Deposition of carbonaceous species over Ag/alumina catalysts for the HC-SCR of NOx under lean conditions: a qualitative and quantitative study†
José R.
Hernández Carucci
a,
Kalle
Arve
a,
Šarka
Bártová
b,
Kari
Eränen
a,
Tapio
Salmi
a and
Dmitry Yu.
Murzin
*a
aLaboratory of Industrial Chemistry and Reaction Engineering, Åbo Akademi University, Biskopsgatan 8, Turku/Åbo, 20500, Finland. E-mail: dmurzin@abo.fi; Tel: +358 225 4985
bDepartment of Chemical Engineering, Institute of Chemical Technology, Prague, Technická 5, 16628 Prague, Czech Republic
First published on 16th August 2011
Abstract
The deposition of carbonaceous species on fresh and aged Ag/Al2O3 catalysts used for the selective catalytic reduction of NOx (HC-SCR) was investigated by a combination of analytical techniques including SEM-EDXA, nitrogen physisorption, GC-MS and TGA. Time-on-stream (TOS) experiments were carried out to determine the level of deactivation over the catalysts. Short-term deactivation was monitored with a MicroGC with a short retention time. In addition, long-term deactivation was also investigated. Hexadecane, a paraffinic component that can be produced by decarboxylation and/or decarbonylation of natural oils and fats, and a standard Finnish commercial diesel fuel were used as the reducing agents. Finally, the influence of hydrogen on the carbonaceous deposition over the catalyst was also investigated. The NO-to-N2 activity improved in the temperature range 200–500 °C as hexadecane and H2 were used as reducing agents. In the case of commercial diesel fuel, a similar positive effect of H2 on the activity was also observed, however, at somewhat higher temperature, e.g., 350–500 °C. The results from the qualitative and quantitative analyses of the carbon deposits on the catalysts are presented and discussed. Indications of decreased activity over the catalyst were found, especially at low temperatures (<350 °C). Nevertheless, only small amounts of carbon were found on the catalytic surface. It is suggested that the deactivation phenomenon could not only be attributed to the deposition of carbonaceous species on the catalysts surface but the dependence of the adsorption kinetics of the reacting species as a function of temperature and partial pressure should also be carefully taken into account.
Introduction
The fragility of the global economy and the political instability of some of the oil-producing countries have led, among other reasons, to a tremendous increase in oil prices during the last few years. Additionally, the implementation of continuously tightening legislation regarding hazardous emissions is a challenge to face for the automotive industry. During the recent years, bio-based fuels have been considered as a partial solution for the possible combustible crisis and their production have been widely encouraged. Lately, a lot of effort has also been put to develop new fuel alternatives based on ethical resources not compromising the world food production. New technologies have been investigated for the production of novel bio-derived fuels, as the governmental as well as societal pressure for implementing biofuels increases. One of the key areas where bio-derived fuels are expected to play a major role is in the automobile industry. Bio-diesel has already been used in traffic and the European Commission has set a target of 5.75% of the total consumption of transportation fuels in the EU to be biofuels.1 In the scenario of pure biodiesel being used as a fuel or merely as a component in the fuel mixture, the understanding of its combustion is crucial if optimization of new technologies has to be achieved. Such green diesels are known for their renewability, biodegradability and moreover, they result in less emissions (PM, SOx, net CO2, VOC) in exhaust gases. Nonetheless, biodiesels have been found to produce the same, or slightly more nitrogen oxides than conventional petrodiesels.2,3 Hence, efforts should be placed on the reduction of these hazardous species. Efforts on biodiesel combustion are still scarce, making it of key importance addressing this topic.For mobile diesel engines, which operate under a large excess of oxygen, selective catalytic reduction using hydrocarbons (HC-SCR) has proven to be an elegant and possible way for reducing nitrogen oxides from mobile sources. It has been tested as one of the most promising deNOx technologies using conventional fuels.4–16 However, only few recent publications have addressed the issue of using biodiesel as reducing agents.17–20 A broad range of catalysts has been tested for the HC-SCR system, being silver supported on alumina one of the most promising catalysts up to date.9,14,15 Nonetheless, the high activity is mainly connected to elevated temperatures (>300 °C), while modern diesel engines could have exhaust gas temperatures of 200 °C or less during most of the NEDC test.14 In this work, the whole temperature range from 150 to 600 °C is evaluated.
A novel technology for the production of biodiesel is decarboxylation and/or decarbonylation of natural oils and fats, yielding long-chained hydrocarbons (alkanes). For example, Neste Oil, the Finnish oil company, has recently developed a process for the production of a biomass-derived diesel, called NExBTL, which consists mostly of C15–C18 paraffins.21 In this work, hexadecane was chosen as the model compound representing biodiesels. Hexadecane is an example of a paraffinic component produced by selectively deoxygenating oil and fat triglycerides, fatty acid esters and fatty acids and it is regarded as a high quality diesel.22 A fully formulated diesel fuel was investigated as well.
Most authors have agreed that coking of the Ag/alumina catalysts is an issue in HC-SCR, and depends much on the nature of the reducing hydrocarbon. Even if there are papers published related to the coking of the Ag/alumina catalyst for HC-SCR, many of the studies do not make the effort to systematically study the coke formation on the catalytic surface for HC-SCR systems. Moreover, quantitative studies for this kind of systems are limited. The objective of this contribution was to qualitatively and quantitatively analyze the fresh and used catalysts for the HC-SCR of NOx applying hexadecane and commercial diesel fuels as reducing agents. The effect of H2 as a co-reducing agent with the hydrocarbon or diesel was also investigated. SEM-EDS, TGA, N2-physisorption and GC-MS were used in order to elucidate and understand the mechanism of carbon deposition on Ag/alumina.
Experimental
Activity tests
The catalyst used in this study was a 1.9 wt% Ag/γ-alumina catalyst prepared by impregnation of a commercial gamma-alumina support (LaRoche Industries Inc.) with a 0.022 M silver nitrate solution followed by drying and calcination.23 The Ag/alumina (particle size 250–500 μm) catalyst was tested in a typical catalytic set-up presented elsewhere.24 In brief, the setup consisted in a fixed-bed quartz reactor inserted in an oven equipped with a K-type thermocouple to precisely control the bed temperature in its centre.The activity tests were carried out using 0.4 g of catalyst. The temperature was varied in the range of 150–600 °C with sampling at 50 °C interval. Such sampling was done at steady-state conditions using GHSV = 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 (residence time 0.034 s) and a total flow rate of 550 cm3 min−1. The gas concentrations were adjusted to simulate a diesel engine exhaust, e.g., 500 ppm NO, 6 vol.% O2, 12 vol.% H2O, 350 ppm CO, 10 vol.% CO2 and 0 or 1 vol.% H2. The C1/NO ratio was kept at 6. Water was introduced using a syringe pump (ALARIS GS) followed by a controlled evaporator mixer (Bronkhorst HI-TEC). All the gases were of high purity (AGA) and were introduced into the reactor by means of mass flow-controllers (Brooks 5850). The addition of hexadecane and diesel took place via a syringe pump (CMA 102/Microdialysis). The properties of the used diesel fuel are presented in Table 1. Oxygen was fed separately into the reactor to avoid oxidation of NO before the catalyst bed. The effluent gas (both NO to N2 and hexadecane to CO + CO2) was analysed by a GC (HP 6890 series) equipped with a GS Q column, a GS molecular sieve column (J&W Scientific) and flame ionization (FI) as well as thermal conductivity (TC) detectors. Fast analyses were performed with an Agilent MicroGC System 3000A series with TC detectors. High purity calibration gases (AGA) were used for calibration of the NOx analyser and the gas chromatographs.
000 h−1 (residence time 0.034 s) and a total flow rate of 550 cm3 min−1. The gas concentrations were adjusted to simulate a diesel engine exhaust, e.g., 500 ppm NO, 6 vol.% O2, 12 vol.% H2O, 350 ppm CO, 10 vol.% CO2 and 0 or 1 vol.% H2. The C1/NO ratio was kept at 6. Water was introduced using a syringe pump (ALARIS GS) followed by a controlled evaporator mixer (Bronkhorst HI-TEC). All the gases were of high purity (AGA) and were introduced into the reactor by means of mass flow-controllers (Brooks 5850). The addition of hexadecane and diesel took place via a syringe pump (CMA 102/Microdialysis). The properties of the used diesel fuel are presented in Table 1. Oxygen was fed separately into the reactor to avoid oxidation of NO before the catalyst bed. The effluent gas (both NO to N2 and hexadecane to CO + CO2) was analysed by a GC (HP 6890 series) equipped with a GS Q column, a GS molecular sieve column (J&W Scientific) and flame ionization (FI) as well as thermal conductivity (TC) detectors. Fast analyses were performed with an Agilent MicroGC System 3000A series with TC detectors. High purity calibration gases (AGA) were used for calibration of the NOx analyser and the gas chromatographs.
| Property | Value |
|---|---|
| Density/kg m−3 | 840 |
| Kinematic viscosity at 40 °C/mm2s−1 | 3.2 |
| Flash point/°C | 75 |
| Cloud point/°C | −6 |
| Cetane number | 53.9–54.5 |
| Lower heating value/MJ kg−1 | 42.7 |
| Stoichiometric air–fuel ratio, kg/kg | 14.6 |
| C/wt% | 85.7–86.5 |
| H2/wt% | 13.3–13.8 |
| N2/mg kg−1 | 28–62 |
| S/mg kg−1 | 51–55 |
| Ash/wt% | <0.001–0.003 |
| Aromatics/wt% | 22.9–28.1 |
| Lubricity/mm | 380 |
Time-on-stream (TOS) activity over the catalyst was studied both at 250 and 450 °C to reveal the level of deactivation at low and high NOx conversion regions. The characterization analyses presented below were made in three different modes: directly after the activity test at 600 °C, after the activity test followed by re-oxidation at 400 °C for 30 minutes and after testing the activity from 150 to 350 °C with a ramping rate of 5 °C min−1. Table 2 presents the experimental matrix with the respective conditions and their number identification through the manuscript.
| N | Catalyst condition after used in HC-SCR | Reductant | H2 (1 vol.%) |
|---|---|---|---|
| 1 | Stopped at 600 °C | Hexadecane | No |
| 2 | Stopped at 600 °C | Hexadecane | Yes |
| 3 | Stopped at 350 °C | Hexadecane | No |
| 4 | Stopped at 350 °C | Hexadecane | Yes |
| 5 | Stopped at 600 °C | Diesel | No |
| 6 | Stopped at 600 °C | Diesel | Yes |
| 7 | Stopped at 350 °C | Diesel | No |
| 8 | Stopped at 350 °C | Diesel | Yes |
| 9 | Fresh | ||
| 10 | TOS 250 °C | Diesel | No |
| 11 | TOS 250 °C | Diesel | Yes |
| 12 | TOS 450 °C | Diesel | No |
| 13 | TOS 450 °C | Diesel | Yes |
| 14 | TOS 250 °C | Hexadecane | No |
| 15 | TOS 250 °C | Hexadecane | Yes |
| 16 | TOS 450 °C | Hexadecane | No |
| 17 | TOS 450 °C | Hexadecane | Yes |
| 18 | Re-oxidized at 400 °C | Hexadecane | No |
| 19 | Re-oxidized at 400 °C | Hexadecane | Yes |
Catalyst characterization
Results
Results from the activity tests
It has been previously shown4,7–10,25 that the nature of the reducing agent dramatically influences the rate of the SCR of NOx. Thus, experimental work using a model compound for understanding the mechanistic phenomena of a single molecule can be used as a starting point. However, in reality, commercial diesel fuels are blends of numerous components, which have varying effects on the performance of the used catalyst in the reduction process.The results from the NO-to-N2 activity tests using hexadecane (n-C16H34), commercial diesel and octane (n-C8H18) as reducing agents are shown in Fig. 1. The amount of injected hydrocarbon was chosen in such a way that the ratio between the carbon atoms and NO in the feed was equal to 6. The diesel fuel was considered as C12, based on a mean molecular weight approach. As it can be seen, octane enhances the SCR of NOx more than hexadecane at higher temperatures, resulting in a maximum reduction activity close to 100% around 450 °C (Fig. 1).
 | ||
Fig. 1
NO-to-N2 conversion over a 1.9 wt% Ag/Al2O3 catalyst. Gas flow: 500 ppm NO, 250 ppm diesel ( ), 375 ppm n-C8H18 (■) or 188 ppm n-C16H34 ( ), 375 ppm n-C8H18 (■) or 188 ppm n-C16H34 ( ), 6 vol.% O2, 12 vol.% H2O and He as balance. GHSV = 60000 h−1. ), 6 vol.% O2, 12 vol.% H2O and He as balance. GHSV = 60000 h−1. | ||
However, for temperatures below 350 °C, the regime in which the diesel exhaust exists most of the time, the hexadecane-SCR is superior compared to the octane-SCR. Moreover, due to the mandatory presence of bio-derived fuels in the gasoline and diesel pools in the near future, the behaviour of these components needs to be investigated and the processes have to be optimized as they are employed. As the commercial diesel fuel was used as the reducing agent the activity over the catalyst was similar to that observed with hexadecane at higher temperatures (>500 °C). Nevertheless, at temperatures below 400 °C, the reduction activity over the catalyst was rather limited in diesel-SCR.
Comparing the activity results recorded using diesel or hexadecane with those obtained using octane, a similar trend is observed: at higher temperatures C8H18 shows better NO reduction, and at lower temperatures the trend is the opposite. Arve et al.17 have proposed for the SCR reaction over the same catalyst that simultaneous oxidation of the reducing agent (HC) and reduction of NO takes place. Hence, the activity of the catalyst would depend on its ability to partially oxidize the hydrocarbon, which is considered as one of the key steps in the SCR reaction.10,24,26 Arve et al.17 proposed that, since the oxidation activities of octane and hexadecane were very similar (Fig. 2), an increased surface coverage of the hydrocarbon-related compounds could be a reason, when using hexadecane instead of octane as a reducing agent, for effectively inhibiting the NO adsorption, thus, resulting in a decreased NO reduction activity. A similar trend was also observed in the oxidation activity of the commercial diesel over the catalyst. However in the temperature range 150–300 °C a fraction of the formulated diesel fuel, i.e., higher alkanes or alkenes, oxidizes easier than octane or hexadecane (Fig. 2).
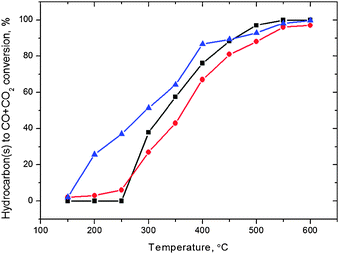 | ||
Fig. 2 Hydrocarbon(s) to CO + CO2 conversion over a 1.9 wt% Ag/Al2O3 catalyst. Gas flow: 500 ppm NO, 250 ppm diesel ( ), 375 ppm n-C8H18 (■) or 188 ppm n-C16H34 ( ), 375 ppm n-C8H18 (■) or 188 ppm n-C16H34 ( ), 6 vol.% O2, 12 vol.% H2O and He as balance. GHSV = 60000 h−1. ), 6 vol.% O2, 12 vol.% H2O and He as balance. GHSV = 60000 h−1. | ||
Fig. 3 compares the NO-to-N2 reduction efficiencies in the absence and presence of 1 vol.% hydrogen in the feed for both hexadecane and diesel as reducing agents. The well-known promoting effect of hydrogen is observed.
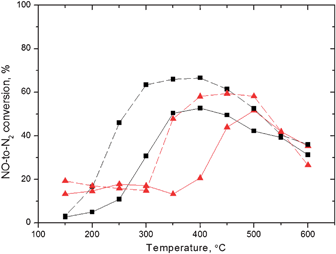 | ||
Fig. 3
NO-to-N2 conversion over a 1.9 wt% Ag/Al2O3 catalyst. Gas flow: 500 ppm NO, 250 ppm diesel ( ) or 188 ppm n-C16H34 (■), 6 vol.% O2, 12 vol.% H2O, 0 (solid) or 1 (dashed) vol.% H2 and He as balance. GHSV = 60000 h−1. ) or 188 ppm n-C16H34 (■), 6 vol.% O2, 12 vol.% H2O, 0 (solid) or 1 (dashed) vol.% H2 and He as balance. GHSV = 60000 h−1. | ||
Maximum conversions close to 50% were achieved in the temperature range 400–450 °C when hexadecane or diesel were used as reducing agents, while at lower temperatures (<300 °C) the NO-to-N2 activity over the catalyst remained below 20% (Fig. 3). Hydrogen is known to enhance both the NO-to-N2 reduction activity and the oxidation of the hydrocarbon to CO and CO2 over Ag/Al2O3 catalysts, when using a range of lower alkanes and alkenes and higher alkanes.27,28 When small amounts of hydrogen were present in the feed, the catalytic activity was dramatically improved at the temperature range 200–500 °C, reaching reduction activities exceeding 70%. This effect was particularly pronounced when using hexadecane. For lower-chained hydrocarbons, similar observations have been found, although the low temperature region (<300 °C) was mostly affected.29, 30
As the commercial diesel was used, the behaviour changed considerably. At low temperatures (below 350 °C) the promotion effect of hydrogen was not observed. However, as the temperature was equal or higher than 350 °C, enhancements in activity were very much pronounced, yielding reduction values close to those obtained by using hexadecane (Fig. 3). Since commercial diesel fuels are complex mixtures of different components, it is likely that the added hydrogen in the mixture acts as a promoter for the production of important intermediates in the SCR system, causing the beneficial effect observed in Fig. 3. On the other hand, it is clear that at the same time some of the components in the diesel could inhibit the reaction, especially at lower temperatures. Identifying these compounds having a negative effect on the HC-SCR over the silver catalyst is of highest interest for the further development of both the HC-SCR catalyst and diesel fuel. As the temperature increases, most of the diesel components are oxidized and the positive effect of adding hydrogen is shown, compared with the case when hexadecane was used as the reducing agent.
Due to the complex nature of fully-formulated diesel fuels, studying the underlying and mechanistic reasons for its behaviour is not straightforward and the understanding of the mechanism of the diesel-SCR is challenging. Still, a description of the catalytic performance and the possible causes for the activity changes are discussed below. Compared to octane-SCR, where the hydrogen enhancing effect seems more pronounced at lower temperatures, when using hexadecane or diesel fuel as a reducing agent co-injected with H2, the latter component seems to have a higher effect on the NO reduction. However, it is possible that due to the high reduction activity at high temperatures (>350 °C) for the octane-SCR, the beneficial effect of hydrogen simply cannot be detected. Moreover, the difference of reactivity of the two alkanes (octanevs.hexadecane) at high temperatures is probably due to the inefficiency of hexadecane to form certain key intermediate(s) necessary for the SCR to take place. These intermediates are more likely formed when lower (and more reactive) alkanes and alkenes are present as reducing agents.
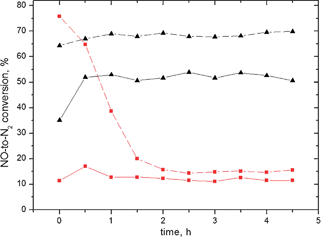 | ||
Fig. 4 Time-on-stream behaviour over the Ag/alumina catalysts at 250 ( ) and 450 (▲) °C in the presence (dashed) or absence (solid) of hydrogen using hexadecane as the reducing agent. Gas flow: 500 ppm NO, 188 ppm n-C16H34, 6 vol.% O2, 12 vol.% H2O, 0 or 1 vol.% H2 and He balance. GHSV = 60000 h−1. ) and 450 (▲) °C in the presence (dashed) or absence (solid) of hydrogen using hexadecane as the reducing agent. Gas flow: 500 ppm NO, 188 ppm n-C16H34, 6 vol.% O2, 12 vol.% H2O, 0 or 1 vol.% H2 and He balance. GHSV = 60000 h−1. | ||
The catalytic activity in the TOS experiments at 450 °C was very stable, with no signs of deactivation after 4.5 hours of the experiment. When hydrogen was present in the feed, an enhancement of the activity of ca. 35% was achieved and no deactivation was observed. However, at 250 °C the pattern was somewhat different. When no hydrogen was introduced into the reactor, the activity seemed to be fairly constant along the time of the experiment. Nonetheless, when 1 vol.% of H2 was fed together with the other gases, a pronounced reduction of the NOx reduction activity was observed during the first hour of the run; after this time, the conversion gradually decreased to the same level as in the experiment without hydrogen (Fig. 4). A similar effect was also demonstrated by Houel et al.31
Houel et al.32 proposed coking as a cause for the decreased activity at low temperatures over the silver catalyst using commercial diesel and long-chain hydrocarbons (dodecane). As shown in Fig. 4 no pronounced decrease on activity, exclusively at 250 °C, was observed over the silver catalyst. To further elucidate this behaviour, the first 30 minutes of the reaction were monitored with a MicroGC with shorter retention times (see ESI†, Fig. S1).
The experiments performed at 450 °C clearly showed that the catalyst reached stable conversion values after 30 minutes of experiment. No signs of deactivation were found at this temperature as shown from the results in Fig. 4. At 250 °C, it seems that the activity peaks at the beginning of the experiment, further stabilize at lower values (Fig. S1, ESI†). Similar results were obtained by Houel et al.32
Additionally, when hydrogen was used as a co-reductant at 250 °C, a sharp decrease in the activity was observed (Fig. S1, ESI†). Nonetheless, the decrease in NO reduction as a function of time was much slower compared to the case where H2 was absent. Such result indicates that hydrogen helps to maintain relatively high activity at the beginning of the experiment, until reaching a point where its effect can no longer be seen, which occurs after approximately 110 minutes of reaction. After this time, the catalytic activities at 250 °C both in the absence and presence of hydrogen are close to each other (Fig. 4).
Based on the experimental data it could be assumed that the initial activity, also at 250 °C, is reasonably high (35–85%) (Fig. S1, ESI†). Subsequently, the activity gradually decreases as the catalyst is poisoned with some strongly adsorbing surface species. Such assumption can be rationalized by the fact that even at such low temperature as 250 °C formation of coke can take place, even if the hydrocarbon would be in the liquid form.33,34 In fact, the results from the GC-MS analyses (Characterization of the carbonaceous species section, Table 3) suggested that there is some carbonaceous deposition on the catalyst’s surface indicating that coke formation can cause problems for the low temperature operation stability of the Ag/alumina catalyst. Most of the deposition found was in the form of longer alkanes and alkenes (up to C19), having relatively high heats of adsorption compared to shorter paraffins.
| Sample number and reaction conditions | Visual inspection | Compounds (GC-MS), the chemical structure of the major compound is given | BET surface area/m2 g−1 | Carbon content (eqn (1), %) |
|---|---|---|---|---|
| (9) Fresh |

|
NP | 187 | 4.1 |
| (1) Reaction stopped at 600 °C, hexadecane, no H2 |

|
|
138 | 4.1 |
| (2) Reaction stopped at 600 °C, hexadecane, with 1 vol.% H2 |

|
|
151 | 4.1 |
| (3) Reaction stopped at 350 °C, hexadecane, no H2 |

|
|
161 | 5.8 |
| (4) Reaction stopped at 350 °C, hexadecane, with 1 vol.% H2 |

|

|
158 | 5.2 |
| (5) Reaction stopped at 600 °C, diesel, no H2 |

|

|
135 | 4.6 |
| (6) Reaction stopped at 600 °C, diesel, with 1 vol.% H2 |

|
NP | 143 | 4.6 |
| (7) Reaction stopped at 350 °C, diesel, no H2 |

|
|
159 | 8.1 |
| (8) Reaction stopped at 350 °C, diesel, with 1 vol.% H2 |

|

|
167 | 7.6 |
| (10) TOS at 250 °C with diesel, no H2 |

|
NM | 144 | NM |
| (18) Re-oxidized at 400 °C for 30 min after catalyst (1) |

|
NP | 160 | NM |
| (19) Re-oxidized at 400 °C for 30 min after catalyst (2) |

|
NP | 164 | NM |
However, it should be kept in mind that the reaction itself is taking place under high concentration of oxygen and water vapour, which effectively should counteract the coking phenomena on the catalyst surface. Furthermore, the catalyst used in this study was a mesoporous Ag/alumina having only small amounts of Lewis acid sites, and no strong Brønsted acid sites.17 Therefore, coking, which is usually associated with long contact time and high acidity of the catalyst, can probably not alone provide a full explanation for the declining activity over the catalyst in the low temperature region. It has previously been demonstrated that the silver catalyst can store a considerable amount of adsorbed nitrate species on the surface, an effect that can significantly be accelerated in the presence of hydrogen.15,35 Thus, taking the storage of nitrates on the Ag/alumina into account, a more logical explanation for the observed decreasing activity at 250 °C would be that at low temperatures, the catalyst surface is first saturated with ad-NOx species, which create a slow dynamic into the system. The adsorption of various species on the catalyst surface or more precisely the balance between the adsorbing species would indeed have the highest importance, regardless if they are of organic nature (hydrocarbons) as suggested by Houel et al.32 or of inorganic origin as reported by Shibata et al.36 and Eränen et al.15 Even if hydrogen can counteract the nitrate poisoning of the surface, as shown in ref. 36, it is possible that the balance between the adsorbed species (ad-NOx and HC) on the catalyst surface (i.e. surface coverage) is changed when a model compound is replaced by commercial diesel because of different steric requirements, adsorption strength, reactivity, etc., leading to a decreased concentration of SCR-active hydrocarbon species on the catalyst surface. Consequently, this modification will result in a decreased reduction activity over the catalyst.
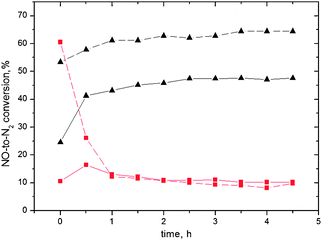 | ||
Fig. 5 Time-on-stream behaviour over the Ag/alumina catalysts at 250 ( ) and 450 (▲) °C in the presence (dashed) or absence (solid) of hydrogen with diesel as a reducing agent. Gas flow: 500 ppm NO, 250 ppm diesel, 6 vol.% O2, 12 vol.% H2O, 0 or 1 vol.% H2 and He balance. GHSV = 60000 h−1. ) and 450 (▲) °C in the presence (dashed) or absence (solid) of hydrogen with diesel as a reducing agent. Gas flow: 500 ppm NO, 250 ppm diesel, 6 vol.% O2, 12 vol.% H2O, 0 or 1 vol.% H2 and He balance. GHSV = 60000 h−1. | ||
As it can be seen, the reduction activity followed the same pattern as when hexadecane was used as the reductant (Fig. 4). Again, the effect of hydrogen in the feed is interesting: there is an increase of activity during the first minutes of the experiment, followed by a sharp decrease, reaching after 45 minutes the same values as in the case where no hydrogen was present. It appears that hydrogen, when present together with the diesel fuel as a reductant, enhanced the activity of the SCR reaction for short periods of time. This was the case at low temperatures, where hydrogen was found to have a promoting effect, results that are in agreement with previous findings.11,15,27,35 For the higher temperature case (450 °C), the situation was similar as when hexadecane was used as a reducing agent and no deactivation of the catalyst was observed.
Characterization of the carbonaceous species
The Ag/alumina catalyst used in this study has been previously characterized by Arve et al.23 However, a detailed analysis of the deposition of carbonaceous species on the catalytic surface when the catalyst is used for the HC-SCR of NOx has not yet been conducted. In this study, the catalyst was exposed to different reaction conditions before characterization. Time-on-stream (TOS) experiments were performed as well. When the catalyst was regenerated by oxidation under a 6 vol.% flow of O2/He during 30 minutes at 400 °C after the activity tests, almost full catalytic activity was regained, as well as an important amount of the initial surface area, determined by nitrogen physisorption. Table 3 presents a summary of the results of most of the characterization techniques employed in this work. The results from the surface analysis are also presented and discussed below.Based on the information received from the visual inspection (Table 3) it can be stated that samples 3, 4, 7 and 8, which were exposed under reaction conditions in the temperature range of 150–350 °C using 5 °C min−1 ramping, had the highest content of carbonaceous deposition using either hexadecane or commercial diesel as reductants. Interestingly, hydrogen seemed to reduce the formation of carbonaceous ad-species at low temperatures when hexadecane was used as a reductant (Table 3, Sample 4). However, when commercial diesel was used as a reducing agent, carbon deposits were still formed (Table 3, Sample 8). When re-oxidizing the samples 1 and 2 (Numbers 18 and 19 in Table 3, respectively) they regained the beige-whitish colour characteristic of an oxidized state. Since the oxidation was done at a relatively low temperature, i.e., 400 °C, and according to the visual inspection of the samples, it is suggested that only soft coke was present on the catalytic surface.
There were no major morphological differences between these samples and more importantly, when compared to the fresh one. Fig. 6 shows a SEM micrograph of sample 1 (reaction stopped at 600 °C with hexadecane) with a confined region quantitatively analyzed for the atomic composition. Fig. S3 shows another SEM picture of the same sample with a higher magnification (see ESI†).
 | ||
| Fig. 6 SEM micrograph of sample 1. 200×. | ||
Additionally, the differences in surface area between sample 1 (reaction stopped at 600 °C, hexadecane as reducing agent) and sample 2 (reaction stopped at 600 °C, hexadecane and hydrogen as reducing agents) were important. H2 has a cleaning effect on the catalytic surface, reducing NOx species, hydrogenating the detrimental aromatic and carbon components or increasing the oxidation rate of the hydrocarbon species present on the catalyst, thus enhancing the reduction process. Moreover, reoxidation of the catalysts (spent catalysts 1 and 2 after SCR experiments in the absence and in the presence of hydrogen respectively) was able to restore the initial surface area (88% and 86% of the surface area of the fresh catalyst, respectively). This suggests that the loss of surface area can be attributed to carbonaceous species deposited on the catalyst surface. Such species can be removed by oxygen treatment at high temperatures.
Thermogravimetric analyses were performed for all the tested catalysts to determine the amount of coke deposits on the surface. The loss of weight as a function of temperature was assumed to represent the mass of carbonaceous species according to eqn (1).
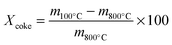 | (1) |
 | ||
| Fig. 7 TGA (dashed) and DTA (solid) of spent Ag/Al2O3 catalyst for sample 7 (Table 3) used for diesel-SCR of NOx, stopped at 350 °C. | ||
The TGA analysis revealed that the highest amount of carbon was deposited on the Ag/alumina surface as commercial diesel was used as the reducing agent and the reaction was stopped at 350 °C (Table 3, samples 7 and 8). The amount of coke was found to be 8.1 and 7.6 wt%, respectively, but was almost totally removed at 400 °C in oxygen. Sample 8 (when H2 was present in the reaction) resulted in slightly lower carbon content. The activities of these catalysts during the SCR were very similar at temperatures below 350 °C. This is interesting as the commercial diesel fuel also contains fractions of hydrocarbons in the C12–C18 range and, according to the model compound experiments (octane, and hexadecane), they should give a positive response to the hydrogen addition. The decrease of the reduction activity at low temperatures when real diesel fuel was used as a reductant could be associated to its high aromatic content (Table 1). This could indicate that the different adsorption strengths of the two hydrocarbon fractions (aromatic vs. paraffinic) in the commercial diesel cause the non-existing hydrogen effect over the catalyst below 350 °C (Fig. 3). In fact, Arve et al.37 proposed that the oxidation activity of aromatic components over silver/alumina catalyst is minimal, leading to high surface coverage at low temperatures (<350 °C). Consequently, it is reasonable to claim that the decreased reduction activity is not only due to the coking effect, but because the strong horizontal adsorption of aromatic and cyclic compounds effectively blocking a large number of active sites. On the other hand, around 350 °C the difference in activity started to be significant. It becomes clear that the hydrogen started promoting the reduction (by either helping the carbon present on the surface to burn, or by hydrogenating aromatics, and hence freeing spaces for the reactants to adsorb). Yet, the amount of carbon found on the sample was not very large and the observed deactivation could not only be attributed to it. Additionally, the nitrogen physisorption results revealed that the surface area of the catalysts 7 and 8 remained relatively high after stopping the reaction (159 and 167 m2 g−1 respectively, N2 physisorption section), suggesting that the catalytic activity was still preserved, and further proved by experiments.
The results from the GC-MS analysis are presented in Table 3 as well. As hexadecane was used as a reducing agent, small amounts of alkanes and alkenes (between C12–C19) were identified by GC-MS in sample 1. The results for sample 2 (hydrogen as co-reductant) were similar to those for sample 1 (Table 3).
The GC-MS analysis revealed that hexadecane was the hydrocarbon dissolved from samples 3 and 4. This suggests the possibility of some coverage of the hydrocarbon on the catalyst at lower temperatures (reaction stopped at 350 °C).
According to Fig. 4 and 6, the most pronounced effect of deactivation was observed at low temperatures (250 °C) for the case when hydrogen was present for both reducing agents (hexadecane and diesel). The same effect, although to a less extent, was also identified in the absence of hydrogen. We have proposed17,19 that since both hexadecane and diesel fuel have high boiling points (287 °C for hexadecane and >250 °C for the heavier fractions of the diesel), even with the small amounts of these components introduced into the reactor, formation of a liquid film on the catalytic surface is still possible, making the diffusion of the reactants through the liquid an extra (and slower) step for the species to adsorb. In this sense, the reduction reaction is inhibited when enough hydrocarbons are covering the catalytic surface. On the other hand, as the temperature rises, hexadecane partially oxidizes and further adsorbs onto the empty sites, thus leading to NO reduction. Problems of the hydrocarbon molecule size have also been found to be limiting the adsorption of the important species, which could be critical for the SCR system.17 It can be expected that due to low temperatures, the proposed condensation over the catalytic surface might occur.
As diesel was used as a reducing agent, the reaction was stopped at high (600 °C, samples 5 and 6) and low temperatures (350 °C, samples 7 and 8). Samples 5 and 6 were relatively clean from carbon species, although sample 5 showed a small peak of cyclohexadecane. For samples 7 and 8, some aromatics and other cyclic components were found in the GC-MS analyses (Table 3). Aromatics were found in sample 7, the one that presented the highest degree of coking according to the TGA studies. In sample 8 (experiment stopped at 350 °C, using diesel as the reducing agent), although no aromatics were found, linear alkanes and alkenes were present. In general, the GC-MS results confirmed the results obtained by the other characterization techniques used in this work. For the re-oxidized catalysts during 30 min at 400 °C (Samples 18 and 19), no peaks were detected in the GC-MS, indicating that the surface was carbon-free. The visual aspect of both catalysts suggested the same. Additionally, measurements of the surface area indicated that after oxidation, the increase on surface area was considerable (from 138 to 160 m2 g−1). The fresh catalyst (sample 9) was also analyzed without the detection of any carbon peaks (Table 3).
Regarding insoluble coke (hard coke), it is difficult to rationalize that it could be present on the surface, since most of the surface area was recovered by oxidation of the catalyst at a relatively low temperature (400 °C). Carbon formation from paraffins is claimed to be much slower than for the case of olefins and aromatics.38 Furthermore, below 500–600 °C the formation of coke from paraffins was found to be negligible.
Additionally, the visual inspection of the catalyst suggested the absence of carbon deposits after oxidation (Table 3).
To summarize, very small amounts of carbon were found on the catalytic surface for the studied conditions by the used characterization methods (Table 3). Even if the catalyst showed a minor sign of deactivation, it could not be solely attributed to the carbon formations. Coke is usually oxidized with oxygen at high temperatures and it is arguable that it can be oxidized easier than the hydrocarbon used as a reductant. Moreover, the catalytic activity could be regained by oxidation of the catalyst at the conditions mentioned above.
Finally, the NOx conversions were found to be similar in successive runs (fresh catalyst and used + re-oxidized catalyst) and with the same selectivities towards molecular nitrogen, suggesting that no other carbon species were covering the catalytic surface.
Conclusions
Detailed quantitative and qualitative characterization of the carbonaceous deposits on the Ag/Al2O3 for the HC-SCR of NOx exposed under various reaction conditions was performed using hexadecane or commercial diesel fuel as reducing agents. Based on the experimental results, the activity over the catalyst at low temperatures (<350 °C) is slightly diminishing in time, which was attributed to the increasing coverage of strongly adsorbed surface species. However, the phenomenon cannot only be attributed to coking of the catalysts, as carbonaceous deposit was not found to be the predominant surface species on more than two of the examined samples. TOS activity tests proved that the catalyst does not deactivate in the long-range even with small carbonaceous deposits on the surface.As hexadecane was used as the reducing agent, the carbon species detected on the catalyst surface by GC-MS were mainly linear paraffins. In addition, in the temperature range of 150–300 °C and when hexadecane was used as the reducing agent, the specie found on the surface was mainly hexadecane. This indicates that hydrocarbons with high boiling point, which are also included in the real diesel formulation, cannot be partially oxidised at temperatures below 300 °C leading to the poor activity observed over the Ag/Al2O3 catalyst.
As diesel fuel was used as a reducing agent, small amounts of aromatics components were detected by the GC-MS analysis. Moreover, the catalysts presented the highest degree of carbonaceous species at low temperatures (350 °C). Nonetheless, the specific surface areas of these samples remained high after the experiments.
Nitrogen physisorption measurements indicated that the decrease of surface area over the tested samples was not very prominent and most of the original surface area could be recovered by re-oxidation of the catalyst at 400 °C for 30 minutes, possibly explaining the regain in catalytic activity. The presence of H2 in the feed helped to keep a higher surface area of the catalyst at the studied reaction conditions. The decrease of surface area was found to be more related with the long exposure of the catalyst to the reaction mixture than with the nature of the reductant and temperature.
Time on stream activity over the catalyst was studied both at 250 and 450 °C revealing that no detectable deactivation occurred over the catalyst either at low or high NOx conversion regions. Even though small amounts of carbon were deposited on the surface, the silver catalyst only showed a minor sign of deactivation at the investigated temperatures. Still, the decrease of the activity over the catalyst was especially pronounced at 250 °C when hydrogen was present and co-fed with either the hydrocarbon or the diesel fuel. This phenomenon is most likely connected to the adsorption and storage of species on the alumina support.
Although coking could in principle occur at temperatures lower than the hydrocarbon or the fuel’s boiling point, it is dubious that this would be a reason for deactivation at those temperatures. The oxidation data showed that at temperatures below 250 °C, there is almost no oxidation of the diesel/hydrocarbon. Other reactions could occur resulting in the blockage of the active sites, especially for the diesel fuel since it contains a significant amount of aromatic and aliphatic components that could cover the surface, reducing the total area available for chemical transformations.
Acknowledgements
Linus Silvander is acknowledged for the SEM-EDXA pictures and measurements and Peter Backman is also acknowledged for the TGA analysis. The financial support from the Graduate School on Chemical Engineering for J. R. Hernández Carucci is gratefully acknowledged. This work is part of the activities at the Åbo Akademi Process Chemistry Centre within the Finnish Centre of Excellence Programme (2006–2011) appointed by the Academy of Finland.References
- Directive 2003/30/EC of the European Parliament and of the Council.
- M. S. Graboski, J. D. Ross and R. L. McCormick, SAE Tech. Pap. Ser., 1996, 961166 Search PubMed.
- W. Marshall, G. L. Schumacher and S. Howell, SAE Tech. Pap. Ser., 1996, 952363 Search PubMed.
- T. Miyadera, Appl. Catal., B, 1993, 2, 199 CrossRef CAS.
- R. Burch and P. J. Millington, Catal. Today, 1996, 29, 37 CrossRef CAS.
- K. A. Bethke and H. H. Kung, J. Catal., 1997, 172, 93 CrossRef CAS.
- F. C. Meunier, J. P. Breen, V. Zuzaniek, M. Olsson and J. R. H. Ross, J. Catal., 1999, 187, 493 CrossRef CAS.
- K. Shimizu, K. Sawabe and A. Satsuma, Catal. Sci. Technol., 2011, 1, 331 CAS.
- K. Eränen, L.-E. Lindfors, A. Niemi, P. Elfving and L. Cider, SAE Paper, 2000, N 2000-01-2813.
- K.-i. Shimizu, J. Shibata, H. Yoshida, A. Satsuma and T. Hattori, Appl. Catal., B, 2001, 30, 151 CrossRef CAS.
- S. Satokawa, J. Shibata, K.-i. Shimizu, A. Satsuma and T. Hattori, Appl. Catal., B, 2003, 42, 179 CrossRef CAS.
- K. Sato, T. Yoshinari, Y. Kintaichi, M. Haneda and H. Hamada, Appl. Catal., B, 2003, 44, 67 CrossRef CAS.
- Satsuma and K.-i. Shimizu, Prog. Energy Combust. Sci., 2003, 29, 71 CrossRef CAS.
- F. Klingstedt, K. Eränen, L.-E. Lindfors, S. Andersson, L. Cider, C. Landberg, E. Jobson, L. Eriksson, T. Ilkenhans and D. Webster, Top. Catal., 2004, 30/31, 27 CrossRef CAS.
- K. Eränen, F. Klingstedt, K. Arve, L.-E. Lindfors and D. Yu. Murzin, J. Catal., 2004, 227, 328 CrossRef.
- K. Arve, F. Klingstedt, K. Eränen, D. Yu. Murzin, L. Čapek, J. Dědeček, Z. Sobalik and B. Wichterlová, Top. Catal., 2004, 30/31, 91 CrossRef CAS.
- K. Arve, J. R. Hernández Carucci, K. Eränen, A. Aho and D. Yu. Murzin, Appl. Catal., B, 2009, 90, 603 CrossRef CAS.
- X. Shi, Y. Yu, H. He, S. Shuai, H. Dong and R. Li, J. Environ. Sci., 2008, 20, 177 CrossRef CAS.
- J. R. Hernández Carucci, A. Kurman, H. Karhu, K. Arve, K. Eränen, J. Wärnå, T. Salmi and D. Yu. Murzin, Chem.–Eng. J., 2009, 154, 34 CrossRef.
- K. Arve, K. Eränen, M. Snåre, F. Klingstedt and D. Yu. Murzin, Top. Catal., 2007, 42–43, 399 CrossRef.
- L. Rantanen, R. Linnaila, P. Aakko and T. Harju, SAE Paper, 2005, No. 2005-01-3771.
- D. Yu. Murzin, I. Kubickova, M. Snåre and P. Mäki-Arvela, European Pat., 05075068.6, 2005 Search PubMed.
- K. Arve, L. Čapek, F. Klingstedt, K. Eränen, L.-E. Lindfors, D. Yu. Murzin, J. Dědecěk, Z. Sobalik and B. Wichterlová, Top. Catal., 2004, 30/31, 91 CrossRef CAS.
- K. Arve, F. Klingstedt, K. Eränen, J. Wärnå, L.-E. Lindfors and D. Yu. Murzin, Chem.–Eng. J., 2005, 107, 215 CrossRef CAS.
- E. F. Iliopoulou, A. P. Evdou, A. A. Lemonidou and I. A. Vasalos, Appl. Catal., A, 2004, 274, 179 CrossRef CAS.
- K. Arve, H. Backman, F. Klingstedt, K. Eränen and D. Yu. Murzin, Appl. Catal., A, 2006, 303, 96 CrossRef CAS.
- R. Burch, J. P. Breen, C. J. Hill, B. Krutzsch, B. Konrad and E. Jobson, Top. Catal., 2004, 30–31, 19 CrossRef.
- M. Richter, U. Bentrup, R. Eckelt, M. Schneider, M.-M. Pohl and R. Fricke, Appl. Catal., B, 2004, 51, 261 CrossRef CAS.
- J. Shibata, Y. Takada, A. Shichi, S. Satokawa, A. Satsuma and T. Hattori, J. Catal., 2004, 222, 368 CrossRef CAS.
- S. Satokawa, Chem. Lett., 2000, 294 CrossRef CAS.
- V. Houel, P. Millington, R. Rajaram and A. Tsolakis, Appl. Catal., B, 2007, 77, 29 CrossRef CAS.
- V. Houel, P. Millington, R. Rajaram and A. Tsolakis, Appl. Catal., B, 2007, 73, 203 CrossRef CAS.
- P. Magnoux, A. Rabeharitsara and H. S. Cerqueira, Appl. Catal., A, 2006, 304, 142 CrossRef CAS.
- R. N. Gimaev, V. Z. Gubaidullin, O. V. Rogacheva, G. F. Davydov and T. D. Danillyan, Khim. Tekhnol. Topl. Masel, 1980, 3, 42 Search PubMed.
- J. Shibata, K.-i. Shimizu, S. Satokawa, A. Satsuma and T. Hattori, Phys. Chem. Chem. Phys., 2003, 5, 2154 RSC.
- K.-i. Shimizu, J. Shibata and A. Satsuma, J. Catal., 2006, 239, 402 CrossRef CAS.
- K. Arve, H. Backman, F. Klingstedt, K. Eränen and D. Yu. Murzin, Appl. Catal., B, 2007, 70, 65 CrossRef CAS.
- D. L. Trimm, Catal. Rev., 1977, 16, 155 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cy00224d |
| This journal is © The Royal Society of Chemistry 2011 |