Ceria-supported ruthenium catalysts for the synthesis of indole via dehydrogenative N-heterocyclization†
Shun
Shimura
a,
Hiroki
Miura
a,
Kenji
Wada
*a,
Saburo
Hosokawa
a,
Seiji
Yamazoe
b and
Masashi
Inoue
*a
aDepartment of Energy and Hydrocarbon Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: wadaken@scl.kyoto-u.ac.jp; Fax: +81-75-383-2479; Tel: +81-75-383-2482
bDepartment of Materials Chemistry, Graduate School of Science and Technology, Ryukoku University, Seta, Otsu 520-2194, Japan
First published on 22nd August 2011
Abstract
Simple heterogeneous Ru/CeO2 catalysts as well as Ru/ZrO2 catalysts were found to be quite effective for the selective direct synthesis of indole via intramolecular dehydrogenative N-heterocyclization of 2-(2-aminophenyl)ethanol, while catalysts supported on SiO2, Al2O3, TiO2, and MgO were less effective. Ru/CeO2 catalysts that were calcined at a relatively low temperature, 200 °C, showed excellent activity and gave indole in a yield over 99% by the reaction at 140 °C for 24 h (Ru catalyst 2.5 mol%). Spectroscopic studies of the Ru/CeO2 catalysts indicated the formation of RuIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species on the surface, which is considered to be transformed into the catalytically-active species at the initial stage of the reaction. Hot filtration tests and an ICP-AES analysis indicated that these Ru/CeO2 catalysts act heterogeneously and that the leaching of ruthenium species into the solution is negligible. These catalysts could be recycled without a significant loss of activity, which suggests that the present oxide-supported catalysts are promising alternatives to conventional homogeneous catalysts.
O species on the surface, which is considered to be transformed into the catalytically-active species at the initial stage of the reaction. Hot filtration tests and an ICP-AES analysis indicated that these Ru/CeO2 catalysts act heterogeneously and that the leaching of ruthenium species into the solution is negligible. These catalysts could be recycled without a significant loss of activity, which suggests that the present oxide-supported catalysts are promising alternatives to conventional homogeneous catalysts.
1. Introduction
The establishment of new environmentally-benign processes for synthesizing organic compounds is an important goal in modern chemistry,1,2 and the use of heterogeneous solid oxide-based catalysts has attracted much attention because of the advantages, such as the ease of preparation (impregnation and calcination) at low cost, high thermal and chemical stabilities, no contamination of the products by metallic species, and excellent recyclability of the catalysts.2 Recently, we developed heterogeneous Ru/CeO2 catalysts3a that are quite effective for transfer-allylation from homoallyl alcohols to aldehydes,3b the direct arylation and alkylation of aromatic C–H bonds,3c,d the addition of carboxylic acids to alkynes,3e and the coupling of alkynes with acrylates.3f These results suggest that the Ru/CeO2 catalysts can be good alternatives to homogeneous, low-valent Ru complex catalysts.On the other hand, benzo-fused N-heterocyclic compounds, particularly indoles, are important chemicals for the synthesis of fine chemicals, pharmaceuticals, and agrochemicals.4,5 Among various methods for the preparation of indoles,5 the N-heterocyclization of 2-(2-aminophenyl)ethanol and its derivatives6–10 is a promising protocol, since such alcohols are easily derived from 2-nitrotoluene derivatives and formaldehyde.8b While the reactions in the presence of supported copper or nickel catalysts 6 or a large amount of nitric acid7 require harsh conditions (e.g. over 200 °C), the synthesis of indoles promoted by ruthenium phosphine complexes,8,9 iridium complexes together with bases,10 or palladium complexes with allyl acetate9 has been reported to proceed smoothly under mild conditions. To the best of our knowledge, however, there have been no previous reports of solid ruthenium catalysts that are effective for the synthesis of indole via N-heterocyclization, whereas a few inorganic solid catalysts have been reported to promote the synthesis of nitrogen-containing compounds.11
Here, we report the development of heterogeneous Ru/CeO2 catalysts3,12 that are effective for the selective synthesis of indole from 2-(2-aminophenyl)ethanol in the absence of any additive. The preparation conditions and ruthenium precursors of the catalysts greatly affected their activities. Remarkably, catalysts prepared using Ru(acac)3 as a Ru precursor followed by calcination at a relatively low temperature, 200 °C, showed excellent activity. These catalysts were recyclable without a significant loss of activity, and the leaching of Ru species was negligible, which suggests that the present heterogeneous catalytic system is quite advantageous from both environmental and practical perspectives. The structures of suitable surface ruthenium species, which converted to catalytically active low-valent ruthenium species during the reactions, are discussed based on the spectroscopic characterization of the catalysts.
2. Experimental section
2.1. Materials
Tris(acetylacetonato)ruthenium(III) (Aldrich), tris(acetylacetonato)iron(III) (Dojin), tris(acetylacetonato)rhodium(III) (Aldrich), tris(acetylacetonato)iridium(III) (Wako), palladium(II) acetate (Nacalai Tesque), aqueous ammonia solution (28%, Nacalai Tesque), potassium hydroxide (Nacalai Tesque), tetrahydrofuran (dehydrated, stabilizer-free, Wako), 2-(2-aminophenyl)ethanol (Aldrich), mesitylene (Nacalai Tesque), naphthalene (Wako), silica (Cabot, Cab-O-Sil), and alumina (Sumitomo Chemical Co., Ltd, AKP-G015; JRC-ALO-8 equivalent) were obtained commercially and used without further purification. Titania (JRC-TIO-4 (P-25)) was obtained from the Catalysis Society of Japan. Ceria was prepared by treating a solution of cerium(III) nitrate hexahydrate (12.6 g, 29 mmol) in 400 cm3 of deionized water with 40 cm3 of a potassium hydroxide solution (3.0 mol dm−3) or 35 cm3 of a 28% aqueous ammonia solution with stirring at room temperature for 2 h. The resulting precipitates were collected by centrifugation and then air-dried overnight at 80 °C. The product was heated in a box furnace at a rate of 10 °C min−1 and maintained at 400 °C for 30 min in air to afford ceria in an excellent ceramic yield. The thus-prepared ceria samples are designated as CeO2(KOH) or CeO2(NH3), where the precipitant used for the preparation of the support is shown in parentheses. Zirconia was prepared by a similar method from zirconium oxynitrate (Nacalai Tesque) using a 3.0 mol dm−3 potassium hydroxide solution, followed by calcination at 500 °C for 30 min in air. Magnesia was prepared from magnesium nitrate (Wako) using a 28% aqueous ammonia solution, and calcined at 400 °C in air. Ceria–zirconia mixed oxides were prepared by treating a solution of cerium(III) nitrate hexahydrate and zirconium oxynitrate in 100 cm3 of deionized water with 200 cm3 of a 28% aqueous ammonia solution (five molar equivalents) with stirring for 1 h at room temperature. The resulting precipitates were collected by centrifugation and then air-dried overnight at room temperature. The product was heated in a box furnace at a rate of 10 °C min−1 and maintained at 500 °C for 30 min in air to afford ceria–zirconia mixed oxide in an excellent ceramic yield.2.2. Physical and analytical measurements
The products of catalytic reactions were analyzed by GC-MS (Shimadzu GC-MS Parvum 2, Zebron ZB-1 capillary column, id 0.25 mm, length 30 m, at 323–523 K) and gas chromatography (Shimadzu GC14APF, Zebron ZB-1 capillary column, id 0.25 mm, length 30 m at 323–523 K). The amount of evolved hydrogen gas was measured by gas chromatography (Shimadzu GC8AlT, Porapak-Q, id 3 mm, length 2 m, at 343 K). Nitrogen adsorption/desorption isotherms were obtained with a computer-controlled automatic gas sorption system (Quantachrome NOVA 4200e). Samples were degassed at 300 °C for 2 h just before the measurements. A thermogravimetry and differential thermal analysis (TG–DTA) study was performed using a Rigaku TG8120 system. The sample (ca. 5 mg) was heated at a rate of 10 °C min−1 under a stream of air (50 cm3 min−1). X-Ray powder diffraction (XRD) analysis was performed using Cu Kα radiation and a carbon monochromator (XD-D1, Shimadzu). Diffuse reflectance infrared Fourier transform (DRIFT) spectra were recorded using a Nicolet Magna-IR 560 FT-IR spectrometer with a DRIFT optical configuration. Temperature-programmed reduction (TPR) was carried out with a flow-type reactor. Hydrogen (1.9 vol% in Ar; atmospheric pressure; 30 cm3 min−1) was passed through a reaction tube containing the catalyst. The tube was heated with an electric furnace at 2 °C min−1, and the amount of H2 consumed was monitored with a TC detector on a Shimadzu 8AIT gas chromatograph. Leaching of ruthenium species from the catalysts during the reaction was investigated by an ICP atomic emission spectroscopic analysis using a Shimadzu ICPS-1000 III analyzer.Ru K-edge X-ray absorption fine structure (XAFS) measurements were performed at the BL01B1 beamline of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI). The storage ring was operated at 8 GeV with a ring current of 98–100 mA. A double-mirror system was used to avoid higher harmonics in the X-ray beam. A Si(311) two crystal monochromator was used. Ru K-edge XAFS spectra were measured in transmission mode using I0 [100% Ar] and I [75% Ar diluted with Kr] ion chambers and in fluorescence mode using an I0 ion chamber [100% Ar] and a Lytle detector [100% Kr]. X-Ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) data were analyzed using the REX2000 program (ver. 2.5; Rigaku Corp.). For EXAFS analyses, the oscillation was first extracted from XAFS data using a spline-smoothing method.13 The XANES spectra were normalized using the absorption intensity at the energy which is ca. 50 eV higher than the absorption edge.
2.3. Preparation of a Ru/support catalyst
Supported catalysts were prepared by the impregnation method. To a solution of Ru(acac)3 (79 mg, 0.198 mmol) in 10 cm3 of THF, 1.0 g of support was added in air at room temperature. After impregnation, the resulting powder was dried at 80 °C in air for a day and calcined in air at the prescribed temperature for 30 min to afford the 2.0 wt% Ru/support catalyst. The thus-prepared catalyst samples are designated as Ru/support-xx catalysts (suffix represents the calcination temperature).2.4. General procedure for the synthesis of indole (2) from 2-(2-aminophenyl)ethanol (1) by the Ru/CeO2 catalyst
All of the reactions were performed using hot stirrers equipped with cooling blocks for refluxing the solution. A typical reaction procedure is as follows: 2-(2-aminophenyl)ethanol 1 (1.0 mmol) and mesitylene (2.0 cm3) were placed in a glass Schlenk tube (20 cm3) in an argon atmosphere together with the Ru/CeO2 catalyst (125 mg, 0.025 mmol as Ru). The reaction mixture was stirred at 140 °C for 18 h, and then rapidly cooled in an ice bath. After the reaction, the solid catalyst was removed by passing it through a 0.45 μm PTFE filter (Millipore Millex LH). For isolation of the products, the remaining solution was concentrated under reduced pressure, and the products were then separated by column chromatography (silica gel, 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hexane/EtOAc, v/v), identified by GC-MS, and quantified by GC using naphthalene as an internal standard.
:1 hexane/EtOAc, v/v), identified by GC-MS, and quantified by GC using naphthalene as an internal standard.
2.5. Hot filtration tests
A 20 cm3 Schlenk tube was charged with 1 (2.0 mmol) and the Ru/CeO2 catalyst (250 mg, 0.050 mmol as Ru) in mesitylene (4.0 cm3) together with an internal standard (naphthalene, ca. 30 mg) in an argon atmosphere. After the reaction was allowed to proceed for 3 h at 140 °C, the mixture was filtered through a 0.45 μm syringe filter (Millipore Millex LH) into another preheated Schlenk tube. The filtrate was stirred at 140 °C, and the conversion and yields of the products were followed by GC analysis.3. Results and discussion
3.1. Effect of Ru/CeO2 catalysts on the synthesis of indole via dehydrogenative N-heterocyclization
The effects of metal oxide-supported Ru catalysts calcined at 400 °C on the synthesis of indole (2) from 2-(2-aminophenyl)ethanol (1) were examined (eqn (1)).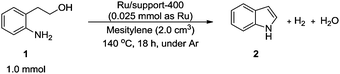 | (1) |
The activity of the catalyst was significantly affected by the support (Table 1), and, of the ruthenium catalysts examined, the ceria-supported catalyst showed the highest activity. The reaction of 1 in mesitylene at 140 °C for 18 h in the presence of the Ru/CeO2(KOH)-400 catalyst selectively gave 2 in a yield of 77% (entry 3). The formation of a stoichiometric amount of hydrogen gas was confirmed by gas chromatographic analysis, and there was no sign of byproducts, which indicated that the present reaction was completely selective for 2. The ruthenium catalyst supported on ceria prepared using aqueous ammonia solution, Ru/CeO2(NH3)-400, showed identical activity under the present conditions (entry 4). The reaction in the absence of the catalyst did not proceed at all, and the ceria support by itself showed a very low catalytic activity (entries 1 and 2). The Ru/ZrO2-400 catalyst showed a moderate activity (entry 5). On the other hand, catalysts supported on ceria–zirconia mixed oxides showed lower activities than those supported on pure ceria or zirconia (entries 6–8). The ruthenium catalysts supported on MgO, TiO2, Al2O3, and SiO2 were not effective (entries 9–12). The trend in the effects of the supports was similar to that observed in previous reactions promoted by supported Ru catalysts.3 Among the other transition metal catalysts supported on ceria, Ir and Rh catalysts showed excellent activities comparable to those of the Ru/CeO2-400 catalysts (entries 14 and 15), while Fe and Pd catalysts were not very effective (entries 13 and 16). Based on a consideration of the very high costs of producing the Ir and Rh catalysts, we optimized the Ru/CeO2 catalysts in the following study.
| Entry | Catalysta | Yield of 2b (%) |
|---|---|---|
| a 2.0 wt% as metal. For supported Ru catalysts, Ru(acac)3 was used as a precursor. b Determined by GLC. | ||
| 1 | None | Trace |
| 2 | CeO2(KOH) | 8 |
| 3 | Ru/CeO2(KOH)-400 | 77 |
| 4 | Ru/CeO2(NH3)-400 | 77 |
| 5 | Ru/ZrO2(KOH)-400 | 45 |
| 6 | Ru/CeO2–ZrO2(80/20)-500 | 39 |
| 7 | Ru/CeO2–ZrO2(50/50)-500 | 15 |
| 8 | Ru/CeO2–ZrO2(20/80)-500 | 15 |
| 9 | Ru/MgO(NH3)-400 | 14 |
| 10 | Ru/TiO2-400 | 6 |
| 11 | Ru/Al2O3-400 | 5 |
| 12 | Ru/SiO2-400 | 5 |
| 13 | Fe/CeO2(KOH)-400 | 12 |
| 14 | Rh/CeO2(KOH)-400 | 70 |
| 15 | Ir/CeO2(KOH)-400 | 76 |
| 16 | Pd/CeO2(KOH)-400 | 63 |
The activities of the ceria-supported catalysts were influenced by ruthenium complexes or salts used as ruthenium precursors for the preparation, as shown in Table 2. Here, CeO2(NH3) was used as a support. Of the catalysts examined, the catalyst prepared using Ru(acac)3 showed the highest activity (Table 2, entry 1), while those prepared using Ru3(CO)12 or RuCl3·nH2O showed slightly lower activities (entries 2 and 3). On the other hand, the catalysts prepared using [RuCl2(p-cymene)]2 and [RuCl2(CO)3]2 showed poor activities (entries 4 and 5). This trend is completely different from those found in the Ru/CeO2-catalyzed chelation-assisted arylation of aromatic C–H bonds3c and the addition of carboxylic acids to alkynes,3e where [RuCl2(p-cymene)]2 and [RuCl2(CO)3]2 were more suitable. These results suggest that suitable ruthenium precursors depend on the type of the reaction. In the following study, Ru(acac)3 was used as a precursor for the catalysts unless otherwise noted.
The calcination temperature of the supported Ru catalysts significantly affected their activities. Table 3 shows the yields of 2 formed by the Ru/CeO2(KOH) or Ru/Al2O3 catalysts calcined at various temperatures. Note that the use of Ru(acac)3 as a homogeneous catalyst resulted in the formation of 2 in a moderate yield (entry 1). Both Ru/CeO2(KOH) and Ru/Al2O3 calcined below 150 °C showed catalytic activities similar to that of Ru(acac)3 (entries 1–3, 10 and 11), probably because Ru species on the surface of these catalysts were not fully oxidized, and basically retained a structure similar to that of Ru(acac)3 (see below). Of the catalysts examined, Ru/CeO2(KOH)-200 showed the highest catalytic activity (entry 4) to afford 2 in a yield of 99% after 24 h. The reaction catalyzed by Ru/CeO2(KOH)-200 proceeded even in air to give 2 in a yield of 61% (entry 5). However, a further increase in the calcination temperature of Ru/CeO2 to above 300 °C gradually decreased the yield of 2 (entries 4, 7–9). On the other hand, the activities of Ru/Al2O3 calcined at above 200 °C were very low (entries 12–15), indicating that ruthenium oxide species on alumina formed by calcination are not active in the present reaction.
| Entry | Ru catalystb | Yield of 2c (%) |
|---|---|---|
| a Reaction conditions were the same as shown in eqn (1). b 2.0 wt% as Ru. Ru(acac)3 was used as a precursor. c Determined by GLC. d Reaction for 24 h. e Reaction in air. | ||
| 1 | Ru(acac)3 | 42 |
| 2 | Ru/CeO2(KOH)-as-imp. | 53 |
| 3 | Ru/CeO2(KOH)-150 | 50 |
| 4 | Ru/CeO2(KOH)-200 | 84 (>99)d |
| 5e | Ru/CeO2(KOH)-200 | 61 |
| 6 | Ru/CeO2(NH3)-200 | 84 |
| 7 | Ru/CeO2(KOH)-300 | 84 |
| 8 | Ru/CeO2(KOH)-400 | 77 |
| 9 | Ru/CeO2(KOH)-600 | 57 |
| 10 | Ru/Al2O3-as-imp. | 40 |
| 11 | Ru/Al2O3-150 | 43 |
| 12 | Ru/Al2O3-200 | 14 |
| 13 | Ru/Al2O3-300 | 7 |
| 14 | Ru/Al2O3-400 | 5 |
| 15 | Ru/Al2O3-600 | 2 |
It is important to investigate whether the reaction actually proceeds on the surface of the solid catalyst.14,15 To examine the contribution of ruthenium species in solution, generated by the so-called “release and capture” mechanism, the effect of removal of the catalysts by hot filtration through a PTFE filter (pore size 0.45 μm) was examined. Fig. 1 shows the time-course of the reaction at 140 °C in the presence of the Ru(2.0 wt%)/CeO2(KOH)-200 catalyst. The formation of 2 was completely stopped by removal of the solid catalyst. According to the ICP-AES analysis, very little of the ruthenium species was found in the solution, 0.000075 mmol (ca. 0.15 mol% of the ruthenium species charged as the fresh catalyst), after the reaction at 140 °C for 18 h catalyzed by Ru/CeO2(KOH)-200. These results clearly indicate that the reaction proceeds on the surface of the catalysts without significant leaching of ruthenium species into the solution. Therefore, the present heterogeneous Ru/CeO2 catalyst is markedly superior to conventional homogeneous catalysts.
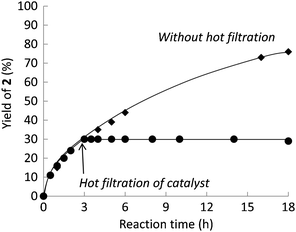 | ||
| Fig. 1 Time-course of the reaction of 1 over Ru/CeO2(KOH)-200. (◆) without filtration of the catalyst and (●) with removal of the catalyst by hot filtration after 3 h. Reaction conditions: 1 (2.0 mmol), Ru/CeO2(KOH)-200 (0.050 mmol as Ru), mesitylene (4.0 cm3), at 140 °C. | ||
One of the major advantages of solid catalysts is their recyclability. After the reaction was performed at 140 °C for 18 h using the Ru/CeO2(NH3)-200 catalyst (Table 3, entry 6), the solid catalyst was separated from the reaction mixture by centrifugation and washed three times with diethyl ether (10 cm3). The resulting solid was calcined at 200 °C for 4 h. The thus-recycled Ru/CeO2(NH3)-200 catalyst afforded 2 in the same yield (84%). Similarly, recycled Ru/CeO2(KOH)-200 gave 2 in a yield of 80%, while the fresh catalyst afforded 2 in 84% yield. These results clearly indicate that the Ru/CeO2 catalysts are recyclable.
3.2. Characterization of supported ruthenium catalysts
To investigate the factors that govern the activity of the catalysts, a series of supported ruthenium catalysts were characterized. The results of a nitrogen gas adsorption study of the fresh catalysts as well as their XRD patterns are summarized in the ESI.†The electronic structure and the geometry of the surroundings of ruthenium ions on supports were investigated by means of the XAFS spectra. The EXAFS oscillations and the Fourier transforms (FT) of the EXAFS spectra are shown in Fig. 2. Spectra of RuO2 and Ru/Al2O33f are also shown for comparison. While the formation of RuO2-like species on Al2O3, SiO2 or TiO2 have been suggested,3f the EXAFS spectra of Ru/CeO2(KOH) and Ru/CeO2(NH3) were different from that of RuO2 in the second shell around 3.0 Å. In the spectra of these catalysts, the FT peak intensities in the second shell were lower than those of other catalysts. Therefore, ruthenium species which have strong interaction with supports are considered to be formed on CeO2.16
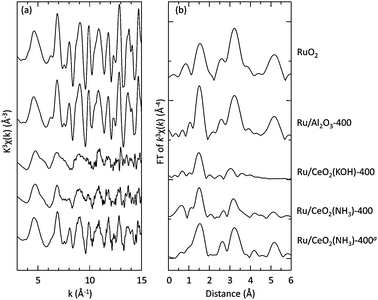 | ||
| Fig. 2 (a) Ru K-edge EXAFS oscillations of 2.0 wt% Ru/support catalysts. Ru(acac)3 was used as a precursor. (b) Ru K-edge FT spectra of the supported-Ru catalysts. aRu3(CO)12 was used as a precursor. | ||
Fig. 3 shows the Ru K-edge XANES spectra of CeO2- or ZrO2-supported Ru catalysts calcined at 400 °C, which were recorded in air at room temperature. As discussed previously,3f,17 a characteristic pre-edge peak was observed at around 22110 eV only in the spectra of Ru/CeO2(KOH)-400, Ru/CeO2(NH3)-400, and Ru/ZrO2-400. This pre-edge peak is assignable to the forbidden transition from the 1s to 4d level of metal ions. However, distortions that break centrosymmetry can permit 5p mixing into the empty 4d orbital, providing an electric dipole allowed character in the metal 4d ← 1s transitions.17,18 For the standard RuO2, which has a symmetrical six-coordinated structure, as well as Ru/Al2O3,19 the pre-edge peak was very weak. Therefore, the presence of the pre-edge peak in the spectra of Ru/CeO2 and Ru/ZrO2 indicates that the Ru species on ceria and zirconia have a structure that is more distorted than that of RuO2.
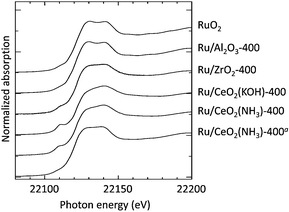 | ||
| Fig. 3 Ru K-edge XANES spectra of 2.0 wt% Ru/support catalysts. Ru(acac)3 was used as a precursor. aRu3(CO)12 was used as a precursor. | ||
Fig. 4 shows the DRIFT spectra of the supported Ru catalysts. Note that both Ru/CeO2(KOH)-400 and Ru/CeO2(NH3)-400 prepared using Ru(acac)3 as well as the CeO2-supported catalyst prepared using Ru3(CO)12 showed a characteristic band at around 980 cm−1, which has been assigned to RuO vibration.17 On the other hand, the other catalysts did not show distinct bands in this region. Although such a peak was not recognized for Ru/ZrO2 in the present study, the formation of similar RuO species has been proposed for zirconia-supported catalysts.20,21 These results suggest that the formation of RuO species in freshly calcined catalysts strongly correlates with the catalytic activity. We deduce that the formation of surface RuO bonds is basically associated with distortion of the coordination around the ruthenium atoms.
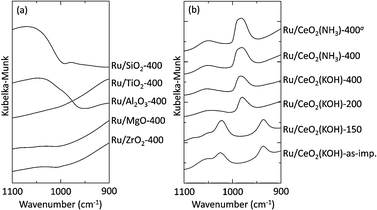 | ||
| Fig. 4 (a) DRIFT spectra of 2.0 wt% Ru/support catalysts. Ru(acac)3 was used as a precursor. (b) DRIFT spectra of Ru/CeO2 calcined at various temperatures. aRu3(CO)12 was used as a precursor. | ||
Note that the XANES spectrum of Ru/CeO2(NH3)-400 prepared using Ru3(CO)12 did not show a distinct pre-edge peak, and there was a relatively strong FT peak at around 3.0 Å in its EXAFS spectrum. On the other hand, its DRIFT spectrum suggests the presence of RuO species. These results indicate the formation of both RuO species and RuO2-like species probably because of the trinuclear nature of the Ru3(CO)12 precursor, which could explain why it has slightly lower catalytic activity than the catalysts prepared using Ru(acac)3 (see Table 2).
The effects of the calcination temperature on the properties of Ru/CeO2(KOH) were investigated. There were no significant changes in the BET surface area with a change in the calcination temperature. The TG–DTA profile of the as-impregnated Ru/CeO2(KOH) shows an exothermic weight decrease at around 200 °C. The extent of the weight decrease is consistent with the estimation based on the oxidative degradation of acetylacetonato ligands (5.6%). The DRIFT spectra of catalysts calcined at various temperatures clearly indicate the changes in the surface species (Fig. 4b).3f The spectra of the as-impregnated Ru/CeO2(KOH) and Ru/CeO2(KOH)-150 are represented as the superposition of the spectra of Ru(acac)3 and CeO2(KOH), indicating that Ru(acac)3 or Ru(acac)3-like species remain intact after impregnation and heat treatment at <150 °C. On the other hand, calcination at >200 °C significantly changed the spectra: peaks due to Ru(acac)3 completely disappeared, and a new band appeared at 980 cm−1, clearly indicating the formation of RuO species by the combustion of surface ruthenium acetylacetonato complexes at >200 °C.
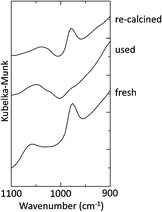
Our previous study using Ru/CeO2 catalysts revealed that surface RuIVO species are easily reduced to low-valent Ru species, which show excellent activities in various organic transformations.3 Therefore, the changes in these RuO species caused by the catalytic run and by re-calcination for regeneration were monitored by DRIFT spectra. As shown in Fig. 5, a characteristic band at 980 cm−1 of the fresh Ru/CeO2(KOH)-200 catalyst disappeared after the catalytic run. This indicates that RuO is not a catalytically active species but rather a precursor for catalytically active species formed during the reaction. The band appeared again after re-calcination of the used catalyst, indicating the regeneration of the RuO species on the surface of the catalyst.
For the present dehydrogenative N-heterocyclization to indole, a pathway via indoline would be possible. The dehydrogenation of indoline in the presence of the Ru/CeO2(KOH)-200 catalyst (2.5 mol% as Ru) at 140 °C for 3 h gave 2 in a yield of 61%. On the other hand, the reaction of 1 under the identical conditions afforded 2 in a yield of 30%, but indoline was not detected at all. This result clearly excludes the possibility that indoline participates in the main catalytic cycle, since consecutive first-order reactions predict the significant formation of indoline (ca. 16% yield) together with 2 in 30% yield. A possible mechanism is shown in Scheme 1. It is quite likely that the first step of the reaction is the hydrogen transfer from an alcohol to the Ru catalyst to form an aldehyde and a hydrido ruthenium species. The intramolecular nucleophilic attack of the amino group to the aldehyde moiety affords a Schiff base, which isomerizes to 2 very rapidly. Release of molecular hydrogen from the hydrido ruthenium species regenerates the catalytically active ruthenium species. Similar mechanisms have been proposed for the reactions promoted by homogeneous ruthenium8b and iridium10 complex catalysts.
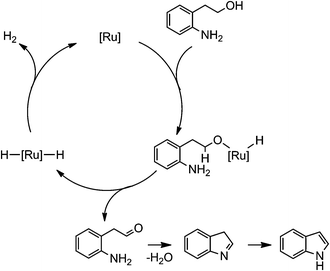 | ||
| Scheme 1 A possible reaction mechanism. | ||
In the view of the possible mechanism discussed above, the redox ability of the surface ruthenium species might be an important factor that governs the catalytic activity. The temperature required for the reduction of RuO species also depends on the calcination temperature of the catalysts. As shown in Fig. 6, the reduction peak of Ru/CeO2(KOH) gradually shifted toward a lower temperature with a decrease in the calcination temperature from 400 °C to 200 °C. This trend is consistent with that of the catalytic activity shown in Table 3; the catalyst bearing more easily reducible RuO species shows higher catalytic activity.
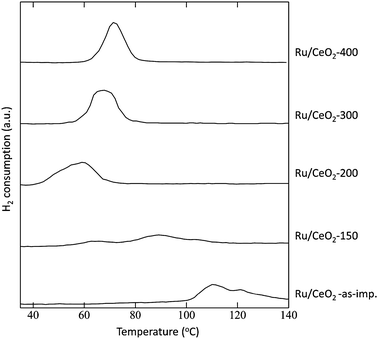 | ||
| Fig. 6 H2-TPR profiles of Ru/CeO2 catalysts calcined at various temperatures. | ||
In the present reaction as well as the previously-reported Ru/CeO2-catalyzed reactions,3 RuIVO species having a distorted coordination environment would be exclusively formed on the ceria or zirconia surface, and this would be a key precursor of the catalytically-active species. However, required properties for surface RuIVO species significantly depend on the types of the reactions. For example, the coupling of alkynes with acrylates proceeded much smoothly over the catalyst calcined at higher temperature, namely 600 °C:3f The catalytic activities positively correlated the degree of the distortion of RuIVO species. Furthermore, not only the surface active species but also a very small amount of soluble ruthenium species was found to be responsible for the coupling reactions.3f On the other hand, Ru/CeO2 calcined at low temperature, 200 °C, showed the highest activity for the present dehydrogenative N-heterocyclization. As shown in H2-TPR profiles, the catalyst having RuIVO species reducible at lower temperature showed higher activity. In addition, the hot filtration test clearly indicated that the reaction proceeded on the solid catalyst. Obviously, the nature of the active species generated in situ from RuIVO species was quite different in both reactions. These results suggest that the strict control of the properties of RuIVO species is very important to generate suitable active ruthenium species.
4. Conclusions
For the synthesis of indole via the dehydrogenative cyclization of amino alcohol, Ru/CeO2 as well as Ru/ZrO2 act as effective heterogeneous catalysts. The activity crucially depends on the nature of the support, and catalysts supported on SiO2, Al2O3, TiO2, and MgO are less effective. The calcination temperature of the catalysts influences the activity, and the Ru/CeO2 catalyst calcined at 200 °C shows the highest activity, to afford indole in a yield over 99% by the reaction at 140 °C for 24 h. RuIVO species interacting with supports are considered to be formed on ceria and zirconia, which act as good precursors for the catalytically active reduced surface ruthenium species.
The present Ru/CeO2 catalyst is recyclable, and the leaching of ruthenium species is negligible. According to the hot filtration test, Ru/CeO2 acts as a truly heterogeneous catalyst. Furthermore, note that the present system does not require any additives. Due to these characteristic features, Ru/CeO2 is quite attractive as an environmentally benign, highly efficient heterogeneous catalyst from synthetic, industrial, and environmental perspectives. Furthermore, extensive and systematic investigation on the intra- and intermolecular reactions between various amines and alcohols by the present catalysts as well as other precious metal catalysts supported on ceria is in progress.
Acknowledgements
This research was supported by a Grant-in-Aid for Scientific Research (No. 21360393) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. This work has been performed with the approval of SPring-8 (Proposal No. 2009B1356). K.W. acknowledges financial support from the Takahashi Industrial and Economic Foundation.Notes and references
- For green chemistry, see: (a) P. T. Anastas and J. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed; (b) R. A. Sheldon and R. S. Downing, Appl. Catal., A, 1999, 189, 163–183 CrossRef CAS; (c) B. M. Trost, Acc. Chem. Res., 2002, 35, 695–705 CrossRef CAS.
- For representative reviews, see: (a) P. Laszlo, Acc. Chem. Res., 1986, 19, 121–127 CrossRef CAS; (b) Y. Izumi and M. Onaka, Adv. Catal., 1992, 38, 245–282 CAS; (c) J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 1996, 25, 303–310 RSC; (d) B. F. Sels, D. E. De Vos and P. A. Jacobs, Catal. Rev. Sci. Eng., 2001, 43, 443–488 CrossRef CAS; (e) S. Kannan, Catal. Surv. Asia, 2006, 10, 117–137 CrossRef CAS; (f) K. Kaneda, Synlett, 2007, 999–1015 CrossRef CAS.
- (a) K. Wada, S. Hosokawa and M. Inoue, Catal. Surv. Asia, 2010, 15, 1–11; (b) H. Miura, K. Wada, S. Hosokawa, M. Sai, T. Kondo and M. Inoue, Chem. Commun., 2009, 4112–4114 RSC; (c) H. Miura, K. Wada, S. Hosokawa and M. Inoue, Chem.–Eur. J., 2010, 16, 4186–4189 CrossRef CAS; (d) H. Miura, K. Wada, S. Hosokawa and M. Inoue, ChemCatChem, 2010, 2, 1223–1225 Search PubMed; (e) M. Nishiumi, H. Miura, K. Wada, S. Hosokawa and M. Inoue, Adv. Synth. Catal., 2010, 352, 3045–3052 CrossRef CAS; (f) H. Miura, S. Shimura, S. Hosokawa, S. Yamazoe, K. Wada and M. Inoue, Adv. Synth. Catal., 2011 Search PubMed , accepted.
- For example, see (a) R. T. Brown, J. A. Joule and P. G. Sammes, in Comprehensive Organic Chemistry, ed. S. D. Barton and W. D. Ollis, Pergamon Press, Oxford, 1979, vol. 4, p. 441 Search PubMed; (b) G. P. Ellis, The Chemistry of Heterocyclic Compounds, Wiley, Chichester, 1992, vol. 47 Search PubMed.
- (a) L. S. Hegedus, Angew. Chem., Int. Ed. Engl., 1988, 27, 1113–1226 CrossRef; (b) U. Pindur and R. Adam, J. Heterocycl. Chem., 1988, 25, 1–8 CAS; (c) T. L. Gilchrist, J. Chem. Soc., Perkin Trans. 1, 1999, 2849–2866 RSC; (d) G. W. Gribble, J. Chem. Soc., Perkin Trans. 1, 2000, 1045–1075 RSC; (e) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875–2911 CrossRef CAS; (f) J. Barluenga, F. Rodríguez and F. T. Fañanás, Chem.–Asian J., 2009, 4, 1036–1048 CrossRef CAS.
- (a) J. Bakke, H. Heikman and E. B. Hellgren, Acta Chem. Scand., Ser. B, 1974, 28, 393–398 CAS; (b) W. Hammerschmidt, A. Baiker, A. Wokaun and W. Fluhr, Appl. Catal., 1986, 20, 305–312 Search PubMed; (c) M. Imanari, H. Iwane and K. Kujira, Japanese Kokai, 88-23861 (Chem. Abstr., 1988, 109, 170230) Search PubMed; (d) M. Imanari, H. Iwane, K. Kujira and T. Seto, Japanese Kokai, 87-114958 (Chem. Abstr., 1987, 107, 154240) Search PubMed; (e) T. Kiyoura and Y. Kogure, Japanese Kokai, 81-5459 (Chem. Abstr., 1981, 95, 97580) Search PubMed.
- T. Morimoto and I. Hashimoto, Japanese Kokai, 77-142063 (Chem. Abstr., 1978, 88, 120988) Search PubMed.
- (a) Y. Tsuji, K.-T. Huh, Y. Yokoyama and Y. Watanabe, J. Chem. Soc., Chem. Commun., 1986, 1575–1576 RSC; (b) Y. Tsuji, S. Kotachi, K.-T. Huh and Y. Watanabe, J. Org. Chem., 1990, 55, 580–584 CrossRef CAS.
- T. Izumi and T. Yokota, J. Heterocycl. Chem., 1992, 29, 1085–1090 CAS.
- K. Fujita, K. Yamamoto and R. Yamaguchi, Org. Lett., 2002, 4, 2691–2694 CrossRef CAS.
- For recent examples, see (a) T. Hara, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, Tetrahedron Lett., 2003, 44, 6207–6210 CrossRef CAS; (b) K. Shimizu, K. Ohshima and A. Satsuma, Chem.–Eur. J., 2009, 15, 9977–9980 CrossRef CAS; (c) J. W. Kim, J. He, K. Yamaguchi and N. Mizuno, Chem. Lett., 2009, 920–921 CrossRef CAS; (d) T. Oishi, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2009, 48, 6286–6288 CrossRef CAS; (e) J. W. Kim, K. Yamaguchi and N. Mizuno, J. Catal., 2009, 263, 205–208 CrossRef CAS.
- For representative examples of organic reactions promoted by heterogeneous CeO2-based catalysts, see: (a) F. Vocanson, Y. P. Guo, J. L. Namy and H. B. Kagan, Synth. Commun., 1998, 28, 2577–2582 CAS; (b) H. Ji, T. Mizugaki, K. Ebitani and K. Kaneda, Tetrahedron Lett., 2002, 43, 7179–7183 CrossRef CAS; (c) K. Ebitani, H. Ji, T. Mizugaki and K. Kaneda, J. Mol. Catal. A: Chem., 2004, 212, 161–170 CrossRef CAS; (d) S. Carrettin, J. Guzman and A. Corma, Angew. Chem., Int. Ed., 2005, 44, 2242–2245 CrossRef CAS; (e) A. Corma, C. Gonzalez-Arellano, M. Iglesias and F. Sanchez, Angew. Chem., Int. Ed., 2007, 46, 7820–7822 CrossRef CAS; (f) Y. Hayashi, S. Hosokawa, S. Imamura and M. Inoue, J. Ceram. Soc. Jpn., 2007, 115, 592–596 CrossRef CAS.
- J. W. Cook, Jr and D. E. Sayers, J. Appl. Phys., 1981, 52, 5024–5031 CrossRef.
- (a) M. D. Smith, A. F. Stepan, C. Ramarao, P. E. Brennan and S. V. Ley, Chem. Commun., 2003, 2652–2653 RSC; (b) S. P. Andrews, A. F. Stepan, H. Tanaka, S. V. Ley and M. D. Smith, Adv. Synth. Catal., 2005, 347, 647–654 CrossRef CAS; (c) U. Kazmaier, S. Hähn, T. D. Weiss, R. Kautenburger and W. F. Maier, Synlett, 2007, 2579–2583 CrossRef CAS.
- (a) N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS; (b) M. Weck and C. W. Jones, Inorg. Chem., 2007, 46, 1865–1875 CrossRef CAS , and references therein.
- S. Hosokawa, H. Kanai, K. Utani, Y. Taniguchi, Y. Saito and S. Imamura, Appl. Catal., B, 2003, 45, 181–187 CrossRef CAS.
- S. Hosokawa, S. Nogawa, M. Taniguchi, K. Utani, H. Kanai and S. Imamura, Appl. Catal., A, 2005, 288, 67–73 CrossRef CAS.
- K. Getty, M. U. Delgado-Jaime and P. Kennepohl, Inorg. Chim. Acta, 2008, 361, 1059–1065 CrossRef CAS.
- C. E. Boman, Acta Chem. Scand., 1970, 24, 116–122 CAS.
- (a) S. Hosokawa, Y. Fujinami and H. Kanai, J. Mol. Catal. A: Chem., 2005, 240, 49–54 CAS; (b) T. Yasueda, S. Kitamura, N. Ikenaga, T. Miyake and T. Suzuki, J. Mol. Catal. A: Chem., 2010, 323, 7–15 CrossRef CAS.
- E. Guglielminotti, F. Boccuzzi, M. Manzoli, F. Pinna and M. Scarpa, J. Catal., 2000, 192, 149–157 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cy00235j |
| This journal is © The Royal Society of Chemistry 2011 |