H. Zhang, H. L. Wu, G. Y. Meng, Q. H. Tang and Y. Wang, Catal. Commun., 2004, 5, 665–669 CrossRef CAS.
- X. Y. Quek, Q. H. Tang, S. Q. Hu and Y. H. Yang, Appl. Catal., A, 2009, 361, 130–136 CrossRef CAS.
- H. J. Zhan, Q. H. Xia, X. H. Lu, Q. Zhang, H. X. Yuan, K. X. Su and X. T. Ma, Catal. Commun., 2007, 8, 1472–1478 CrossRef CAS.
- M. L. Kantam, B. P. C. Rao, R. S. Reddy, N. S. Sekhar, B. Sreedhar and B. M. Choudary, J. Mol. Catal. A: Chem., 2007, 272, 1–5 CrossRef CAS.
- S. N. Rao, K. N. Munshi and N. N. Rao, J. Mol. Catal. A: Chem., 2000, 156, 205–211 CrossRef CAS.
- Q. H. Tang, Y. Wang, J. Liang, P. Wang, Q. H. Zhang and H. L. Wan, Chem. Commun., 2004, 440–441 RSC.
- Z. Opre, J. D. Grunwaldt, M. Maciejewski, D. Ferri, T. Mallat and A. Baiker, J. Catal., 2005, 230, 406–419 CrossRef CAS.
- Z. Opre, D. Ferri, F. Krumeich, T. Mallat and A. Baiker, J. Catal., 2007, 251, 48–58 CrossRef CAS.
- Z. Opre, T. Mallat and A. Baiker, J. Catal., 2007, 245, 482–486 CrossRef CAS.
- J. Sebastian, K. M. Jinka and R. V. Jasra, J. Catal., 2006, 244, 208–218 CrossRef CAS.
- K. M. Jinka, J. Sebastian and R. V. Jasra, J. Mol. Catal. A: Chem., 2007, 274, 33–41 CrossRef CAS.
- M. V. Patil, M. K. Yadav and R. V. Jasra, J. Mol. Catal. A: Chem., 2007, 277, 72–80 CrossRef CAS.
- M. V. Lock and B. F. Sagar, J. Chem. Soc. B, 1966, 690–696 RSC.
- B. F. Sagar, J. Chem. Soc. B, 1967, 1047–1061 RSC.
- B. F. Sagar, J. Chem. Soc. B, 1967, 428–429 RSC.
- S. K. Dhar, J. Abdelaziz, P. Cozzi, P. Jasien, C. Mason and R. Zalenas, J. Chem. Soc., Chem. Commun., 1984, 8–9 Search PubMed.
- J. Runge, Z. Chem., 1977, 17, 265–266 CAS.
- P. L. Timmanagoudar, G. A. Hiremath and S. T. Nandibewoor, J. Indian Chem. Soc., 1997, 74, 296–298 CAS.
- R. S. Drago and R. Riley, J. Am. Chem. Soc., 1990, 112, 215–218 CrossRef CAS.
- H. Alper and M. Harustiak, J. Mol. Catal., 1993, 84, 87–92 CrossRef CAS.
- D. E. Patton and R. S. Drago, J. Chem. Soc., Perkin Trans. 1, 1993, 1611–1615 RSC.
- O. B. Ryan, D. E. Akporiaye, K. H. Holm and M. Stocker, Stud. Surf. Sci. Catal., 1997, 108, 369–376 CAS.
- Y. W. Chen and J. L. Reymond, Tetrahedron Lett., 1995, 36, 4015–4018 CrossRef CAS.
- K. Yamaguchi, K. Ebitani and K. Kaneda, J. Org. Chem., 1999, 64, 2966–2968 CrossRef CAS.
- V. D. Makwana, Y. C. Son, A. R. Howell and S. L. Suib, J. Catal., 2002, 210, 46–52 CrossRef CAS.
- K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657–10666 CrossRef CAS.
- Z. Opre, J. D. Grunwaldt, T. Mallat and A. Baiker, J. Mol. Catal. A: Chem., 2005, 242, 224–232 CrossRef CAS.
