Imidazolium-derived organosilicas for catalytic applications
Amàlia
Monge-Marcet
ab,
Roser
Pleixats
*a,
Xavier
Cattoën
b and
Michel
Wong Chi Man
*b
aChemistry Department, Universitat Autònoma de Barcelona, 08193-Cerdanyola del Vallès, Barcelona, Spain. E-mail: roser.pleixats@uab.cat; Fax: +34 93 581 1265; Tel: +34 93 581 2067
bInstitut Charles Gerhardt Montpellier (UMR 5253 CNRS-UM2-ENSCM-UM1), 8 rue de l'école normale, 34296-Montpellier, France. E-mail: michel.wong-chi-man@enscm.fr; Fax: +33 467147212; Tel: +33 467147219
First published on 19th September 2011
Abstract
Imidazolium salts are applied in different fields of research and have found a wide variety of applications owing to their distinctive properties. This review exclusively focuses on the various methods (grafting, surfactant-assisted sol–gel synthesis, bridged silsesquioxanes formation) described in recent years for the covalent immobilization of imidazolium salts on silica supports via C–Si bonds and on the catalytic applications of the resulting silica-supported imidazolium salts and NHC complexes. C–C bond formation reactions such as cross-coupling, olefin metathesis or organocatalyzed Knoevenagel condensations as well as functionalization reactions are described. This review is intended to help in inspiring future developments and interest in the growing field of catalysis by supported imidazolium salts and NHC complexes based on organosilicas.
 Amàlia Monge-Marcet | Amàlia Monge-Marcet was born in 1985 in Barcelona (Spain). In 2007 she received the Degree in Chemistry from the Universitat Autònoma de Barcelona (UAB) and her Master degree from the same University in 2008. She is now pursuing her PhD under a cotutelle regime between UAB and ICG Montpellier working on silica-supported catalysts. |
 Roser Pleixats | Roser Pleixats graduated in Chemistry and obtained her doctorate degree from the Universitat Autònoma de Barcelona (UAB). After a postdoctoral stay at the Université des Sciences et Techniques du Languedoc (USTL) in Montpellier with Prof. R. J. P. Corriu and E. Colomer, she returned to UAB where she became Associate Professor in 1991 and Full Professor of Organic Chemistry in 2004. Her current research focuses on organometallic chemistry, catalyst recycling, metal nanoparticles and hybrid silica materials for catalysis (cross-coupling, metathesis, organocatalysis). |
 Xavier Cattoën | Xavier Cattoën was born in 1978 in Poitiers. He graduated from ENS Cachan then completed his PhD (2004) under the supervision of Prof. G. Bertrand and Dr D. Bourissou, carrying out his research at the University of Toulouse and at the University of California, Riverside. After a post-doctoral stay at ICIQ (Tarragona, Spain) in M. A. Pericàs' group, he joined the CNRS in 2007 at ICG Montpellier where he is developing structured hybrid materials for applications in photonics and catalysis. |
 Michel Wong Chi Man | Michel Wong Chi Man was born in Black River (Mauritius). He received his PhD from the University of Montpellier (1987). After spending one year at the University of Heidelberg (Germany) as an AvH post-doctoral fellow he joined the CNRS in 1990. In 2004 and in 2006, he worked as visiting scientist at the UAB (Spain) and at ANSTO (Australia) respectively. His current research interests focus on sol–gel chemistry for nanostructured hybrid materials with implementation in several fields: molecular recognition, photonics and supported catalysis. He co-authored more than 100 publications and 3 patents. |
1. Introduction
During the past decades, imidazolium salts and their derivatives have attracted considerable attention in several fields of research. Those that behave as ionic liquids (IL) at room temperature have been used as green alternatives to volatile organic solvents not only on the laboratory scale but also in industrial processes.1 Indeed they are appropriate solvents for organometallic catalysis.2 However, their non-innocent behavior as solvents under some reaction conditions have to be taken into account.3,4 Thus, imidazolium and dihydro-imidazolium salts (A and B, Fig. 1 [For convenience in the figures and schemes of this review, a dashed line in the five-membered ring (C) indicates that the structure can correspond either to an imidazole or to a dihydroimidazole framework.]) can form N-heterocyclic carbenes D (NHC) by deprotonation at the C-2 position.5–7 Besides the fundamental interests,8,9 much attention has been paid to such species which can behave as ligands for all transition metals. Due to their original electronic properties, they are much more than phosphine mimics.10 Indeed they are considered as pure σ-donor ligands with a very poor π-acceptor character and are hence capable of conferring electron density to metallic centres, forming NHC–metal complexes (E).11 Owing to the strong metal–carbon bond in these complexes, the NHC ligand is particularly not labile which can be useful for immobilizing these types of complexes. While imidazolium salts have become major tools in organometallic catalysis, their uses have recently been extended to organocatalysis, in particular for the polymerization of lactide and glycolide.12,13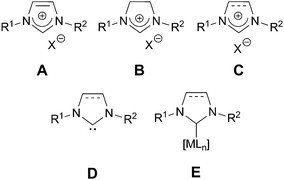 | ||
| Fig. 1 General structure of imidazolium salts (A), dihydroimidazolium salts (B); convenient representation for both imidazolium and dihydroimidazolium salts (C), free NHC (D) and NHC complexes (E). | ||
It soon appeared interesting to immobilize these species on insoluble supports among which silica is the most commonly used.14 Imidazolium-based hybrid organic–inorganic silicas can be classified into three families: (1) ionogels,15,16 (2) Supported Ionic Liquid Films (SILFs)17 and (3) covalently bonded organosilicas.18 Ionogels consist of a bi-continuous phase of ionic liquid and a silica network. In this composite, the IL preserves its liquid behavior but remains embedded in the material. Consequently the existing ionic mobility in these solid materials can be exploited for applications; for example they might be used as solid electrolytes for lithium batteries.19 Moreover, they keep their solvent properties and hence organometallic catalysis can be performed within these solids.20–22 SILFs17,23,24 are formed by a thin layer of IL adsorbed on the surface of a silica powder. Unlike other solid supports, such as alumina, silica exhibits a noticeable interaction with the imidazolium salts, leading to a thermostability decrease of the IL in the final material.25 For catalytic purposes, the adsorption of an IL film on a silica surface has the advantage to considerably increase the interfacial surface contact compared with the bulk IL phase and to enhance the exchange rate between the IL containing the catalytic species and the phase containing the reactants.26 The covalently bonded imidazolium-based silicas can be prepared from imidazolium-derived organotriethoxysilanes either by grafting methods or by the sol–gel process.18 In these cases, the imidazolium cations are strongly anchored within the solid support while the anions can still be exchanged following application for anionic separation.27,28 Even if some organosilane precursors exhibit IL behaviour, the denomination of immobilized/supported IL for the corresponding organosilicas is misleading,17 the term i-silica29 being more appropriate to define these solid materials.
In this review, only the covalently bonded imidazolium-based organosilicas will be discussed together with the corresponding covalently anchored NHC–metal complexes. In the first part the syntheses of the precursors and hybrid materials will be described and in the second part, the application of these materials in various reactions will be reviewed.
2. Synthesis of the organosilicas
2.1 Synthesis of the imidazolium-based silylated precursors
 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
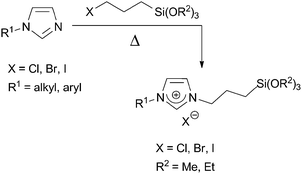
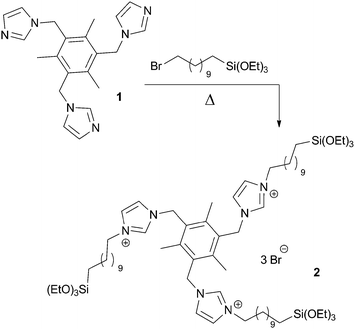
In a similar fashion, alkyl halides reacted with the commercial N-(3-trialkoxysilylpropyl)imidazole or N-(3-trialkoxysilylpropyl)imidazoline under comparable conditions to afford the corresponding salts (Scheme 3). Compounds featuring linear alkyl chains, benzyl or 2,4,6-trimethylbenzyl groups31 and functionalized alkyl chains (with amino or sulfonic acid32 functions) could be obtained by this way. In related examples, bis(alkylsilylated) imidazolium salts 433 or the mesitylene-bis(imidazolinium) salt 334 were obtained (Scheme 3).
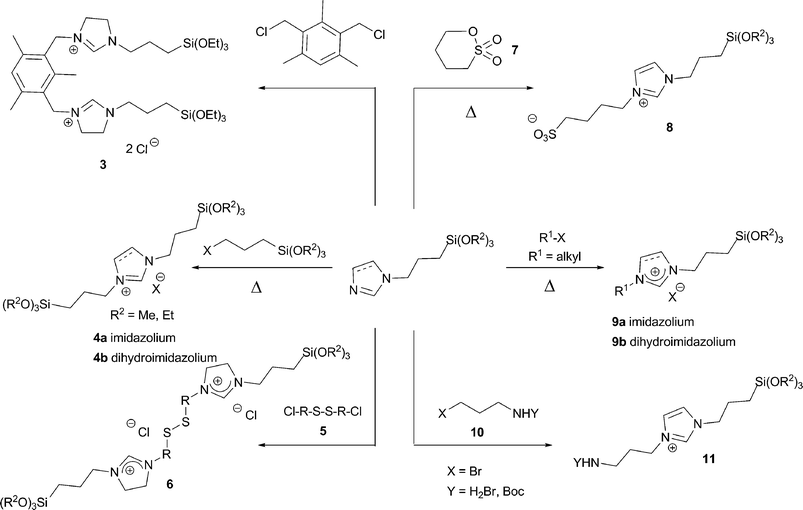 | ||
| Scheme 3 | ||
The most widely used NHC complexes in homogeneous catalysis feature aryl substituents on the nitrogen atoms (2,4,6-trimethylphenyl- or 2,6-diisopropylphenyl-). Unfortunately, it is impossible to incorporate such substituents by nucleophilic substitution. Thus, several groups have carried out more elaborated syntheses to incorporate the trialkoxysilyl substituent either at the 4 position of the aryl group, or at the backbone of a dihydroimidazolium ring. In the first approach, an imidazolium salt 12 featuring 4-halogenoaryl substituents undergoes a rhodium-catalyzed silylation reaction (Scheme 4).35–37 This reaction can be performed with aryl bromides but occurs under milder conditions if aryl iodides are used. In the second approach, initially developed by Grubbs et al.,38 a dihydroimidazolium salt 14 featuring two allyl groups in its backbone (pure meso diastereomer) can undergo a classical hydrosilylation reaction with trichlorosilane, followed by an alcoholysis of the chlorosilane moieties. The competitive reduction of the dihydroimidazolium ring is decreased by this two-step approach, compared with the direct hydrosilylation reaction with triethoxysilane.39
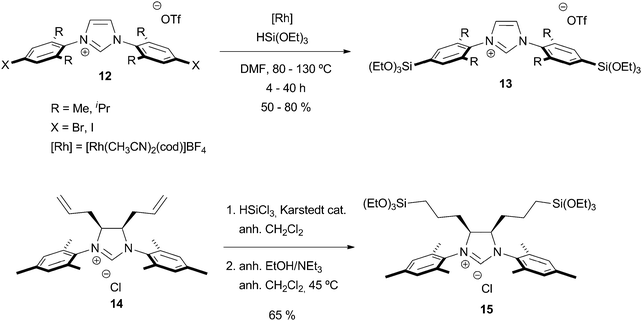 | ||
| Scheme 4 | ||
It is worth noting that these two approaches lead to important structural differences in the materials: while the silylated moieties are directed towards the concave part of the imidazolium salt in 13, their incorporation at the backbone of the dihydroimidazolium ring in 15 results in geometries similar to the one observed in the non-silylated species. Indeed, an important effect of the location of the silylated groups on the catalytic properties was evidenced for olefin metathesis reactions (see Section 3.4).38
The formation of NHC–metal complexes can be performed in two steps, involving first the deprotonation of the imidazolium salt to form a NHC, followed by the complexation (Scheme 5). This synthesis pathway is typically used to form sol–gel precursors based on Grubbs' catalysts. For non-bulky dihydroimidazolium salts, the deprotonation yields the so-called Wanzlick dimers40 that act as masked carbenes and yield NHC complexes upon reaction with metallic precursors such as [RhCl(cod)]241 or [RuCl2(p-cymene)]2.31 The deprotonation can also be performed simultaneously with the complexation step: this method is typically used to generate NHC–Pd(II) complexes from imidazolium halides and palladium acetate. Complexes of general formula [(NHC)2PdX2] are usually formed by this way.42 The most general way to produce NHC–metal complexes bearing trialkoxysilylated groups consists in the formation of a silver complex by simultaneous deprotonation and complexation of the imidazolium salt using silver oxide followed by a transmetallation step.43 This method is used for the formation of Au(I),44,45 Rh(I),46,47 Ir(I)47 and Pd(II)45,48 complexes.
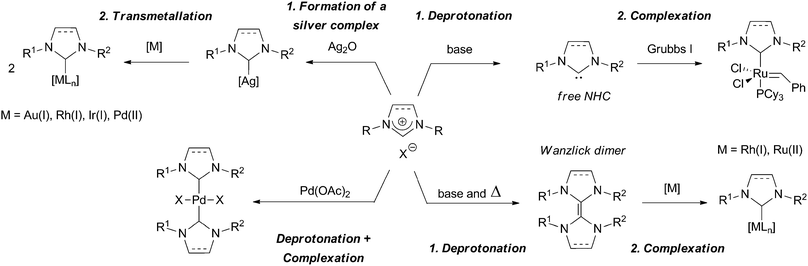 | ||
| Scheme 5 | ||
2.2 Synthesis of the materials
Organosilicas containing covalently bonded imidazolium salts are obtained from organotrialkoxysilanes by different types of syntheses. The challenges associated with these syntheses are (i) the control of the textural properties (surface area, pore size and shape…); (ii) the control of the imidazolium loading and of the homogeneity of the distribution; (iii) the preservation of the organometallic fragment during the synthesis, which is especially true in the case of some NHC complexes which can be water- or air-sensitive, and/or thermally deteriorated.The grafting method consists in the reaction of an organotrialkoxysilane with the surface of a previously activated silica material (Scheme 6). This reaction that is typically carried out at rather high temperatures (65–110 °C) results in materials with an ill-controlled spatial distribution of the organics within the material. The main advantages reside in the simplicity of the process, and in the preservation of the initial texture of the solid (despite a slight decrease in the specific surface area). Starting materials such as commercial, amorphous silicas, mesostructured silicas (such as the hexagonally-structured MCM-41 and SBA-15, or the cubic SBA-1649), silica nanoparticles,50 or magnetite–silica particles51 have been used, in conjunction with silylated imidazolium salts, or NHC complexes. Despite the high temperature commonly used, sensitive complexes such as the Grubbs II52 catalysts can be successfully anchored. Less sensitive Au–,53 Pd–42 and Rh–46 NHC complexes were also incorporated on silica materials. A better control of the spatial homogeneity of the material can be obtained when the organosilicas are synthesized using the sol–gel process from molecular precursors (Scheme 6). Simple sol–gel co-condensation reactions implying an organotrialkoxysilane and a tetraalkoxysilane yield materials with ill-defined morphologies and mesostructures, unless the synthesis is performed in the presence of surfactants. By carefully tuning the synthesis conditions,35 regularly mesostructured organosilicas containing imidazolium moieties can be obtained, usually using the triblock copolymer P123 as a surfactant under acidic conditions, which most often results in the formation of a hexagonal structure (Scheme 6). However, using P123 in conjunction with F127, it is possible to master the formation of a 3D cubic structure.37 Examples of organosilicas containing NHC complexes obtained by the sol–gel route are scarce,31,39,54 the need of water during the synthesis being a limiting factor.
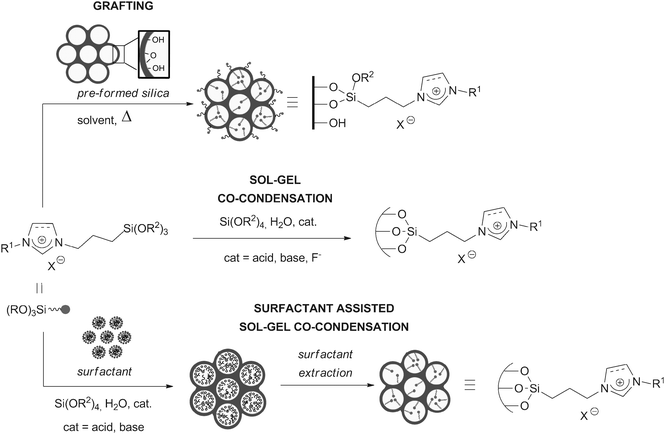 | ||
| Scheme 6 | ||
Starting from bridged organotrialkoxysilanes, it is also possible to obtain pure, inherently homogeneous silica materials called bridged silsesquioxanes, which usually lack porosity (Scheme 7).55,56 Notably, hierarchically macro- and nanostructured i-silica bridged silsesquioxanes could be obtained under acidic conditions.30 Another noticeable example consists in the formation of a bridged silsesquioxane from a [bis(NHC)PdI2] complex.57,58 Moreover, mesostructured bridged silsesquioxanes, called PMOs, can be obtained in the presence of surfactants.59–61 This type of materials can also be prepared by a co-condensation method in the presence of an imidazolium-containing bridged organosilane yielding “hybrid PMO” materials (Scheme 7).36 The high hydrophobicity of such materials might result in modified properties in catalysis.
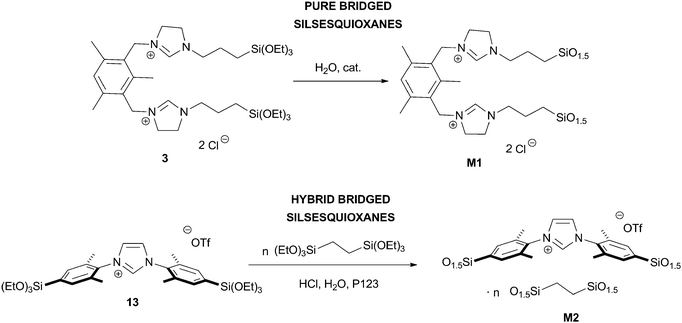 | ||
| Scheme 7 | ||
Instead of starting from organotrialkoxysilanes containing an imidazolium or NHC–[MLn] moiety, some materials have been synthesized from functionalizable organosilicas. Most often, materials containing pending chlorobenzyl fragments were functionalized with mesityl62–64 or methyl imidazole65,66 to form the corresponding imidazolium salt on the surface of the silica material (Scheme 8). Alternatively, core–shell magnetite–silica nanoparticles were grafted with propylimidazole and subsequently alkylated with 2-picolyl chloride.51 Another approach consists in the nucleophilic substitution of a surface Si–Cl function with a hydroxyl group linked with a NHC–metal complex.67 However, as the Si–O bonds are labile, such materials might undergo leaching in the presence of water or alcohols.
 | ||
| Scheme 8 | ||
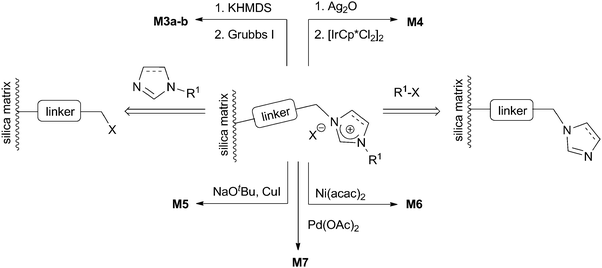
More often, metallic complexes are formed at the surface of imidazolium-containing materials, using the previously described syntheses for the NHC–metal complexes on the silylated precursors (Scheme 5). However, by this method, it is difficult to prove the formation of a metal–NHC complex. Interestingly, a 13C-labelled, silica-anchored imidazolium salt was synthesized, and the successful metallation by silver perfluoro tert-butoxide followed by transmetallation with [IrCp*Cl2]2 was monitored by 13C-NMR spectroscopy, evidencing the possible metallation of imidazolium salts by such a sequence on a silica surface.64 Other examples include the formation of supported Cu(I)–,65,66 Pd(II)–NHC36 complexes and Grubbs II catalysts63,68 by deprotonation with a strong base (NaOt-Bu, KOt-Bu or KHMDS) followed by complexation (Scheme 8). The formation of Ni(II)–51 and Pd(II)–NHC34,37,54,69,70 complexes by simultaneous deprotonation and complexation with a metal–base complex ([Ni(acac)2] or [Pd(OAc)2]) was also described, although the structure of the complexes was never demonstrated.
3. Application in catalysis
This part is devoted to reports dealing with reactions mediated by NHC complexes anchored to silica, followed by those catalyzed by covalently anchored imidazolium salts.3.1 Hydrogenation reactions
Several silica-supported metal-based catalysts containing imidazolium-derived moieties have been used in the hydrogenation of different functional groups (Fig. 2).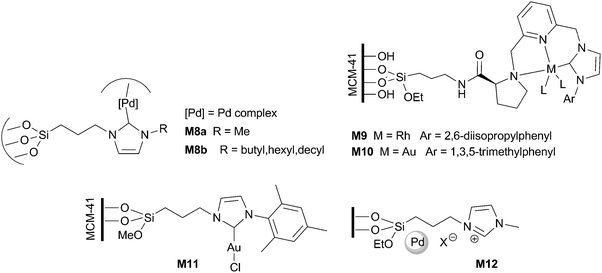 | ||
| Fig. 2 Supported catalysts for hydrogenation reactions. | ||
Mesoporous organosilicas bearing Pd(II)–NHC complexes (M8) were recently found to be very active in the hydrogenation of alkenes and allylic alcohols, reaching TOF values of about 6000 h−1. The selectivity was improved by the use of scCO2. According to XPS and HRTEM experiments, no Pd(0) nanoparticles were formed in the reaction and Pd(II) has been suggested as the main reactive species.54 Asymmetric hydrogenation of diethyl (E)-2-benzylidenesuccinate and diethyl itaconate has been recently achieved by chiral Rh and Au pincer complexes immobilized on MCM-41 (M9 and M10, respectively).46 The same authors have previously reported the hydrogenation of diethyl benzylidenesuccinate and diethyl citraconate by a Cl–Au–NHC complex supported in MCM-41 (M11).53 In Table 1 comparative data of heterogeneous catalysts are given for one of the substrates. In this case, materials M9 and M10 gave higher TOFs than related non-supported homogeneous catalysts and similar or higher ee. For diethyl itaconate the activity was superior (TOF values between 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 33000 h−1), but the enantioselectivity was much inferior (ee < 20%). Catalysts M9 and M11 could be reused in five consecutive runs without significant loss of activity and selectivity. A hot filtration test proved that the catalytic activity did not arise from leached homogeneous species, but from the reactive supported catalyst.71 The integrity of both the organometallic moiety and the inorganic support remained unchanged upon recycling.
000 and 33000 h−1), but the enantioselectivity was much inferior (ee < 20%). Catalysts M9 and M11 could be reused in five consecutive runs without significant loss of activity and selectivity. A hot filtration test proved that the catalytic activity did not arise from leached homogeneous species, but from the reactive supported catalyst.71 The integrity of both the organometallic moiety and the inorganic support remained unchanged upon recycling.
Selective hydrogenation of cinnamaldehyde to hydrocinnamaldehyde was achieved by supported Pd nanoparticles on imidazolium salts-modified silica gel M12.72 Depending on the counteranion, NHC–Pd complexes (X = Cl) or Pd nanoparticles (X = NO3−, BF4−, PF6−) were formed from the original Pd(OAc)2. TOF values were also anion-dependent, achieving a maximum of >33000 h−1 for X = NO3− and which is about 1000 times higher than those corresponding to the analogous biphasic system (IL + Pd source). The supported catalyst was recycled for 9 consecutive runs without decreasing activity (0.5 mol% Pd, xylene, 80 °C, 20 min for full conversion).
3.2 Hydroformylation reactions
Fig. 3 summarizes the silica-supported rhodium-based catalysts containing imidazolium-derived moieties which have been used in hydroformylation reactions.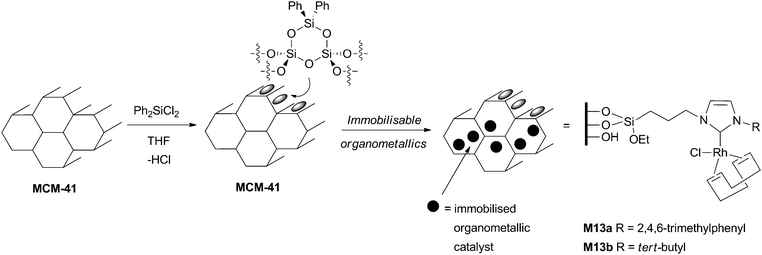
Very recently, Rh(I) complexes have been immobilized inside the channels of MCM-41 after site selective pre-silylation of external surface silanol groups with Ph2SiCl2, and these materials (M13a–b) have been used in the hydroformylation of 1-octene in toluene.47 According to the authors they exhibit comparable TOF values to those of related homogeneous systems. The system was reusable for 8 runs and low rhodium leaching was observed (20 ppm).
3.3 H/D exchange reactions
Thieuleux and coworkers64 have used Ir–NHC containing mesostructured silicas M4 (Fig. 4) as catalysts for the H/D exchange reaction between [D4]methanol and acetophenone. When compared with homogeneous analogues slightly longer reaction times were required for the supported catalyst, which could be recycled up to three runs. No leaching data were provided. Later on, they reported a more complete study broadening the scope of the reaction of C–H activation to other substrates such as 9-acetylanthracene, benzophenone and diethyl ether.62 They conclude that the substituents on the NHC ligand have an important influence on the reaction rate in the case of large substrates (M4b better than M4a) and that for heterogeneous systems the nature of the first and second coordination spheres around the metal seems to play a key role (size of the NHC ligand, proximity of the active center to the surface due to the length of the tether and the size of the passivating SiMe3 groups). In contrast, in the mesoporous range investigated, with pores large enough for easy diffusion of substrates, the rate did not seem to be correlated with the pore size of the material.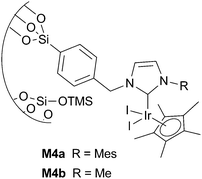 | ||
| Fig. 4 Supported Ir complexes. | ||
3.4 Olefin metathesis reactions
Several silica-supported versions of Ru–NHC alkylidene complexes for metathesis reactions have been described in the literature.73,74 One of the first reports is due to Hoveyda et al.,75 who attached a Grubbs–Hoveyda II type complex to a sol–gel monolith through the chelating styrenic ligand. This article and others presenting the same approach will not be considered here and we will restrict our comments to those works where the attachment to the silica support is made through the NHC ligand (permanently immobilized catalysts). The materials M3, M14–M19 represented in Fig. 5 were tested in the ring-closing metathesis reaction of several substrates. Comparative results for 2,2′-diallylmalonates and for N,N′-diallyl-p-toluenesulfonamide are summarized in Tables 2 and 3, respectively.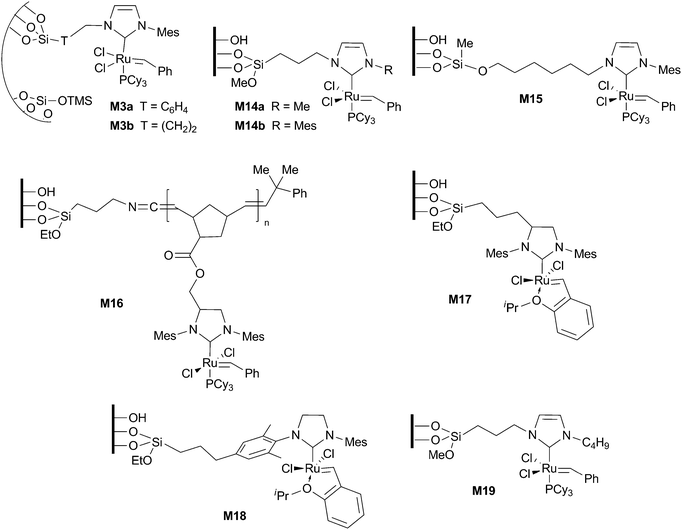 | ||
| Fig. 5 Supported ruthenium complexes for metathesis (T = linker). | ||
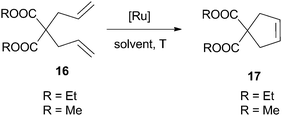
| Entry | Catalyst | Ru [mol%] | R | Solvent/T [°C]/t [h] | Conv. [%] 1st cycle | Ru leaching |
|---|---|---|---|---|---|---|
| a [Substrate] = 0.04 M. b Product isolated by filtration and further purified by chromatography on silica gel. c Below detection limit (ICP-OES). | ||||||
| 1a,b | M14a 68 | 5 | Me | CH2Cl2/rt/3 | 90 | — |
| 2 | M15 67 | 5 | Et | CH2Cl2/reflux/24 | 98 | 250 ppm |
| 3a | M16 76 | 0.2 | Et | 1,2-DCB/50/2 | 43 | c |
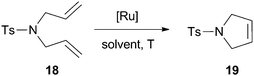
| Entry | Catalyst | Ru [mol%] | Solvent/T [°C]/t [h] | Conv. [%] 1st cycle | Ru leaching |
|---|---|---|---|---|---|
| a [Substrate] = 0.04 M. b Product isolated by filtration and further purified by chromatography on silica gel. c [Substrate] = 0.1 M, isolation of the product by filtration. d [Substrate] = 0.2 M. e Below detection limit (ICP). | |||||
| 1a,b | M14a 68 | 5 | CH2Cl2/rt/3 | 90 | 1.2 ppm |
| 2a,b | M14b 68 | 5 | CH2Cl2/rt/3 | 85 | — |
| 3c | M17 38 | 0.4 | Benzene/30 °C/2 | 95 | <5 ppb |
| 4c | M18 38 | 0.4 | Benzene/30 °C/2 | 85 | <5 ppb |
| 5d | M19 52 | 5 | CH2Cl2/45 °C/0.6 | 98 | e |
In materials M14a–b68 the Ru–NHC moiety was linked to mesostructured cellular foam (MCF) through the N-alkyl substituent. The previously grafted imidazolium salt was converted to the NHC–Ru complex on the solid material. Good results were achieved for the formation of disubstituted olefins (Tables 2 and 3), but lower conversions were obtained for trisubstituted alkenes (30%). M14a was recycled up to 6 runs in the RCM of N,N′-diallyl-p-toluenesulfonamide and it was used in a flow microreactor system. Material M15 was obtained by reaction of previously functionalized commercial silica with a N-hydroxyalkyl-containing second generation Grubbs-type ruthenium complex.67 It was assayed in several dienes, leading to the formation of disubstituted, trisubstituted and tetrasubstituted olefins after one day in refluxing dichloromethane (98, 95 and 64% conversions, respectively). It could be reused for 3 cycles (entry 2 of Table 2).
The immobilization of a NHC precursor onto porous and non-porous silica through Ru or Mo-initiated oligomerization, followed by their conversion to the Ru–alkylidene complexes, afforded materials M16, which were used in the RCM of diethyl 2,2′-diallyl malonate in dichlorobenzene at 50 °C (entry 3 of Table 2).76 Similar TON values (75–80) were obtained for both porous and non-porous materials. However, the reaction with other less reactive compounds such as 1,7-octadiene, N,N-diallyltrifluoroacetamide, diallyl ether and diallyldiphenylsilane gave very low TONs (<5). The authors suggest that the stirred batch set-up with this type of support is highly diffusion controlled. With less reactive substrates the reaction is too slow and decomposition of ruthenium alkylidene species takes place.
Grubbs et al. have recently described the preparation of materials M17 and M18 by grafting two triethoxysilyl functionalized Grubbs–Hoveyda II type-complexes to commercial silica.38 They were tested in the RCM of N,N-diallyl-p-toluenesulfonamide (entries 3 and 4 of Table 3), among other dienes and olefins (RCM and CM), achieving remarkable TOF values (106 and 119 h−1 for M17 and M18 respectively). In all cases, M18 was less active than M17. Although the reason remains unknown, the authors speculate that the difference can arise from an interaction between the ruthenium active site and the silica surface that is present in the case of M18 but it is blocked by the steric bulk of the NHC ligand in the case of M17. This catalyst could be reused up to 7 cycles (0.75 mol% of Ru) in the formation of the trisubstituted alkene diethyl 3-methylcyclopent-3-ene-1,1-dicarboxylate.
Materials M16, M17 and M18 were mentioned by Vougioukalakis and Grubbs in a recent review as efficient strategies to recover the catalyst and reduce product contamination.77
Heterogeneous Ru–NHC complexes M3a–b, obtained by surface organometallic chemistry on hybrid mesostructured materials, have been tested in the cross-metathesis of ethyl oleate.63 Conversions of 50% (thermodynamic equilibrium) were reached in 5 h with 0.01 mol% Ru, under neat (solvent-free, only the catalyst and neat ethyl oleate) conditions at 40 °C. High initial TOF values of 65 min−1 for M3b and 30 min−1 for M3a were observed, which were independent of the Ru loadings (0.3–1.0 wt%). This is consistent with a single-site behaviour in the hybrid materials. M3a presented TON values of 17000, higher than the TON value of 4000 for the Grubbs I catalyst under the same conditions. Recycling experiments up to five cycles were performed using 0.25 mol% of Ru, no traces of ruthenium being detected in the liquid fractions (<50 ppm detection limit).
3.5 Formation of furan rings by Ru–NHC complexes
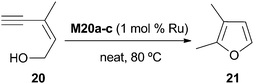
Immobilized NHC–Ru(II) complexes M20a–c (Fig. 6) prepared by co-condensation with tetraethoxysilane (TEOS) have been applied to the formation of 2,3-dimethylfuran from (Z)-3-methylpent-2-en-4-yn-1-ol (Scheme 9).31M20c gave the best results and it was recycled for 5 runs (TOF of 93 h−1 for the first cycle). No data of leaching were provided.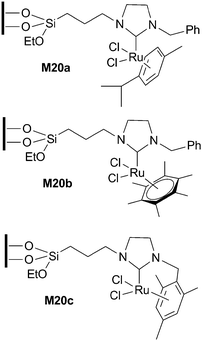 | ||
| Fig. 6 Supported ruthenium complexes. | ||
 | ||
| Scheme 9 | ||
3.6 Palladium-catalyzed reactions
The importance of palladium-catalyzed cross-coupling reactions was recently highlighted by the Nobel committee who awarded R. Heck, A. Suzuki and E.-I. Negishi, for their pioneering work on C–C bond formation methodologies. These Pd-catalyzed reactions have become a widely used chemical tool, but suffer from the high cost of this metal. Furthermore, Pd traces are undesired in the final pharmaceutically active compounds due to toxicity issues. Therefore, the immobilization of catalytic systems is the subject of intense research.78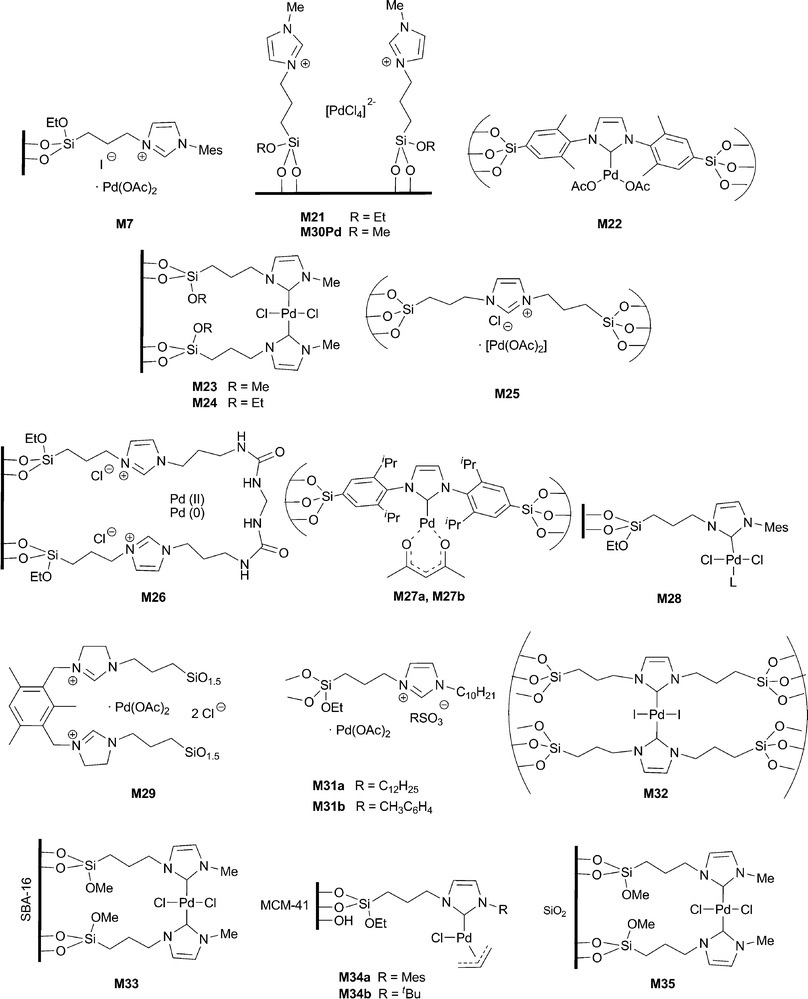 | ||
| Fig. 7 Supported Pd catalysts for Suzuki, Heck and Sonogashira reactions. | ||
In Table 4 we summarize some comparative results in the Suzuki cross-coupling between phenylboronic acid and p-substituted aryl halides.

| Entry | Cat. | Pd [mol%] | X | R | Solvent/T (°C) | Base | t/h | Yield [%] |
|---|---|---|---|---|---|---|---|---|
| a 0.5 eq. TBABr was added. b Microwave heating. | ||||||||
| 1 | M21 79 | 0.1 | Br | OMe | EtOH/H2O (1/1)/rt | K2CO3 | 1 | 97 |
| 2 | M21 79 | 0.1 | Br | OMe | EtOH/H2O (1/1)/50 | K2CO3 | 0.5 | 99 |
| 3 | M21 79 | 0.001 | Br | OMe | EtOH/H2O (1/1)/50 | K2CO3 | 1 | 84 |
| 4 | M22 36 | 0.25 | Br | OMe | i PrOH/80 | t-BuOK | 3 | 81 |
| 5 | M22 36 | 0.5 | Cl | OMe | i PrOH/80 | t-BuOK | 24 | 72 |
| 6 | M22 36 | 0.5 | Cl | COCH3 | i PrOH/80 | t-BuOK | 18 | 98 |
| 7 | M23 80 | 0.5 | Br | OMe | DMF/H2O (1/1)/rt | K2CO3 | 2 | 98 |
| 8 | M23 80 | 0.5 | Br | H | DMF/H2O (1/1)/rt | K2CO3 | 2 | 99 |
| 9 | M24 81 | 0.1 | Br | OMe | DMF/H2O (1/1)/65 | Na2CO3 | 0.6 | 95 |
| 10 | M25 69 | 0.2 | Br | OMe | H2O/75 | K2CO3 | 5 | 95 |
| 11 | M25 69 | 1 | Cl | OMe | H2O/90a | K2CO3 | 20 | 55 |
| 12 | M26 50 | 5 | Br | COCH3 | DMF/130 | Cs2CO3 | 18 | 99 |
| 13 | M27a 37 | 0.5 | Cl | OMe | i PrOH/80 | t-BuOK | 24 | 68 |
| 14 | M27a 37 | 0.5 | Cl | COCH3 | i PrOH/80 | t-BuOK | 20 | 89 |
| 15 | M27a 37 | 0.25 | Br | COCH3 | i PrOH/80 | t-BuOK | 6 | 92 |
| 16 | M27b 82 | 0.32 | Cl | COCH3 | i PrOH/80 | t-BuOK | 5 | 87 |
| 17 | M28 45 | 10 | I | OMe | Xylene/130 | K3PO4 | 24 | 60 |
| 18 | M7 34 | 0.2 | Br | OMe | DMF/H2O (95/5)/110 | K2CO3 | 1 | 99 |
| 19 | M29 34 | 0.2 | Br | OMe | DMF/H2O (95/5)/110 | K2CO3 | 0.5 | 96 |
| 20 | M30Pd 83 | 1.2 | Br | OMe | m-Xylene/110 | K2CO3 | 24 | 74 |
| 21 | M31a 70 | 2 | I | OMe | i PrOH/H2O (1/1)/50 | Na2CO3 | 2 | 89 |
| 22 | M31a 70 | 2 | Br | OMe | i PrOH/H2O (1/1)/50 | Na2CO3 | 6 | 91 |
| 23 | M31a 70 | 2 | Br | COCH3 | i PrOH/H2O (1/1)/50 | Na2CO3 | 6 | 93 |
| 24 | M31b 70 | 2 | I | OMe | i PrOH/H2O (1/1)/50 | Na2CO3 | 2 | 89 |
| 25 | M32 57 | 0.037 | Br | OMe | DMF/H2O (1/2)/100b | K2CO3 | 0.16 | 90 |
| 26 | M33 49 | 0.01 | Br | OMe | EtOH/H2O (1/1)/50 | K3PO4 | 10 | 99 |
| 27 | M33 49 | 0.01 | Br | COCH3 | EtOH/H2O (1/1)/50 | K3PO4 | 7 | 87 |
| 28 | M34a 47 | 1.4 | I | H | H2O/rt | KOH | 12 | 95 |
| 29 | M34b 47 | 1.4 | I | H | H2O/rt | KOH | 12 | 93 |
Palladium salts were introduced into the pore channels of imidazolium-functionalized SBA-15. The resulting palladium catalyst M21 was used in the Suzuki coupling with aryl bromides in water–ethanol mixtures at room temperature or 50 °C (entries 1–3 of Table 4). Aryl chlorides did not react. In the reaction with p-bromoanisole, reusability of the catalyst was proven up to 6 cycles, a decrease of activity being observed after the third run due to some palladium leaching.79
Following a co-condensation method in acidic medium in the presence of a template, hybrid PMO M2 bearing bridged imidazolium moieties was synthesized (Scheme 7), which was further treated with potassium tert-butoxide and Pd(OAc)2 to yield M22.36 XPS spectroscopy showed the presence of Pd(0) and Pd(II) species on the solid surface, but Pd(0) clusters were not observed by TEM on the freshly prepared material. This catalyst was active for the Suzuki coupling of various arylboronic acids with aryl bromides and aryl and benzyl chlorides in isopropanol at 80 °C, using t-BuOK as base (entries 4–6 of Table 4). The catalyst was reused in the reaction between p-chloroacetophenone and phenylboronic acid for 8 runs, and the activity was observed to decrease from the fifth reaction cycle, although 90% yield could be obtained in the eighth cycle with longer reaction time. Pd(0) nanoparticles located inside the nanochannels were observed by TEM in the reused material M22, whose ordered mesostructures remained almost intact even after eight runs in a strong basic medium.
Immobilized NHC–Pd complex M23 was obtained by grafting the previously synthesized disilylated homogeneous Pd bis-carbene complex on silica particles (38–60 μm) and applied to the Suzuki coupling between various aryl iodides and bromides and phenylboronic acid at room temperature in a DMF–H2O 1/1 mixture (entries 7 and 8 of Table 4). In the case of aryl chlorides it was not effective and the reaction with p-chloroanisole gave only 7% yield of the corresponding biphenyl. The reusability of the heterogeneous catalyst was tested with bromobenzene as substrate (4 cycles with almost no decrease in activity).80
A similar immobilized NHC–Pd complex, M24, was prepared by Jin and co-workers by a strategy in which the steps were reversed. The grafting of the triethoxysilylated imidazolium chloride onto silica gel was followed by treatment with Pd(OAc)2 in DMSO at 60 °C.81 The supported catalyst was active in the Suzuki reaction for aryl iodides, bromides and chlorides in DMF–H2O 1/1. Reactions conducted with aryl iodides and bromides were performed at 65 °C with 0.1 mol% of catalyst (entry 9 of Table 4), whereas the more challenging chlorides required higher loading and temperature (1 mol%, 85 °C), modest yields being obtained. Competitive homocoupling of phenylboronic acid occurred to a remarkable extent. Reusability was checked for iodobenzene and p-nitrobromobenzene (6 cycles without significant loss of activity).
Karimi et al. have recently described the preparation of a palladium-supported mesostructured organosilica based on an alkyl-imidazolium salt, M25.69 This material was obtained by co-condensation of the corresponding bridged organosilane with tetramethoxysilane in the presence of a non-ionic surfactant under acidic conditions, followed by treatment with Pd(OAc)2 in DMSO at 60 and 100 °C. In contrast to the case of M22, no external base was added. This catalyst was successfully tested for the Suzuki coupling with aryl iodides, bromides and chlorides in water at 60, 75 or 90 °C depending on the substrate (entries 10 and 11 in Table 4). By using the coupling reaction of phenylboronic acid and 4-bromobenzaldehyde as a model reaction, recovery and recycling could be successfully achieved in four reaction runs without significant loss of activity. This system was found to operate at least partially through a homogeneous mechanism (hot filtration test, Hg and three-phase poisoning tests), suggesting that the supported palladium catalyst serves as a reservoir for active palladium species released from the solid catalyst under the reaction conditions. TEM and N2-sorption analyses of the recovered material indicated that the highly ordered mesostructures had survived and that regularly distributed Pd(0) nanoparticles were formed.
In another example, palladium acetate was captured inside a molecular band composed of imidazolium chloride and urea moieties grafted onto spherical silica particles. Suzuki coupling between phenylboronic acid and 4-bromoacetophenone was carried out with the material M26 in DMF at 130 °C (entry 12 of Table 4).50 Although no Pd(0) particles were present in the freshly prepared material, TEM analysis of the recovered catalyst evidenced the formation of nano-sized palladium particles of 15 nm and XPS measurements indicated the presence of Pd(II) and Pd(0) species. Despite 26% of the palladium was leached out after the first run, M26 could be reused four more times.
The cubic mesoporous material M27a was synthesized by acid-catalyzed co-condensation of the corresponding disilylated monomer with TEOS in the presence of a mixture of P123 and F127 as template and subsequent treatment with Pd(acac)2 in dioxane at 100 °C.37 XPS analysis showed the presence of Pd(II) species on the solid surface. This material successfully catalyzed the Suzuki reaction of several arylboronic acids with various aryl bromides and chlorides in isopropanol at 80 °C (entries 13–15 of Table 4). The recyclability was investigated in the reaction of p-chloroacetophenone with phenylboronic acid, eight consecutive runs being performed. From the fifth cycle slightly longer reaction times were needed to complete the reaction. The TEM analysis of the recovered catalyst revealed that the mesoporous structure roughly survived the first reaction cycle and that Pd nanoparticles were formed. Using a reverse micelle strategy, the same group has very recently reported an improved version of this catalyst where the imidazolium-containing silica forms a shell around a magnetic Fe3O4 core, M27b.82 They have also applied this catalyst to Suzuki coupling of aryl chlorides (entry 16 of Table 4) with the added advantage of easy recycling by the use of a magnetic field to separate the catalyst (six runs).
Heterogenized Pd–NHC complexes on silica gel and MCM-41 by grafting procedures, M28, were tested on Suzuki reactions with aryl iodides as substrates in xylene at 130 °C (entry 17 of Table 4) achieving moderate yields and recycling up to four runs.45
Catalytic systems M7 and M29 constituted by mixtures of palladium acetate and hybrid silica materials were prepared by a template assisted hydrolytic co-condensation from the corresponding monosilylated imidazolium and disilylated dihydroimidazolium salts. They were active and recoverable in Suzuki coupling, in a DMF–H2O 95/5 mixture at 110 °C (for activated and deactivated aryl bromides, entries 18 and 19 of Table 4) or at 150 °C (for p-chloroacetophenone).34 They were efficiently reused up to 5 runs for the reaction between p-bromoacetophenone and phenylboronic acid. Recycling was also possible with p-chloroacetophenone, although with lower conversions. In situ formation of palladium nanoparticles was observed by TEM of the recovered catalyst. A hot-test revealed that a homogeneous pathway is mainly operating in the catalysis. The Pd content determined by ICP in the final products was found to be 6.4 and 22.9 ppm for M7 and M29, respectively.
Imidazolium salts immobilized on the silica surface by grafting were subsequently treated with palladium(II) chloride in refluxing acetonitrile to afford M30Pd, which was active and reusable for Suzuki coupling with aryl bromides in m-xylene (entry 20 of Table 4). Recycling up to 3 runs for reaction of bromobenzene and phenylboronic acid was possible; low yields were obtained with chlorobenzene.83
Pd–NHC complexes supported on silica nanoparticles (10 nm) M31a–b were prepared by grafting of the silylated imidazolium chloride on colloidal silica followed by anion exchange and further treatment of the solid with Pd(OAc)2 in toluene at 50 °C. The activity was examined for the Suzuki reaction with aryl iodides and bromides in isopropanol–water 1/1 at 50 °C (entries 21–24 of Table 4).70M31a was reused for 5 runs in the coupling between 4-iodotoluene and phenylboronic acid. It was much less effective with chlorobenzene, low yields being obtained.
Pd–NHC organosilica M32 was obtained by the sol–gel method from the corresponding tetrasilylated Pd–bis carbene complex.57,58 This material had been previously synthesized and used in the Heck reaction (see below).58 Its activity was explored in the Suzuki reaction of various aryl iodides and bromides.57 High TOF values were achieved in DMF–H2O (1/2) by microwave activation at 100 °C (entry 25 of Table 4). Recyclability was claimed but the substrates and number of runs were not specified. Aryl chlorides were not tested.
Trimethoxysilyl derivatives of Pd–NHC complexes and imidazolium salts were grafted on the internal surface of mesostructured SBA-16 to afford the supported catalyst M33.49 It provided good performances for the Suzuki coupling with aryl bromides as substrates in ethanol–water (1/1) at 50 °C (entries 26 and 27 of Table 4). Pd(0) nanoparticles were formed during the catalytic reaction as revealed by TEM and XPS. Its recyclability (10 runs for the reaction of bromobenzene with phenylboronic acid) was much better than that of the catalyst prepared from amorphous silica. This can be attributed to an efficient prevention of the agglomeration of Pd nanoparticles immobilized into the nanocages of SBA-16.
Very recently, Green et al. have immobilized (η3-C3H5)(NHC)PdCl complexes by covalent attachment to chemically modified MCM-41 (presilylation of the outer surface) affording M34a–b. These catalysts were investigated in the Suzuki coupling with aryl iodides in water at room temperature in the presence of KOH as a base and were recycled up to seven runs for the reaction of iodobenzene with phenylboronic acid (entries 28 and 29 of Table 4).47
It has also been reported that heterogenized Au–NHC complexes on MCM-41, M11, catalyze the Suzuki cross-coupling between iodobenzene and aryl boronic acids in xylene at 130 °C, using K3PO4 as base and 20 mol% of catalyst. Moderate yields and reuse for 4 consecutive runs were described.45
| Entry | Cat. | Pd [mol%] | X | R1 | R2 | Solvent/T (°C) | Base | t/h | Yield [%] |
|---|---|---|---|---|---|---|---|---|---|
| a Microwave heating. | |||||||||
| 1 | M31b 70 | 2 | I | OMe | Bu | DMF/100 | K2CO3/NEt3 | 2 | 97 |
| 2 | M32 58 | 1.3 | I | H | Me | DMF/130 | NEt3 | 12 | 95 |
| 3 | M32 58 | 1.3 | Br | H | Me | DMF/130 | NEt3 | — | 82 |
| 4 | M32 58 | 0.037 | I | OMe | Me | DMF/120a | NEt3 | 0.25 | 96 |
| 5 | M32 58 | 0.037 | Br | OMe | Me | DMF/120a | NEt3 | 0.25 | 87 |
| 6 | M33 49 | 0.01 | Br | COCH3 | Me | NMP/130 | NaOAc | 11.5 | 76 |
| 7 | M35 42 | 0.05 | I | OMe | Bu | DMF/100 | K2CO3/NEt3 | 7 | 92 |
| 8 | M35 42 | 0.01 | I | OMe | Bu | NMP/140 | NaOAc | 10 | 92 |
| 9 | M35 42 | 0.01 | Br | OMe | Me | NMP/140 | NaOAc | 24 | 94 |
| 10 | M35 42 | 0.01 | Br | OMe | Bu | NMP/140 | NaOAc | 24 | 91 |
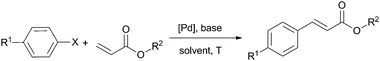
Supported catalyst M31b was effective in the Heck reaction between aryl iodides and bromobenzene with n-butyl acrylate in DMF at 100 °C (entry 1 of Table 5), but no recycling was reported.70
Hybrid silica M32 was tested in the Heck reaction of aryl iodides and bromides with styrene and methyl acrylate in DMF at 130 °C using NEt3 as base (entries 2 and 3 of Table 5) and could be reused for at least five cycles for the coupling between iodobenzene and methyl acrylate.58 Later on, the efficiency of this catalyst was improved using microwave activation at 120 °C (entries 4 and 5 of Table 5).57 Yang et al. found that M33 was also active in the Heck reaction between aryl bromides and styrene or methyl acrylate in NMP at 130 °C in the presence of NaOAc (entry 6 of Table 5). It was reused for eight cycles in the reaction of styrene with 4-bromobenzaldehyde although a decrease in activity was observed (reaction times increased from 16 to 49 h).49 Karimi and Enders42 performed a simultaneous covalent anchoring of the Pd–NHC complex and an imidazolium salt on commercial silica. The resulting catalyst M35 was investigated for the reaction between acrylates and various aryl iodides and bromides under different conditions (entries 7–10 of Table 5). Four cycles were achieved for the reaction of bromobenzene with methyl acrylate (yields from 95% to 89%). In situ formation of Pd(0) nanoparticles was observed. Based on the hot filtration test, they claimed for a heterogeneous nature of the catalysis, contrary to other groups' findings. Moreover, Çetinkaya et al. reported the immobilization of saturated NHC–Pd complexes on amorphous silica for the Heck reaction of aryl iodides and bromides with butyl acrylate and styrene (DMF, 140 °C, Na2CO3) with high TOFs at lower Pd loadings. A homogeneous pathway is suggested from leached Pd species. It appeared to be reusable for 10 cycles for the reaction of 4-iodotoluene with styrene, longer reaction times being required after the sixth cycle. For 4-bromoacetophenone, the gradual deactivation upon recycling was more pronounced (from 95% at 2 h to 65% at 28.5 h on the eighth cycle).48
 | ||
| Scheme 10 | ||
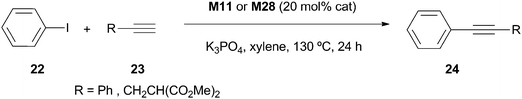
Remarkably, both materials were more active than the corresponding homogeneous systems. However, the reported GC yields were low and the amounts of catalysts were high (TOF values of 0.07 h−1 for M11 and 0.08 h−1 for M28 in the case of phenylacetylene as substrate). Both catalysts were reused up to four cycles for the reaction of iodobenzene and phenylacetylene without significant loss of activity.
Although a significant number of studies have appeared in recent years on palladium catalysts based on imidazolium-derived organosilicas for C–C bond forming reactions, a successful recyclable catalytic system able to operate with less reactive but less expensive aryl chlorides is still a challenge. As previously mentioned, only few studies exist on Suzuki coupling that describe efficient and reusable catalysts for aryl chlorides. These refer to mesostructured organosilicas prepared by template-assisted sol–gel methodologies.
3.7 Addition of phenylboronic acid to benzaldehydes
Çetinkaya et al. applied the Rh–NHC complex grafted to amorphous mesoporous silicate, M36, as a catalyst for the addition of phenylboronic acid to benzaldehydes (Scheme 11), although no recycling was mentioned. The required reaction times were much higher than those reported for the homogeneous silylated complex.41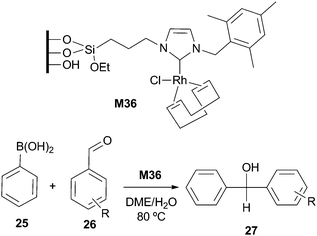 | ||
| Scheme 11 | ||
3.8 Kharasch reaction
Silica immobilized metal ion-containing imidazolium salts were tested in the Kharasch addition reaction between CCl4 and styrene (Scheme 12).83,84 The best performances were obtained with [CuCl4]2− counteranions (M30Cu), 98% conversions being achieved in 20 h with 1 mol% of catalyst (95% selectivity, 93% yield). The system was recycled up to 6 runs with a slight decrease of activity.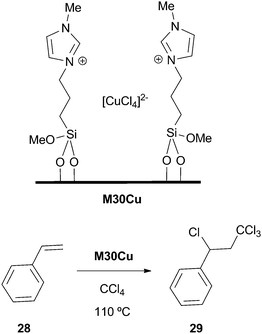 | ||
| Scheme 12 | ||
3.9 C–S cross-coupling reactions
Recently, Jun and Lee et al. described recyclable NHC–Ni complexes immobilized on magnetite/silica nanoparticles, M6a–b, for the C–S cross-coupling of aryl iodides with thiols (Scheme 13).51 In the reaction of 4-iodoacetophenone with thiophenol, M6a and M6b gave 81 and 92% isolated yields in 10 h. A slight enhancement of activity was generally found for the catalysts containing a pyridyl group. The catalyst M6b was easily recovered by an external magnet and three successful consecutive cycles were performed in the mentioned reaction. Leaching of Ni species was detected from the filtrates after the fourth run (14% by ICP-AES).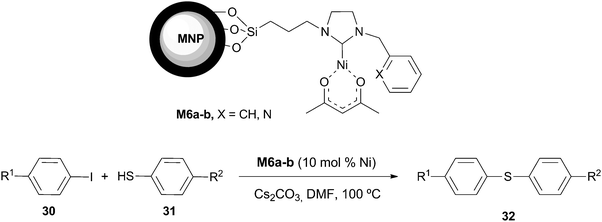 | ||
| Scheme 13 | ||
3.10 Oxidation processes
 | ||
| Scheme 14 | ||
The combination of Pd(OAc)2 and imidazolium-derived organosilica M38, prepared by a post-grafting route, also afforded catalytic systems for the selective aerobic oxidation of benzyl alcohol to benzaldehyde (Fig. 8).85 A NHC–Pd complex was formed, which was transformed into Pd(0) nanoparticles by H2 reduction at 200 °C. Compared with the supported Pd complexes, the immobilized Pd nanoparticles exhibited much higher activity and reusability for the title reaction (3 cycles vs. 8 cycles) (0.5 mol% Pd, K2CO3, 1 atm O2, toluene, 80 °C). According to the authors, no leaching of Pd species could be detected. A reference catalyst prepared by an analogous approach from Pd(OAc)2 and non-functionalized SBA-15 proved to be unstable, with a considerable Pd leaching occurring in the oxidation process. These results suggest that the presence of the imidazolium moieties in the material plays an important role in the formation of an active and stable supported Pd nanoparticles catalyst.
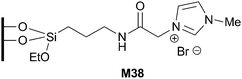 | ||
| Fig. 8 Imidazolium-derived organosilica used in alcohol oxidations. | ||
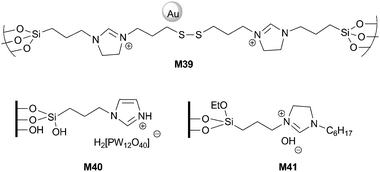 | ||
| Fig. 9 Supported imidazolium salts for olefin epoxidation. | ||
Yu and co-workers studied the catalytic performances of gold nanoparticles (1.8 nm) stabilized in a mesostructured hybrid silica (SBA-15 type, M39) in the epoxidation of styrene with tert-butyl hydroperoxide.86 Under the conditions used (2 wt% Au, benzene, 82 °C, 12 h), 95% of styrene conversion and 75% of selectivity to styrene oxide were obtained, benzaldehyde and phenyl acetaldehyde being detected as side products. This system displays higher activity and selectivity than classical gold nanocatalysts supported on alumina or magnesium oxide (particle size 3.2 and 7.9 nm, respectively). The activity slightly decreased (95 to 85% conversion) after reuse of M39 for eight times.
Phosphotungstic acid (PTA) immobilized onto imidazole functionalized fumed silica (M40) was an efficient catalyst for the epoxidation of a variety of olefins (limonene, cis-cyclooctene, 1-octene, norbornene, trans-2-octene, 1-methyl-1-cyclohexene) with aqueous H2O2 in acetonitrile at 60 °C. Recycling was possible up to 3 runs with cyclooctene as a model substrate.87
Mizuno et al. reported the catalytic activity of the silica supported dihydroimidazolium salt M41, which was prepared by grafting, for the epoxidation of some α,β-unsaturated cyclic ketones with H2O2 as an oxidant.88 The reactions were performed in heptane at 32 °C with 1.4 to 2.8 mol% of catalyst, full conversions being achieved within 1–4 h. No recycling was mentioned for this reaction.
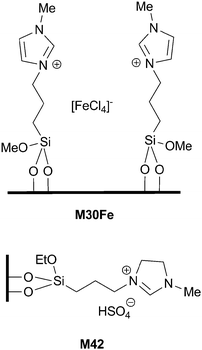 | ||
| Fig. 10 Supported catalysts for oxidation reactions. | ||
The Baeyer–Villiger reaction of several cyclic ketones to afford the corresponding lactones has been performed with imidazolium based organosilica M42 as a catalyst (Fig. 10), using 68% H2O2 as an oxidant in dichloromethane at 50 °C.90 Recycling up to 4 consecutive runs was possible with cyclopentanone as a model substrate.
3.11 Supported NHC–Cu catalyzed reactions
Silica-immobilized NHC–Cu(I) complex M5 (R = Me) (Scheme 15) has been used as an efficient catalyst in the three component coupling reaction of aldehydes, alkynes and amines (A3-coupling) under solvent-free conditions at room temperature. It was reused for ten cycles in the reaction of phenylacetylene, formaldehyde and piperidine with good isolated yields of the corresponding propargylamine (95–88%).65 The same group also applied this type of supported complexes M5 to the [3+2] cycloaddition of organic azides and terminal alkynes (click-type reaction) to yield regiospecifically 1,4-disubstituted 1,2,3-triazoles (Scheme 15). Silica supported material M5 (R = CH2Ph) was efficiently recycled for ten runs in the reaction between benzyl azide and phenylacetylene under neat conditions at room temperature (93–87%).66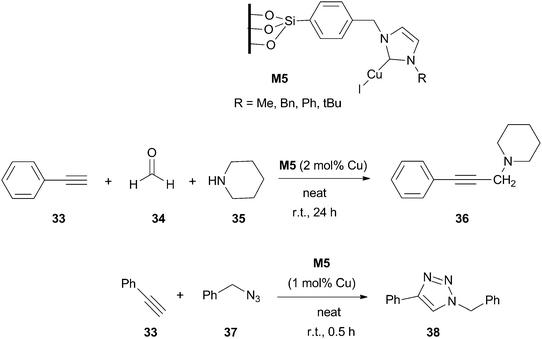 | ||
| Scheme 15 | ||
3.12 Organocatalysis: metal-free reactions
The previously reported silica material M41 has also been applied as an organocatalyst in the cyanosilylation of cyclic and acyclic ketones with trimethylsilyl cyanide (2.8 mol% of catalyst, DCM, 32 °C, 0.5–2 h) and it has been reused in a second run.88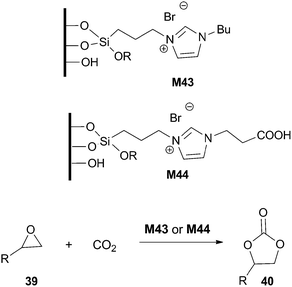
Higher activity and selectivity in the formation of cyclic carbonates were obtained with material M44 bearing a carboxyl group; recycling of the catalyst was performed for 5 runs without loss of activity. A mechanistic proposal has been made to explain the acceleration effect of the COOH group.93
 | ||
| Fig. 11 Silica-supported Knoevenagel catalysts. | ||
| Entry | Cat. | [mol%] | X | R | Solvent/T [°C] | t/h | Yield [%] |
|---|---|---|---|---|---|---|---|
| 1 | M45 98 | 0.8 | H | CN | −/100 | 6 | 98 |
| 2 | M45 98 | 0.8 | OMe | CN | −/100 | 16 | 100 |
| 4 | M1 98 | 0.8 | H | CN | −/100 | 6 | 98 |
| 5 | M1 98 | 1.2 | OMe | CN | −/100 | 4 | 99 |
| 6 | M1 98 | 1.2 | H | COOEt | −/130 | 2 | 100 |
| 7 | M46 99,100 | 0.8 | H | CN | −/100 | 3.5 | 93.5 |
| 8 | M46 99,100 | 0.8 | OMe | CN | −/100 | 3.5 | 89.6 |
| 9 | M46 99,100 | 0.8 | OMe | COOEt | −/100 | 6 | 79.1 |
| 10 | M47a 101 | 0.45 | H | CN | H2O/25 | 1 | 99 |
| 11 | M47a 101 | 0.45 | OMe | CN | H2O/25 | 1 | 91 |
| 12 | M47a 101 | 4.45 | OMe | COOEt | H2O/25 | 1 | 88 |
| 13 | M47b 101 | 0.43 | H | CN | H2O/25 | 1 | 99 |
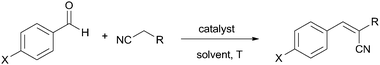
Hybrid silica materials M45 and M1 containing imidazolium and dihydroimidazolium salts in different dilutions have been prepared using template-based sol–gel methodologies.98 These materials were found to be active and reusable catalysts in the Knoevenagel condensations depicted in Table 7 under solvent-free conditions at 100 °C. Five consecutive cycles were performed with the same batch of catalysts. The immobilized catalysts showed higher activities for this transformation than other related homogeneous bis-imidazolium salts and were much more active than non-functionalized mesostructured MCM-41, which presented some residual activity. These facts suggest a certain degree of cooperativity between the surface of the inorganic matrix and the active sites and underline the crucial role of the (dihydro)imidazolium salt. Better performance was observed from the hybrid catalysts with higher organic loading, showing that the predominant factor is the concentration of the organics rather than the porosity of the materials under these neat conditions. Examples provided in entries 1 and 2 of Table 7 refer to M45 prepared with 10 equivalents of TEOS. The non-porous bridged silsesquioxane M1, obtained by the sol–gel condensation of the disilylated dihydroimidazolium salt 3, gave the best results in the reaction of a variety of aromatic aldehydes and active methylene compounds such as malononitrile and ethyl cyanoacetate. This better catalytic performance might be attributed to a higher density of active sites and to cooperative effects between neighboring centres.
An imidazolium-functionalized SBA-15-type material M46 prepared by co-condensation with TEOS has also been tested as a recyclable catalyst in the same reactions under neat conditions (some examples are depicted in entries 7–9 of Table 7).99,100 Shen and co-workers have reported similar results for a related material obtained by co-condensation of the same monomer with TEOS under acidic conditions without template assistance.97
Task-specific basic imidazolium salts immobilized on mesoporous silicas SBA-15 and MCM-41 by grafting (materials M47a–b) have recently been described as reusable catalysts for Knoevenagel condensations in aqueous media (examples in entries 10–13 of Table 7).101 In this case, the basic amino group is the true catalytic moiety. However, they found much better activity and reusability (eight runs without decreasing activity) in comparison with the amino-functionalized mesostructured silicas.
3.13 Miscellaneous
Silica–polymer core–shell microspheres with acidic imidazolium–styrene copolymer shells have been prepared and used as recyclable catalysts for acetalization of aldehydes with methanol.102A silica grafted dual acidic imidazolium salt effectively catalyzed the one-pot synthesis of amidoalkyl naphthols by the multicomponent condensation of aldehydes with 2-naphthol and amides under solvent-free conditions. Moreover, the catalyst could be recycled six times without a significant loss of catalytic activity.32
Dehydration of fructose to 5-hydroxymethylfurfural has been recently achieved by an imidazolium hydrogen sulfate derivative grafted on silica nanoparticles, the heterogeneous catalyst being reused up to 6 runs.103
4. Conclusions
Over the past decade, numerous reports on organosilicas containing covalently anchored imidazolium salts or metal–NHC complexes and their application in catalysis have appeared. This reflects the considerable interest in metal–NHC complexes and in ionic liquids chemistry, although the materials that are reported lose their liquid nature and behave as supported imidazolium salts. Despite the numerous reports, few synthetic approaches have been developed, most of the imidazolium salts being obtained via nucleophilic substitution of an imidazole with an alkyl halide. Currently on-going synthetic methodologies such as the CuAAC click reactions at the stage of the sol–gel precursors104,105 or as a post-functionalization method106–108 will open new pathways to such materials. Moreover, while only few types of materials have been investigated (the MCM-41 and SBA-15 grafted ones being the most widely studied), original mesostructures, like 3D-cubic systems37,49 (SBA-16 type) and new materials, such as hybrid PMOs36 remain almost unexplored.Imidazolium– and NHC–metal-containing organosilicas offer a wide range of applications in catalysis, and proved to be very versatile scaffolds. Owing to the strong metal–NHC bond, Ru and Ir complexes enable a good control on the metal leaching, which is of prime importance in organic synthesis (especially for pharmaceutics). Furthermore, confinement effects contribute to the improvement of the metallic centres' stability. In palladium chemistry, it has been shown that the supported imidazolium moieties are appropriate matrices to stabilize nanoparticles, which act as a reservoir of palladium catalysts. Robust systems with excellent catalytic properties have been described and the activation of aryl chlorides at low temperature still remains as a major goal. Besides, the applications are not only restricted to metal-catalyzed reactions: metal-free supported imidazolium salts also catalyze organic reactions such as the cycloaddition of CO2 with epoxides or the Knoevenagel reactions. Future developments are foreseen in more challenging organocatalyzed reactions such as the recent asymmetric benzoin condensations mediated by chiral triazolylidenes,109,110 which still constitute an open field of research.
Acknowledgements
We acknowledge financial support from Ministerio de Ciencia e Innovación (MICINN) and Ministerio de Educación y Ciencia (MEC) of Spain (Projects CTQ2009-07881/BQU and a grant to A. M.-M.), Consolider Ingenio 2010 (Project CSD2007-00006), Generalitat de Catalunya (Project SGR2009-01441), the Agence Nationale de la Recherche (CP2D-MESORCAT).References
- Y. Chauvin, S. Einloft and H. Olivier, Ind. Eng. Chem. Res., 1995, 34, 1149–1155 CrossRef CAS.
- V. I. Pârvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef.
- S. Chowdhury, R. S. Mohan and J. L. Scott, Tetrahedron, 2007, 63, 2363–2389 CrossRef CAS.
- J. Dupont and J. Spencer, Angew. Chem., Int. Ed., 2004, 43, 5296–5297 CrossRef CAS.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–91 CrossRef CAS.
- M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- A. Igau, H. Grützmacher, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 1988, 110, 6463–6466 CrossRef CAS.
- J. Vignolle, X. Cattoën and D. Bourissou, Chem. Rev., 2009, 109, 3333–3384 CrossRef CAS.
- W. A. Herrmann and C. Kocher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162–2187 CrossRef CAS.
- T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef.
- G. W. Nyce, S. Csihony, R. M. Waymouth and J. L. Hedrick, Chem.–Eur. J., 2004, 10, 4073–4079 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS.
- Z. Ma, J. H. Yu and S. Dai, Adv. Mater., 2010, 22, 261–285 CrossRef CAS.
- K. Anderson, S. Cortiñas Fernández, C. Hardacre and P. C. Marr, Inorg. Chem. Commun., 2004, 7, 73–76 CrossRef CAS.
- J. Le Bideau, L. Viau and A. Vioux, Chem. Soc. Rev., 2011, 40, 907–925 RSC.
- C. P. Mehnert, Chem.–Eur. J., 2005, 11, 50–56 CrossRef.
- B. Gadenne, P. Hesemann and J. J. E. Moreau, Chem. Commun., 2004, 1768–1769 RSC.
- T. Echelmeyer, H. W. Meyer and L. van Wullen, Chem. Mater., 2009, 21, 2280–2285 CrossRef CAS.
- S. J. Craythorne, K. Anderson, F. Lorenzini, C. McCausland, E. F. Smith, P. Licence, A. C. Marr and P. C. Marr, Chem.–Eur. J., 2009, 15, 7094–7100 CrossRef CAS.
- S. Volland, M. Gruit, T. Regnier, L. Viau, O. Lavastre and A. Vioux, New J. Chem., 2009, 33, 2015–2021 RSC.
- A. C. Marr and P. C. Marr, Dalton Trans., 2011, 40, 20–26 RSC.
- A. Riisager, R. Fehrmann, M. Haumann and P. Wasserscheid, Top. Catal., 2006, 40, 91–102 CrossRef CAS.
- C. P. Mehnert, R. A. Cook, N. C. Dispenziere and M. Afeworki, J. Am. Chem. Soc., 2002, 124, 12932–12933 CrossRef CAS.
- L. Rodríguez-Pérez, Y. Coppel, I. Favier, E. Teuma, P. Serp and M. Gómez, Dalton Trans., 2010, 39, 7565–7568 RSC.
- A. Zamboulis, N. Moitra, J. J. E. Moreau, X. Cattoën and M. Wong Chi Man, J. Mater. Chem., 2010, 20, 9322–9338 RSC.
- B. Lee, H. J. Im, H. M. Luo, E. W. Hagaman and S. Dai, Langmuir, 2005, 21, 5372–5376 CrossRef CAS.
- R. Ciriminna, P. Hesemann, J. J. E. Moreau, M. Carraro, S. Campestrini and M. Pagliaro, Chem.–Eur. J., 2006, 12, 5220–5224 CrossRef CAS.
- T. P. Nguyen, P. Hesemann and J. J. E. Moreau, Microporous Mesoporous Mater., 2011, 142, 292–300 CrossRef CAS.
- M. Trilla, X. Cattoën, C. Blanc, M. Wong Chi Man and R. Pleixats, J. Mater. Chem., 2011, 21, 1058–1063 RSC.
- B. Çetinkaya, N. Gürbüz, T. Seçkin and I. Özdemir, J. Mol. Catal. A: Chem., 2002, 184, 31–38 CrossRef.
- Q. A. Zhang, J. Luo and Y. Y. Wei, Green Chem., 2010, 12, 2246–2254 RSC.
- C. S. J. Cazin, M. Veith, P. Braunstein and R. B. Bedford, Synthesis, 2005, 622–626 CAS.
- M. Trilla, G. Borja, R. Pleixats, M. Wong Chi Man, C. Bied and J. J. E. Moreau, Adv. Synth. Catal., 2008, 350, 2566–2574 CrossRef CAS.
- T. P. Nguyen, P. Hesemann, P. Gaveau and J. J. E. Moreau, J. Mater. Chem., 2009, 19, 4164–4171 RSC.
- H. Q. Yang, X. J. Han, G. A. Li, Z. C. Ma and Y. J. Hao, J. Phys. Chem. C, 2010, 114, 22221–22229 CAS.
- H. Q. Yang, G. A. Li, Z. C. Ma, J. B. Chao and Z. Q. Guo, J. Catal., 2010, 276, 123–133 CrossRef CAS.
- D. P. Allen, M. M. Van Wingerden and R. H. Grubbs, Org. Lett., 2009, 11, 1261–1264 CrossRef CAS.
- A. Monge-Marcet, R. Pleixats, X. Cattoën and M. Wong Chi Man, unpublished results.
- H. W. Wanzlick and E. Schikora, Angew. Chem., 1960, 72, 494–494 CrossRef CAS.
- I. Özdemir, N. Gürbüz, T. Seçkin and B. Çetinkaya, Appl. Organomet. Chem., 2005, 19, 633–638 CrossRef.
- B. Karimi and D. Enders, Org. Lett., 2006, 8, 1237–1240 CrossRef CAS.
- H. M. J. Wang and I. J. B. Lin, Organometallics, 1998, 17, 972–975 CrossRef CAS.
- S. Carrettin, M. C. Blanco, A. Corma and A. S. K. Hashmi, Adv. Synth. Catal., 2006, 348, 1283–1288 CrossRef CAS.
- A. Corma, C. González-Arellano, M. Iglesias, S. Pérez-Ferreras and F. Sánchez, Synlett, 2007, 1771–1774 CrossRef CAS.
- C. del Pozo, A. Corma, M. Iglesias and F. Sánchez, Organometallics, 2010, 29, 4491–4498 CrossRef CAS.
- S. Dastgir, K. S. Coleman and M. L. H. Green, Dalton Trans., 2011, 40, 661–672 RSC.
- Ö. Aksin, H. Türkmen, L. Artok, B. Çetinkaya, C. Y. Ni, O. Büyükgüngor and E. Özkal, J. Organomet. Chem., 2006, 691, 3027–3036 CrossRef.
- H. Q. Yang, X. J. Han, G. Li and Y. W. Wang, Green Chem., 2009, 11, 1184–1193 RSC.
- J. Y. Shin, B. S. Lee, Y. Jung, S. J. Kim and S. G. Lee, Chem. Commun., 2007, 5238–5240 RSC.
- H. J. Yoon, J. W. Choi, H. Kang, T. Kang, S. M. Lee, B. H. Jun and Y. S. Lee, Synlett, 2010, 2518–2522 CAS.
- L. Li and J. L. Shi, Adv. Synth. Catal., 2005, 347, 1745–1749 CrossRef CAS.
- A. Corma, E. Gutiérrez-Puebla, M. Iglesias, A. Monge, S. Pérez-Ferreras and F. Sánchez, Adv. Synth. Catal., 2006, 348, 1899–1907 CrossRef CAS.
- G. Liu, M. Q. Hou, T. B. Wu, T. Jiang, H. L. Fan, G. Y. Yang and B. X. Han, Phys. Chem. Chem. Phys., 2011, 13, 2062–2068 RSC.
- D. A. Loy and K. J. Shea, Chem. Rev., 1995, 95, 1431–1442 CrossRef CAS.
- K. J. Shea, J. J. E. Moreau, D. A. Loy, R. J. P. Corriu and B. Boury, in Functional Hybrid Materials, ed. P. Gomez-Romero and C. Sanchez, WILEY-VCH, Weinheim, 2004, pp. 50–85 Search PubMed.
- V. Polshettiwar and R. S. Varma, Tetrahedron, 2008, 64, 4637–4643 CrossRef CAS.
- V. Polshettiwar, P. Hesemann and J. J. E. Moreau, Tetrahedron Lett., 2007, 48, 5363–5366 CrossRef CAS.
- B. J. Melde, B. T. Holland, C. F. Blanford and A. Stein, Chem. Mater., 1999, 11, 3302–3308 CrossRef CAS.
- T. Asefa, M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 402, 867–871 CAS.
- S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611–9614 CrossRef CAS.
- T. K. Maishal, M. Boualleg, M. Bouhrara, C. Copéret, E. Jeanneau, L. Veyre and C. Thieuleux, Eur. J. Inorg. Chem., 2010, 5005–5010 CrossRef CAS.
- I. Karamé, M. Boualleg, J. M. Camus, T. K. Maishal, J. Alauzun, J. M. Basset, C. Copéret, R. J. P. Corriu, E. Jeanneau, A. Mehdi, C. Reyé, L. Veyre and C. Thieuleux, Chem.–Eur. J., 2009, 15, 11820–11823 CrossRef.
- T. K. Maishal, J. Alauzun, J.-M. Basset, C. Copéret, R. J. P. Corriu, E. Jeanneau, A. Mehdi, C. Reyé, L. Veyre and C. Thieuleux, Angew. Chem., Int. Ed., 2008, 47, 8654–8656 CrossRef CAS.
- M. Wang, P. H. Li and L. Wang, Eur. J. Org. Chem., 2008, 2255–2261 CrossRef CAS.
- P. H. Li, L. Wang and Y. C. Zhang, Tetrahedron, 2008, 64, 10825–10830 CrossRef CAS.
- S. Prühs, C. W. Lehmann and A. Fürstner, Organometallics, 2004, 23, 280–287 CrossRef.
- K. H. Park, S. Kim and Y. K. Chung, Bull. Korean Chem. Soc., 2008, 29, 2057–2060 CrossRef CAS.
- B. Karimi, D. Elhamifar, J. H. Clark and A. J. Hunt, Chem.–Eur. J., 2010, 16, 8047–8053 CAS.
- S. Tandukar and A. Sen, J. Mol. Catal. A: Chem., 2007, 268, 112–119 CrossRef CAS.
- J. Schwartz, Acc. Chem. Res., 1985, 18, 302–308 CrossRef CAS.
- Y. Kume, K. Qiao, D. Tomida and C. Yokoyama, Catal. Commun., 2008, 9, 369–375 CrossRef CAS.
- C. Copéret and J.-M. Basset, Adv. Synth. Catal., 2007, 349, 78–92 CrossRef.
- M. R. Buchmeiser, Chem. Rev., 2009, 109, 303–321 CrossRef CAS.
- J. S. Kingsbury, S. B. Garber, J. M. Giftos, B. L. Gray, M. M. Okamoto, R. A. Farrer, J. T. Fourkas and A. H. Hoveyda, Angew. Chem., Int. Ed., 2001, 40, 4251–4256 CrossRef CAS.
- M. Mayr, M. R. Buchmeiser and K. Wurst, Adv. Synth. Catal., 2002, 344, 712–719 CrossRef CAS.
- G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS.
- V. Polshettiwar, C. Len and A. Fihri, Coord. Chem. Rev., 2009, 253, 2599–2626 CrossRef CAS.
- P. Han, H. M. Zhang, X. P. Qiu, X. L. Ji and L. X. Gao, J. Mol. Catal. A: Chem., 2008, 295, 57–67 CrossRef CAS.
- S. M. Lee, H. J. Yoon, J. H. Kim, W. J. Chung and Y. S. Lee, Pure Appl. Chem., 2007, 79, 1553–1559 CrossRef CAS.
- H. Qiu, S. M. Sarkar, D.-H. Lee and M.-J. Jin, Green Chem., 2008, 10, 37–40 RSC.
- H. Q. Yang, Y. W. Wang, Y. Qin, Y. Z. Chong, Q. Z. Yang, G. Li, L. Zhang and W. Li, Green Chem., 2011, 13, 1352–1361 RSC.
- T. Sasaki, M. Tada, C. Zhong, T. Kume and Y. Iwasawa, J. Mol. Catal. A: Chem., 2008, 279, 200–209 CrossRef CAS.
- T. Sasaki, C. M. Zhong, M. Tada and Y. Iwasawa, Chem. Commun., 2005, 2506–2508 RSC.
- Y. Z. Hou, X. T. Ji, G. Liu, J. Y. Tang, J. Zheng, Y. Liu, W. X. Zhang and M. J. Jia, Catal. Commun., 2009, 10, 1459–1462 CrossRef CAS.
- Y. Jin, P. J. Wang, D. H. Yin, J. F. Liu, H. Y. Qiu and N. Y. Yu, Microporous Mesoporous Mater., 2008, 111, 569–576 CrossRef CAS.
- L. T. Aany Sofia, A. Krishnan, M. Sankar, N. K. K. Raj, P. Manikandan, P. R. Rajamohanan and T. G. Ajithkumar, J. Phys. Chem. C, 2009, 113, 21114–21122 Search PubMed.
- K. Yamaguchi, T. Imago, Y. Ogasawara, J. Kasai, M. Kotani and N. Mizuno, Adv. Synth. Catal., 2006, 348, 1516–1520 CrossRef CAS.
- S. V. Sirotin, I. F. Moskovskaya, Y. G. Kolyagin, A. V. Yatsenko and B. V. Romanovsky, Russ. J. Phys. Chem. A, 2011, 85, 390–396 CrossRef CAS.
- A. Chrobok, S. Baj, W. Pudlo and A. Jarzebski, Appl. Catal., A, 2009, 366, 22–28 CrossRef CAS.
- M. M. Dharman, H. J. Choi, S. W. Park and D. W. Park, Top. Catal., 2010, 53, 462–469 CrossRef.
- M. K. Lee, H. L. Shim, M. M. Dharman, K. H. Kim, S. W. Park and D. W. Park, Korean J. Chem. Eng., 2008, 25, 1004–1007 CrossRef CAS.
- L. Han, H. J. Choi, S. J. Choi, B. Y. Liu and D. W. Park, Green Chem., 2011, 13, 1023–1028 RSC.
- H. L. Shim, M. K. Lee, K. H. Kim, D. W. Park and S. W. Park, Polym. Adv. Technol., 2008, 19, 1436–1440 CrossRef CAS.
- L. Han, S. W. Park and D. W. Park, Energy Environ. Sci., 2009, 2, 1286–1292 CAS.
- S. Udayakumar, M. K. Lee, H. L. Shim, S. W. Park and D. W. Park, Catal. Commun., 2009, 10, 659–664 CrossRef CAS.
- G. Q. Lai, J. J. Peng, J. Y. Li, H. Y. Qiu, J. X. Jiang, K. Z. Jiang and Y. J. Shen, Tetrahedron Lett., 2006, 47, 6951–6953 CrossRef CAS.
- M. Trilla, R. Pleixats, M. Wong Chi Man and B. Bied, Green Chem., 2009, 11, 1815–1820 RSC.
- Y. Liu, J. J. Peng, S. R. Zhai, J. Y. Li, J. J. Mao, M. J. Li, H. Y. Qiu and G. Q. Lai, Eur. J. Inorg. Chem., 2006, 2947–2949 CrossRef CAS.
- J. Y. Li, J. J. Peng, H. Y. Qiu, J. X. Jiang, J. R. Wu, Y. Ni and G. Q. Lai, Chin. J. Org. Chem., 2007, 27, 483–487 CAS.
- H. H. Zhao, N. Y. Yu, Y. Ding, R. Tan, C. Liu, D. H. Yin, H. Y. Qiu and D. L. Yin, Microporous Mesoporous Mater., 2010, 136, 10–17 CrossRef CAS.
- Y. Yamada, K. Qiao, Q. X. Bao, D. Tomida, D. Nagao, M. Konno and C. Yokoyama, Catal. Commun., 2009, 11, 227–231 CrossRef CAS.
- K. B. Sidhpuria, A. L. Daniel-da-Silva, T. Trindade and J. A. P. Coutinho, Green Chem., 2011, 13, 340–349 RSC.
- N. Moitra, J. J. E. Moreau, X. Cattoën and M. Wong Chi Man, Chem. Commun., 2010, 46, 8416–8418 RSC.
- K. Bürglová, N. Moitra, J. Hodačová, X. Cattoën and M. Wong Chi Man, J. Org. Chem., 2011, 76, 7326–7333 CrossRef.
- J. Nakazawa and T. D. P. Stack, J. Am. Chem. Soc., 2008, 130, 14360–14361 CrossRef CAS.
- B. Malvi, B. R. Sarkar, D. Pati, R. Mathew, T. G. Ajithkumar and S. Sen Gupta, J. Mater. Chem., 2009, 19, 1409–1416 RSC.
- A. K. Ganai, R. Bhardwaj, S. Hotha, S. Sen Gupta and B. L. V. Prasad, New J. Chem., 2010, 34, 2662–2670 RSC.
- D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS.
- Z. Zhou, Q. Meng, A. Seifert, A. Wagener, Y. Sun, S. Ernst and W. Thiel, Microporous Mesoporous Mater., 2009, 121, 145–151 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |