Exploring the role of phosphorus substituents on the enantioselectivity of Ru-catalysed ketone hydrogenation using tridentate phosphine-diamine ligands†
Scott D.
Phillips
a,
Kristian H. O.
Andersson
b,
Nina
Kann
*b,
Michael T.
Kuntz
c,
Marcia B.
France
*c,
Piotr
Wawrzyniak
a and
Matthew L.
Clarke
*a
aSchool of Chemistry, University of St Andrews, St Andrews, Fife, UK KY16 9ST. E-mail: mc28@st-andrews.ac.uk; Fax: +44 (0) 1334 463808; Tel: +44 (0) 1334 463850
bDepartment of Chemical and Biological Engineering, Chalmers University of Technology, SE-412 96, Gothenburg, Sweden
cDepartment of Chemistry, Washington and Lee University, Lexington, VA 24450, USA
First published on 5th September 2011
Abstract
Methods to produce a range of new phosphine-diamine ligands from phosphino-aldehydes have been developed and a hypothesis that larger P-substituents would increase the enantioselectivity towards the (S) isomer in Ru-catalysed ketone hydrogenation of acetophenone has been examined; the successful validation of this hypothesis is further evidence that the mechanism of these catalysts involves a secondary amine assisted reduction.
The reduction of ketones to alcohols is a key technology for practical asymmetric synthesis. In early studies, the ketone had to contain a second co-ordinating group,1 but the development of ruthenium diphosphine/diamine catalysts such as [RuCl2(Xyl-BINAP)(DPEN)] by the Noyori group enabled highly efficient reduction of a range of so-called unfunctionalised non-chelating ketones, and some functionalised ketones as well.2 In addition to a wide range of catalysts of type A, a few groups have attempted to develop catalysts that can hydrogenate unreactive or problematic ketones and/or to explore different ligand designs and thus provide a further expansion to this most important asymmetric synthesis method.3 Our approach has been to use a different catalyst design (Fig. 1), that uses a tridentate ligand (B) in place of a diphosphine and a diamine (A).4
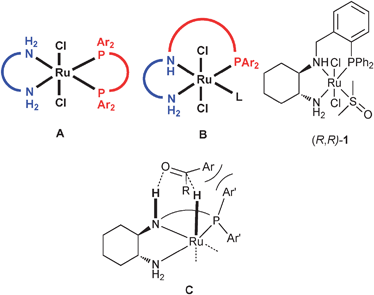 | ||
| Fig. 1 Schematic of Noyori Catalysts, and the St Andrews tridentate systems including, (C): a working model for a disfavoured transition state for hydride attack if PAr′2 groups are bulky. | ||
These catalysts show unusual reactivity and high enantioselectivity in the hydrogenation of low-reactivity bulky ketones and bulky heterocyclic ketones, but we note are completely unselective in the reduction of simple acetophenones.
Our initial design of these catalysts envisaged that the primary amine terminus of the ligand would bind to ketones in the same way as the primary amine functionality does in a Noyori catalyst. However, a recent mechanistic study suggested to us that the secondary amine part of our catalysts may be more significant, and could bind the ketone substrate prior to and during hydride attack.4c If this were the case, then reconsidering the interaction of a ketone substrate with the secondary amine suggests that the phosphorus substituents on catalysts related to 1, hitherto thought to be remote from the action, would be in close proximity to the ketone substrate. Specifically bulky PAr′2 groups (fragment C, Fig. 1) might occupy the same space required for the ketone Ar group; If the transition state represented by schematic C is disfavoured, this would enable the selective production of the (S) enantiomer of phenethyl alcohol from acetophenone. We therefore set out to test this hypothesis, since we felt it could be the start of developing a system with very broad scope. We communicate the methods used to produce such catalysts and the successful validation of this hypothesis here.
We elected to explore both P-chiral and simpler bulky P-aryl phosphorus ligands. While chiral aminophosphine ligands are widely used in asymmetric synthesis,5 examples of the corresponding P-chirogenic versions are less common.6–8P,N,N-type ligands of this type, as desired for our purposes, are virtually unknown7b although examples of both P-chiral P,N,P-ligands9 as well as a P,N,N,P-macrocycle have been reported.10 The phenylanisylmethylphosphine (PAMP) scaffold11 was selected as the phosphine component in the construction of new P-chiral P,N,N-type ligand (Scheme 1). We have earlier reported a synthetic route to P-chiral P,N- and P,N,N,P-type aminophosphines employing microwave-assisted reductive amination,7a and initially employed this method in the synthesis of our target ligands, starting from (S)-2-((2-methoxyphenyl)(phenyl)phosphino)acetaldehyde. The desired ligands were formed, but the synthesis was in this particular case low yielding and difficult to reproduce, most likely due to the sensitive nature of the ligand and the imine precursor. We thus instead opted for a somewhat longer but more robust approach via the corresponding P,N,N-amide 4. (S)-2-(Boronato(2-methoxyphenyl)(phenyl)-phosphino)acetic acid ((S)-2),12 prepared in one step from borane-protected (S)-PAMP, was treated with N-hydroxysuccinimide under standard conditions13 forming active ester (S)-2 in 71% yield. Dropwise addition of (S)-3 to an excess of (1R,2R)-diaminocyclohexane over 3 h produced the desired intermediate P,N,N-amide (R,R),(SP)-4 in 64% yield. This was reduced to the desired P,N,N-borane complex, (R,R),(SP)-5 using borane·THF complex (Scheme 1 and ESI†). The corresponding (S,S),(SP) diastereomer of 5 was prepared in the same manner starting from (S)-3 and (1R,2R)-diaminocyclohexane.
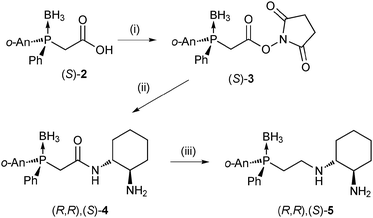 | ||
| Scheme 1 Synthesis of a P-chiral P,N,N-type ligand. Reagents and conditions: (i) N-hydroxysuccinimide (2 eq.), DCC (2 eq.), CH2Cl2, r.t., 3 h (71%); (ii) (1R,2R)-diaminocyclohexane (2.5 eq.), CH2Cl2, r.t., 3 h (64%); (iii) BH3·THF (10 eq.), THF, 0 °C to r.t., 16 h. | ||
A workable synthesis for the simpler P-Aryl ligands also needed to be developed, since only the xylyl and phenyl substituted phosphine benzaldehydes were commercially available. The route shown in Scheme 2 has considerable flexibility, since compound 7 should react with a variety of nucleophiles. With the phosphine-benzaldehydes in hand, the formation of the P,N,N ligands was attempted. We note here that the syntheses of phosphine-diamines derived from di-primary amines can be rather challenging, depending on the diamine and aldehyde used. It was possible to obtain pure Ru complexes 19-22 after chromatographic purification, although in the case of 18 using impure ligand (see ESI†).
![Synthesis of diarylphosphinobenzaldehydes. General conditions: (i) 6 (1 eq.), nBuLi (1.1 eq.), THF,−78 °C, 2 h; PCl3 (10 eq.), THF,−78 °C, 0.5 h (ii) 7 (1 eq.), ArMgX or ArLi (2 eq.), THF,−78 °C→rt, 2 h (iii) 8, 9 or 10 (1eq.), p-TsOH·H2O (0.1 eq.), acetone, Δ, 2 h. (iv) 11, 12, 14 or 13 (1 eq.), (R,R)-diaminocyclohexane (3 eq.), EtOH, 45 °C → rt, 2 h; NaBH4 (4 eq.), EtOH, rt, 8 h; (v) 15, 16, 17 or 18 (1 eq.), [RuCl2(DMSO)4] (1 eq.), THF, μw, 120 °C, 20 min.](/image/article/2011/CY/c1cy00253h/c1cy00253h-s2.gif) | ||
| Scheme 2 Synthesis of diarylphosphinobenzaldehydes. General conditions: (i) 6 (1 eq.), nBuLi (1.1 eq.), THF,−78 °C, 2 h; PCl3 (10 eq.), THF,−78 °C, 0.5 h (ii) 7 (1 eq.), ArMgX or ArLi (2 eq.), THF,−78 °C→rt, 2 h (iii) 8, 9 or 10 (1eq.), p-TsOH·H2O (0.1 eq.), acetone, Δ, 2 h. (iv) 11, 12, 14 or 13 (1 eq.), (R,R)-diaminocyclohexane (3 eq.), EtOH, 45 °C → rt, 2 h; NaBH4 (4 eq.), EtOH, rt, 8 h; (v) 15, 16, 17 or 18 (1 eq.), [RuCl2(DMSO)4] (1 eq.), THF, μw, 120 °C, 20 min. | ||
Developing an in situ screening method such that a ligand or its borane complex could be used directly in combination with a Ru precursor in catalysis seemed a useful objective to allow us to test ligands prepared on a small scale. Pleasingly, using the parent borane-protected ligand directly with the ruthenium precursor [RuCl2(DMSO)4] gives a catalyst that gives the same enantioselectivity and yields in asymmetric hydrogenation of 1,1′dimethylpropiophenone as the purified pre-catalysts, 1 (Table 1, Entries 2 and 3). We therefore screened the P-chiral phosphine boranes, 4 and 5in situ. Significantly, (R,R),(S)-5 does give greater selectivity to the (S) enantiomer in the reduction of acetophenone relative to 1, although the magnitude of enantioselectivity is quite poor (Table 1, Entry 4). Moving on to the bulky ligands that were achiral at phosphorus, tested as their isolated complexes, a further increase in the selectivity towards the (S) enantiomer was observed in the hydrogenation of acetophenone, with complex 22 delivering significant selectivity towards (S)-phenethyl alcohol (60% ee, Table 1, Entry 11). There are absolutely no synthetic advantages in this process for preparing (S)-phenethyl alcohol in moderately high e.e; a range of Noyori catalysts, providing higher base: catalyst ratio's are used, have been shown to give near perfect results (∼99% e.e., > 99% conversion, high T. O. F., under milder conditions!). However, there are substrate classes that either give results that are less synthetically useful, or some substrates that do not reduce at all,2b,3d,j,n,q,t,4 maintaining a strong impetus for developing alternative types of catalyst that could have different scope. Moreover, this strongly supports the hypothesis we have examined and opens the way towards rational design of catalysts to match substrates in this system. This is also a rare example of a completely unselective catalyst being tuned to give very significant selectivity by a rational modification rather than random screening. In the case of the bulkier substrate, this modification creates a further bias for the phenyl ring to avoid the P-Aryl rings, and a slight but meaningful increase in enantioselectivity to 85% ee was recorded (we assume that the bulbous but relatively short tBu group can fit in the pocket between the Ru and the PAr2 groups but the longer phenyl group does not). Catalysts 19 and 20 were examined to assess electronic effects on the productivity of these catalysts; this revealed that the electron-withdrawing catalyst is essentially inactive, while electron donating 20 is slightly less active than 1. Whether this results from electronic effects on various stages of the catalytic cycle or off-cycle events is unknown. Given that the very bulky catalyst 22 is also significantly less active than 1, then there is clearly a compromise between selectivity and activity that must be reached for each substrate.
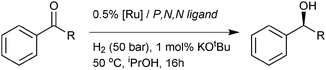
| Entrya | Catalyst/Ligand | Ketone R = | Conversion [%]b | ee [%]c |
|---|---|---|---|---|
| a Unless otherwise indicated, reactions were carried out using 0.33 mmol mL−1 of ketone, 0.5 mol% [RuCl2(DMSO)4], 0.7 mol% protected ligand and conditions in the equation. b Conversions were determined by 1H NMR analysis of the crude reaction mixtures (all peaks could be assigned). c see experimental for e.e. determinations and configurations. d Using preformed catalyst, 0.5 mol% catalyst 1, (or 21, or 22), 1 mol% KOtBu, iPrOH, H2 (50 bar), 50 °C, 16 h. e 0.5 mol% [RuCl2(DMSO)4], 0.7 mol% original PPh2 ligand converted to BH3 adduct and 1 mol% KOtBu. f 70 °C. g 70 °C, 24 h. | ||||
| 1d | (R,R)-1 | CH3 | >99 | 0 |
| 2d | (R,R)-1 | C(CH3)3 | >99 | 74 (S) |
| 3e | (R,R)-1 | C(CH3)3 | >99 | 74 (S) |
| 4 | (R,R),(SP)-5 | CH3 | >99 | 29 (S) |
| 5 | (R,R),(SP)-5 | C(CH3)3 | >99 | 74 (S) |
| 6 | (S,S),(SP)-5 | CH3 | >99 | 13 (S) |
| 7 | (S,S),(SP)-5 | C(CH3)3 | >99 | 67 (S) |
| 8d | (R,R)-21 | CH3 | >99 | 35 (S) |
| 9d | (R,R)-21 | C(CH3)3 | >99 | 77 (S) |
| 10d | (R,R)-22 | CH3 | 24 | 56 (S) |
| 11d,f | (R,R)-22 | CH3 | 84 | 60 (S) |
| 12d,g | (R,R)-22 | CH3 | >99 | 58 (S) |
| 13d | (R,R)-22 | C(CH3)3 | 5 | 83 (S) |
| 14d,g | (R,R)-22 | C(CH3)3 | 21 | 85 (S) |
In summary, we have proposed a model for hydride transfer in the tridentate Ru hydrogenation catalysts, developed synthetic routes to new bulky P,N,N ligands, and validated the model by examining their performance in the hydrogenation of two substrates where enhanced selectivity towards the (S) enantiomer of alcohol was predicted and obtained. These new members of this Ru/P,N,X family, along with this new understanding should prove useful in further studies directed towards hydrogenations in which other catalysts are completely unreactive. Any important advances will be reported in due course.
The authors would like to thank the EPSRC for funding this project and the use of the National Mass Spectrometry service. The authors also thank the Washington and Lee University Lenfest Grant (MBF), R.E. Lee Research Fellowship (MTK), the Swedish Reseach Council (KHOA) and the Associated Colleges of the South Andrew W. Mellon Faculty Renewal Grant (MBF).
Notes and references
- M. Kitamura, T. Ohkuma, S. Inoue, N. Sayo, H. Kumobayashi, S. Akutagawa, T. Ohta, H. Takaya and R. Noyori, J. Am. Chem. Soc., 1988, 110, 629 CrossRef CAS.
- (a) T. Ohkuma, S. Hashaguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 2675 CrossRef CAS; (b) R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40 CrossRef CAS; (c) The handbook of homogeneous hydrogenation, ed. C. J. Elsevier and J. G. De Vries, Wiley-VCH, Weinheim, 2007 Search PubMed.
- Some recent catalysts are based on significantly different co-ordination environments, and developments using such catalysts can be anticipated in the future. See: (a) Y. J. Xu, N. W. Alcock, G. J. Clarkson, G. Docherty, G. Woodward and M. Wills, Org. Lett., 2004, 6, 4105 CrossRef CAS; (b) Y. Xu, G. C. Clarkson, G. Docherty, C. L. North, G. Woodward and M. Wills, J. Org. Chem., 2005, 70, 8079 CrossRef CAS; (c) Q. Jing, X. Zhang, H. Sun and K. L. Ding, Adv. Synth. Catal., 2005, 347, 1193 CrossRef CAS; (d) T. Ohkuma, K. Tsutsumi, N. Utsumi, N. Arai, R. Noyori and K. Murata, Org. Lett., 2007, 9, 255 CrossRef CAS; (e) F. Naud, C. Malan, F. Spindler, C. Ruggeberg, A. T. Schmidt and H. U. Blaser, Adv. Synth. Catal., 2006, 348, 47 CrossRef CAS; (f) R. W. Guo, A. J. Lough, R. H. Morris and D. T. Song, Organometallics, 2004, 23, 5524 CrossRef CAS; (g) K. Abdur-Rashid, R. W. Guo, A. J. Lough, R. H. Morris and D. T. Song, Adv. Synth. Catal., 2005, 347, 571 CrossRef CAS; (h) H. Huang, T. Okuno, K. Tsuda, M. Yoshimura and M. Kitamura, J. Am. Chem. Soc., 2006, 128, 8716 CrossRef CAS; (i) T. Ohkuma, C. A. Sandoval, R. Srinivasan, Q. Lin, Y. Wei, K. Muñiz and R. Noyori, J. Am. Chem. Soc., 2005, 127, 8288 CrossRef CAS; (j) N. Arai, K. Suzuki, S. Sugizaki, H. Sorimachi and T. Ohkuma, Angew. Chem., Int. Ed., 2008, 47, 1770 CrossRef CAS; (k) Ir system using P,N ligands: J.-B. Xie, J.-H. Xie, X.-Y. Liu, W.-L. Kong, S. Li and Q.-L. Zhou, J. Am. Chem. Soc., 2010, 132, 4538 CrossRef CAS; (l) J. E. D. Martins, D. J. Morris and M. Wills, Tetrahedron Lett., 2009, 50, 688 CrossRef CAS; (m) Y. Xu, G. F. Docherty, G. Woodward and M. Wills, Tetrahedron: Asymmetry, 2006, 17, 2925 CrossRef CAS; (n) T. Ohkuma, N. Utsumi, K. Tsutsumi, K. Murata, C. Sandoval and R. Noyori, J. Am. Chem. Soc., 2006, 128, 8724 CrossRef CAS; (o) A. C. Marr, M. Niewenhuyzen, C. L. Pollock and G. C. Saunders, Organometallics, 2007, 26, 2659 CrossRef CAS; (p) L. A. Saudan, C. M. Saudan, C. Debrieux and P. Wyss, Angew. Chem., Int. Ed., 2007, 46, 7473 CrossRef CAS; (q) W. W. N. M. O, A. J. Lough and R. H. Morris, Chem. Commun., 2010, 46, 8240 RSC; (r) Os systems: W. Barratta, M. Ballico, G. Chelucci, K. Siega and P. Rigo, Angew. Chem., Int. Ed., 2008, 47, 4362 CrossRef; (s) Fe systems: R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282 RSC; (t) I. C. Lennon and J. A. Ramsden, Org. Process Res. Dev., 2005, 9, 110 CrossRef CAS; (u) C. J. Cobley and j. P. Hensche, Adv. Synth. Catal., 2003, 345, 195 CrossRef CAS.
- (a) M. L. Clarke, M. B. Diaz-Valenzuela and A. M. Z. Slawin, Organometallics, 2007, 26, 16 CrossRef CAS; (b) M. B. Diaz-Valenzuela, S. D. Phillips, M. B. France, M. E. Gunn and M. L. Clarke, Chem.–Eur. J., 2009, 15, 1227 CrossRef CAS; (c) S. D. Phillips, J. A. Fuentes and M. L. Clarke, Chem.–Eur. J., 2010, 16, 8002 CAS; (d) a manuscript on the hydrogenation of another class of ketone that is not effectively reduced with Noyori-type catalysts, is in preparation.
- For recent reviews on P,N-ligands and their applications, see (a) P. J. Guiry and C. P. Saunders, Adv. Synth. Catal., 2004, 346, 497 CrossRef CAS; (b) I. D. Kostas, Curr. Org. Synth., 2008, 5, 227 CrossRef CAS; (c) D. Amoroso, T. W. Graham, R. Guo, C.-W. Tsang and K. Abdur-Rashid, Aldrichimica Acta, 2008, 41, 15 CAS.
- For examples of P-chiral P,N-ligands, see (a) H. Yang, N. Lugan and R. Mathieu, Organometallics, 1997, 16, 2089 CrossRef CAS; (b) S. R. Gilbertson, D. G. Genov and A. L. Rheingold, Org. Lett., 2000, 2, 2885 CrossRef CAS; (c) W. J. Tang, W. M. Wang and X. M. Zhang, Angew. Chem., Int. Ed., 2003, 42, 943 CrossRef CAS; (d) H. Danjo, M. Higuchi, M. Yada and T. Imamoto, Tetrahedron Lett., 2004, 45, 603 CrossRef CAS; (e) T. Imamoto, K. Sugita and K. Yoshida, J. Am. Chem. Soc., 2005, 127, 11934 CrossRef CAS; (f) X.-M. Sun, M. Koizumi, K. Manabe and S. Kobayashi, Adv. Synth. Catal., 2005, 347, 1893 CrossRef CAS.
- (a) M. J. Johansson, K. H. O. Andersson and N. Kann, J. Org. Chem., 2008, 73, 4458 CrossRef CAS; (b) G. Brenchley, E. Merifield, M. Wills and M. Fedouloff, Tetrahedron Lett., 1994, 35, 2791 CrossRef CAS; (c) A P-chiral N,P,N ligand: A. P. Duncan and J. L. Leighton, Org. Lett., 2004, 6, 4117 CrossRef CAS.
- For recent reviews on the synthesis and applications of P-chiral ligands, see (a) A. Grabulosa, J. Granell and G. Muller, Coord. Chem. Rev., 2007, 251, 25 CrossRef CAS; (b) D. S. Glueck, Chem.–Eur. J., 2008, 14, 7108 CrossRef CAS; (c) Phosphorus Ligands in Asymmetric Catalysis, ed. A. Börner, Wiley-VCH, Weinheim, 2008 Search PubMed; (d) J. S. Harvey and V. Gouverneur, Chem. Commun., 2010, 46, 7477 RSC; (e) A. Grabulosa, P-Stereogenic Ligands in Enantioselective Catalysis, RSC, Cambridge, 2011 Search PubMed; (f) P. O'Brien, Chem. Commun., 2008, 655 RSC.
- H. Lam, P. N. Horton, M. B. Hursthouse, D. J. Aldous and K. K. Hi, Tetrahedron Lett., 2005, 46, 8145 CrossRef CAS.
- M. Ranocchiari and A. Mezzetti, Organometallics, 2009, 28, 1286 CrossRef CAS.
- W. S. Knowles, M. J. Sabacky, B. D. Vineyard and D. J. Weinkauff, J. Am. Chem. Soc., 1975, 97, 2567 CrossRef CAS.
- A. Ohashi, S. Kikuchi, M. Yasutake and T. Imamoto, Eur. J. Org. Chem., 2002, 2535 CrossRef CAS.
- J. D. G. Correia, A. Domingos and I. Santos, Eur. J. Inorg. Chem., 2000, 1523 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and spectroscopic data. See DOI: 10.1039/c1cy00253h |
| This journal is © The Royal Society of Chemistry 2011 |