- S. E. J. Hackett, R. M. Brydson, M. H. Gass, I. Harvey, A. D. Newman, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2007, 46, 8593–8596 CrossRef CAS.
- X. M. Yang, X. N. Wang and J. S. Qiu, Appl. Catal., A, 2010, 382, 131–137 CrossRef CAS.
- M. Ilyas and M. Sadiq, Chem. Eng. Technol., 2007, 30, 1391–1397 CrossRef CAS.
- F. Lopez-Linares, O. Colmenares, E. Catari and A. Karam, React. Kinet. Catal. Lett., 2005, 85, 139–144 CrossRef CAS.
- X. B. Zuo, B. Subramaniam and D. H. Busch, Ind. Eng. Chem. Res., 2008, 47, 546–552 CrossRef CAS.
- M. Ilyas and M. Sadiq, Catal. Lett., 2009, 128, 337–342 CrossRef CAS.
- I. Hermans, J. Peeters, L. Vereecken and P. A. Jacobs, ChemPhysChem, 2007, 8, 2678–2688 CrossRef CAS.
- V. I. Parvulescu, D. Dumitriu and G. Poncelet, J. Mol. Catal. A: Chem., 1999, 140, 91–105 CrossRef CAS.
- S. Velu, N. Shah, T. M. Jyothi and S. Sivasanker, Microporous Mesoporous Mater., 1999, 33, 61–75 CrossRef CAS.
- D. Dumitriu, R. Barjega, L. Frunza, D. Macovei, T. Hu, Y. Xie, V. I. Parvulescu and S. Kaliaguine, J. Catal., 2003, 219, 337–351 CrossRef CAS.
- A. V. Shijina and N. K. Renuka, React. Kinet. Catal. Lett., 2008, 94, 261–270 CrossRef CAS.
- X. Q. Wang, J. P. Wu, M. W. Zhao, Y. F. Lv, G. Y. Li and C. W. Hu, J. Phys. Chem. C, 2009, 113, 14270–14278 CrossRef CAS.
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS.
- G. B. Shul'pin, J. Mol. Catal. A: Chem., 2002, 189, 39–66 CrossRef CAS.
- T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2363 CrossRef CAS.
- I. Hermans, P. A. Jacobs and J. Peeters, Chem.–Eur. J., 2006, 12, 4230–4240.
- T. Punniyamurthy and L. Rout, Coord. Chem. Rev., 2008, 252, 134–154 CAS.
- I. Hermans, J. Peeters and P. A. Jacobs, Top. Catal., 2008, 48, 41–48 CrossRef CAS.
- B. P. C. Hereijgers and B. M. Weckhuysen, J. Catal., 2010, 270, 16–25 CrossRef CAS.
- C. Beck, T. Mallat and A. Baiker, Catal. Lett., 2001, 75, 131–136 CrossRef CAS.
- B. Subramaniam, C. J. Lyon and V. Arunajatesan, Appl. Catal., B, 2002, 37, 279–292 CrossRef CAS.
- T. Seki, J. D. Grunwaldt and A. Baiker, Ind. Eng. Chem. Res., 2008, 47, 4561–4585 CrossRef CAS.
- T. Seki and M. Baiker, Chem. Rev., 2009, 109, 2409–2454 CrossRef CAS.
- S. Keskin, D. Kayrak-Talay, U. Akman and O. Hortacsu, J. Supercrit. Fluids, 2007, 43, 150–180 CrossRef CAS.
- F. Jutz, J. M. Andanson and A. Baiker, Chem. Rev., 2011, 111, 322–353 CrossRef CAS.
- T. Mallat, Z. Bodnar, A. Baiker, O. Greis, H. Strübig and A. Reller, J. Catal., 1993, 142, 237 CrossRef CAS.
- P. Gallezot, Catal. Today, 1997, 37, 405–418 CrossRef CAS.
- J. H. J. Kluytmans, A. P. Markusse, B. F. M. Kuster, G. B. Marin and J. C. Schouten, Catal. Today, 2000, 57, 143–155 CrossRef.
- H. van Bekkum, in Carbohydrates as Organic Raw Materials, ed. F. W. Lichtenthaler, VCH, Weinheim, 1991, p. 289 Search PubMed.
- P. Vinke, D. de Wit, A. T. J. W. de Goede and H. van Bekkum, Stud. Surf. Sci. Catal., 1992, 72, 1–20 CrossRef CAS.
- F. S. Xiao, J. M. Sun, X. J. Meng, R. B. Yu, H. M. Yuan, J. N. Xu, T. Y. Song, D. Z. Jiang and R. R. Xu, J. Catal., 2001, 199, 273–281 CrossRef CAS.
- A. Baiker, Chem. Rev., 1999, 99, 453–473 CrossRef CAS.
- J. R. Hyde, P. Licence, D. Carter and M. Poliakoff, Appl. Catal., A, 2001, 222, 119–131 CrossRef CAS.
- R. S. Oakes, A. A. Clifford and C. M. Rayner, J. Chem. Soc., Perkin Trans. 1, 2001, 917–941 RSC.
- W. Leitner, Acc. Chem. Res., 2002, 35, 746–756 CrossRef CAS.
- G. P. Li, H. F. Jiang and J. H. Li, Prog. Chem., 2001, 13, 455–460 Search PubMed.
- S. Campestrini and U. Tonellato, Curr. Org. Chem., 2005, 9, 31–47 CrossRef CAS.
- M. Wei, G. T. Musie, D. H. Busch and B. Subramaniam, J. Am. Chem. Soc., 2002, 124, 2513–2517 CrossRef CAS.
- B. Minder, T. Mallat, K. H. Pickel, K. Steiner and A. Baiker, Catal. Lett., 1995, 34, 1–9 CrossRef CAS.
- J. M. Bonello, R. M. Lambert, N. Künzle and A. Baiker, J. Am. Chem. Soc., 2000, 122, 9864–9865 CrossRef CAS.
- M. von Arx, T. Mallat and A. Baiker, Top. Catal., 2002, 19, 75–87 CrossRef CAS.
- D. Ferri, T. Bürgi and A. Baiker, Phys. Chem. Chem. Phys., 2002, 4, 2667–2672 RSC.
- M. Burgener, D. Ferri, J. D. Grünwaldt, T. Mallat and A. Baiker, J. Phys. Chem. B, 2005, 109, 16794–16800 CrossRef CAS.
- T. Bürgi and A. Baiker, Acc. Chem. Res., 2004, 37, 909–917 CrossRef.
- T. Mallat, E. Orglmeister and A. Baiker, Chem. Rev., 2007, 107, 4863–4890 CrossRef CAS.
- S. Ichikawa, M. Tada, Y. Iwasawea and T. Ikariya, Chem. Commun., 2005, 924–926 RSC.
- J. Z. Xu and J. T. Yates, Surf. Sci., 1995, 327, 193–201 CrossRef CAS.
- Y. Sakurai, T. Suzaki, K. Nakagawa, N. Ikenaga, H. Aota and T. Suzuki, Chem. Lett., 2000, 526–527 CrossRef CAS.
- B. M. Reddy, S. C. Lee, D. S. Han and S. E. Park, Appl. Catal., B, 2009, 87, 230–238 CrossRef CAS.
- K. Saito, K. Okuda, N. Ikenaga, T. Miyake and T. Suzuki, J. Phys. Chem. A, 2010, 114, 3845–3854 CrossRef CAS.
- A. K. Fazlur-Rahman, J. C. Tsai and K. M. Nicholas, J. Chem. Soc., Chem. Commun., 1992, 1334–1335 RSC.
- W. M. Nelson, in Ionic Liquids-Industrial Applications for Green Chemistry, ed. R. D. Rogers and K. R. Seddon, ACS Symposium Series, 2002, pp. 30–41 Search PubMed.
- N. Isambert, M. D. S. Duque, J. C. Plaquevent, Y. Genisson, J. Rodriguez and T. Constantieux, Chem. Soc. Rev., 2011, 40, 1347–1357 RSC.
- S. Zhu, R. Chen, Y. Wu, Q. Chen, X. Zhang and Z. Yu, Chem. Biochem. Eng. Q., 2009, 23, 207–211 Search PubMed.
- M. Petkovic, K. R. Seddon, L. P. N. Rebelo and C. S. Pereira, Chem. Soc. Rev., 2011, 40, 1383–1403 RSC.
- C. Pretti, C. Chiappe, I. Baldetti, S. Brunini, G. Monni and L. Intorre, Ecotoxicol. Environ. Saf., 2009, 72, 1170–1176 CrossRef.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS.
- V. Farmer and T. Welton, Green Chem., 2002, 4, 97–102 RSC.
- B. M. Choudary, V. Bhuma and N. Narender, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1997, 36, 278–280.
- L. Menini, M. C. Pereira, L. A. Parreira, J. D. Fabris and E. V. Gusevskaya, J. Catal., 2008, 254, 355–364 CrossRef CAS.
- M. K. Lajunen, T. Maunula and A. M. P. Koskinen, Tetrahedron, 2000, 56, 8167–8171 CrossRef CAS.
- Y. Hayashi, M. Takeda, Y. Miyamoto and M. Shoji, Chem. Lett., 2002, 414–415 CrossRef CAS.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS.
- C. Burda, X. B. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS.
- P. Barbaro, V. Dal Santo and F. Liguori, Dalton Trans., 2010, 8391–8402 Search PubMed.
- K. Mori and H. Yamashita, Phys. Chem. Chem. Phys., 2010, 12, 14420–14432 RSC.
- L. D. Pachon and G. Rothenberg, Appl. Organomet. Chem., 2008, 22, 288–299 CrossRef.
- S. Szabo and I. Bakos, Int. J. Corros., 2010, 1–20 Search PubMed.
- K. Schwabe and A. Satsco, J. Electroanal. Chem., 1966, 11, 308–309 Search PubMed.
- T. Mallat and A. Baiker, Top. Catal., 1999, 8, 115–124 CrossRef CAS.
- D. V. Tripkovic, D. Strmcnik, D. van der Vliet, V. Stamenkovic and N. M. Markovic, Faraday Discuss., 2008, 140, 25–40 Search PubMed.
- P. J. M. Dijkgraaf, H. A. M. Duisters, B. F. M. Kuster and K. van der Wiele, J. Catal., 1988, 112, 337–344 CAS.
- J. W. Nicoletti and G. M. Whitesides, J. Phys. Chem., 1989, 93, 759–767 CrossRef CAS.
- K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2003, 42, 1480–1483 CrossRef CAS.
- T. Mallat and A. Baiker, Catal. Today, 1995, 24, 143–150 CrossRef CAS.
- J. G. de Vries, Dalton Trans., 2006, 421–429 RSC.
- I. Favier, M. Gomez, G. Muller, M. R. Axet, S. Castillon, C. Claver, S. Jansat, B. Chaudret and K. Philippot, Adv. Synth. Catal., 2007, 349, 2459–2469 CrossRef CAS.
- D. Astruc, Inorg. Chem., 2007, 46, 1884–1894 CrossRef CAS.
- J. Durand, E. Teuma and M. Gomez, Eur. J. Inorg. Chem., 2008, 3577–3586 CrossRef CAS.
- S. Jansat, M. Gomez, K. Philippot, G. Muller, E. Guiu, C. Claver, S. Castillon and B. Chaudret, J. Am. Chem. Soc., 2004, 126, 1592–1593 CrossRef CAS.
- M. Dieguez, O. Pamies, Y. Mata, E. Teuma, M. Gomez, F. Ribaudo and P. van Leeuwen, Adv. Synth. Catal., 2008, 350, 2583–2598 CrossRef CAS.
- S. Reimann, T. Mallat and A. Baiker, J. Catal., 2007, 252, 30–38 CrossRef CAS.
- S. Reimann, T. Mallat and A. Baiker, J. Catal., 2008, 254, 79–83 CrossRef CAS.
- S. Reimann, J. D. Grunwaldt, T. Mallat and A. Baiker, Chem.–Eur. J., 2010, 16, 9658–9668 CrossRef CAS.
- B. E. Bent, Chem. Rev., 1996, 96, 1361–1390 CrossRef CAS.
- S. Reimann, J. Stotzel, R. Frahm, W. Kleist, J. D. Grunwaldt and A. Baiker, J. Am. Chem. Soc., 2011, 133, 3921–3930 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |