An attempt to achieve the direct hydrogenolysis of tetrahydrofurfuryl alcohol in supercritical carbon dioxide†
Received
11th April 2011
, Accepted 3rd August 2011
First published on 24th August 2011
Abstract
The use of supercritical carbon dioxide (scCO2) in the hydrogenolysis of tetrahydrofurfuryl alcohol was investigated over a Rh supported MCM-41 catalyst (metal loading ≈ 1%). Without any additive, tetrahydrofurfuryl alcohol was converted to 1,5-pentanediol with high conversion (80.2%) and selectivity (91.2%) under mild reaction conditions (temperature = 80 °C and H2 pressure = 4 MPa). The effects of different variables like CO2 and H2 pressure, temperature and the reaction time were also optimized. A strong influence of CO2 pressure on the conversion was observed. The conversion was increased with pressure due to the enhanced solubility of tetrahydrofurfuryl alcohol in CO2 and reached the maximum at 14 MPa, as the single phase (CO2–H2–substrate) was formed, but the selectivity of 1,5-pentanediol remained unaltered. On the other hand, H2 pressure also changed the conversion as well as the selectivity. Temperature was found to play an important role in enhancing the catalytic efficiency; conversion of the substrate was increased along with the temperature, but the selectivity of 1,5-pentanediol dropped after reaching 120 °C. The Rh catalyst exhibited strikingly different product distribution under the solvent-less conditions compared to scCO2. In addition, instead of CO2, when the reaction was carried out in H2O, the conversion and the selectivity of 1,5-pentanediol decreased substantially. However, addition of only 7 MPa of CO2 modified the conversion and the product selectivity. Under similar reaction conditions, hydrogenolysis of THFA over a Pd catalyst provides a maximum of 50% conversion and the product distribution was different in comparison with Rh catalysts.
1. Introduction
Biomass is a self-renewing energy source and an important part of the fuel and energy research because of the increased awareness of alternative fuels that is spreading all over the world.1 The liquid product obtained after the pyrolysis of lignocellulosic biomass is a complex mixture containing a wide variety of oxygenated compounds, with various chemical functional groups and the overall oxygen content is in the range of 35–60 wt%.2–4 To improve the stability of the obtained liquid product, these oxygenated compounds need to be removed.5 Hence, there is an intrinsic challenge to convert these compounds to various value-added chemicals. Among them, furfural and its derivatives present many interesting possibilities as starting materials due to their high oxygen content.6,7Tetrahydrofurfurylalcohol (THFA) is one of the promising compounds obtained by the hydrogenation of furfural and an important building block in the synthesis of polyols. Polyols, especially diols, have direct use as monomers in the polyester industry. After oxidation and amination, diols could serve as precursors of polyamides and polyurethenes.8,9
Development of a new strategy for the conversion of furfural derivatives to polyols is highly challenging especially using heterogeneous catalysts. According to the literature, there are few results reported on the formation of diols. Adkins and Connor conducted the direct hydrogenation of furfuryl alcohol over a copper-chromite catalyst at 175 °C and achieved high conversion, but only 30% of 1,5-pentanediol (1,5-PD) was obtained.10 In another route, 70% of 1,5-PD was formed through a multi-step reaction viaδ-valeraldehyde; however, this method required a number of steps for isolation and purification.11 Koso et al. successfully achieved high yield of 1,5-PD (yield = 77%) by the hydrogenolysis of THFA using a Rh–ReOx/SiO2 catalyst at 120 °C, and a H2 pressure of 8 MPa.12 Furthermore, the same group observed that instead of Re, Mo has a drastic effect on the formation of 1,5-PD, and is more active in comparison to ReOx under the similar reaction conditions.13 Very recently Xu et al. claimed that instead of THFA, furfural would be the better candidate for direct conversion to 1,5-PD in ethanol over a Pd/Co2AlO4 catalyst under the mild conditions of 130 °C, 1–1.5 MPa of H2 pressure,14 but the obtained yield of 1,5-PD was ∼30%. Thus, still it is desirable to design an improved method for the direct conversion of furfural compounds to diols.
In this work, we have attempted to build up a method for hydrogenolysis of THFA using heterogeneous catalysts in supercritical carbon dioxide (scCO2). The main interest behind the use of scCO2 is that it could dissolve a large amount of reactant gases such as H2, which allows kinetic control of reactions as opposed to limiting of reaction rates by the transport of the gaseous reactant across the gas–liquid interface. In addition, CO2 has tuneable physico-chemical properties depending on the pressure and temperature. Moreover, scCO2 is a well established ecologically benign, energy conserving, waste reducing (economical) and highly efficient reaction medium for metal catalyzed reactions.15
The hydrogenolysis of THFA was conducted over a metal supported mesoporous catalyst. Mesoporous material seems to be an ideal host for metal nanoparticles due to its very high surface area to immobilize catalytically active species.16 Compared to other conventional supports, mesoporous material exhibits superior catalytic activity and significant enhancement of selectivity in scCO2 to the selective hydrogenation of different organic compounds.17
2. Experimental
2.1. Materials
THFA (98%; Wako Pure Chemicals) was used as-received. Carbon dioxide (>99.99%) was supplied by Nippon Sanso Co. Ltd. 1% Pd/C, 1% Pd/Al2O3, 1% Rh/C and 1% Rh/Al2O3 were from Aldrich. Rh and Pd supported on MCM-41 were synthesized in our lab.
2.2.
Catalyst characterization techniques
The powder X-ray diffraction pattern was recorded on a Rigaku-RAD-X system using monochromatized Cu Kα radiation (λ = 1.542 Å). In general, the diffraction data were collected using continuous scan mode with a scan speed of 2° min−1 over the scan range of 2θ = 1.5–15° and for wide-angle (2θ = 30–90°) measurements.
The transmission electron microscopy (TEM) image of the material was recorded on a Philips Tecnai operating at 200 kV. The powder was suspended in ethanol by an ultrasonic method. A drop of this solution is placed on a grid with a holey carbon copper film and then allowed to dry, covered by a watch glass.
2.3.
Catalytic activity
The hydrogenolysis of THFA was studied at 80 °C over the Rh/MCM-41 catalyst. All reactions were carried out in a 50 ml stainless steel batch reactor placed in a hot air circulating oven and the details are given elsewhere.17 Briefly, 0.1 g of the catalyst and 0.4 g of the reactant were introduced into the reactor. After the required temperature was attained, H2, followed by CO2, was charged into the reactor using a high-pressure liquid pump and then compressed to the desired pressure. The liquid product was separated from the catalyst simply by filtration and identified by NMR and GC-MS, followed by quantitative analysis using a GC (HP 6890 N) equipped with a capillary column (DB-WAX) and a flame ionization detector. For all results reported, the selectivity is given as follows:
2.4. Phase behaviour
The phase behaviour of THFA was studied in a 10 ml high pressure view cell fitted with a sapphire window. The cell was placed over a magnetic stirrer for stirring the content and connected to a pressure controller, to regulate the pressure inside the view cell. In addition, a temperature controller was also used to maintain the desired temperature of 80 °C. The substrate was introduced into the view cell at a constant H2 pressure of 4 MPa while CO2 pressure was varied between 7–14 MPa and the phase behaviour was monitored. The reaction mixture exhibited bi-phase condition (substrate and CO2–H2) at PCO2 < 12 MPa, but with increasing CO2 pressure the solubility of THFA increases and a single phase (substrate–CO2–H2) was reached (Fig. 1).
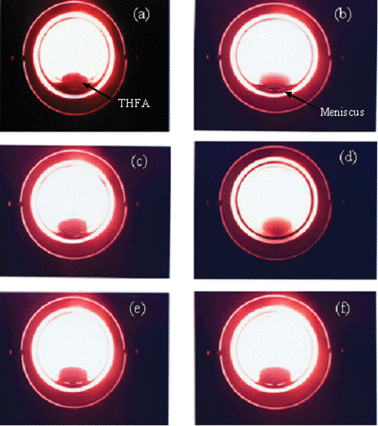 |
| | Fig. 1 Phase behaviour of THFA in CO2 at the fixed pressure of H2 (PH2 = 4 MPa). (a) THFA in the view cell and in the presence of CO2 (b) 8 MPa, (c) 10 MPa, (d) 12 MPa cloud point, (e) 12 MPa and (f) 14 MPa. | |
3. Results and discussion
3.1.
Catalyst characterization
3.1.1.
Powder X-ray diffraction
.
Fig. 2 represents the XRD pattern of calcined Rh/MCM-41 and only Si-MCM-41 for comparison. Both the patterns resemble each other and exhibited a single peak in the low angle region (1 0 0) along with the other Bragg peaks, and confirmed the hexagonal structure. After the incorporation of Rh, d100 space changed from 4.1 nm to 4.4 nm along with decreased intensity of the peak. The XRD patterns of the higher angle region of the calcined Rh/MCM-41 sample (Fig. 1; inset c) exhibited a single peak at 2θ = 34.3° indicating the presence of Rh2O3 particles with orthorhombic symmetry.18 To determine the crystallite size using Scherrer's equation19 seems to be unsuitable due to the presence of a smaller peak in the higher angle region.
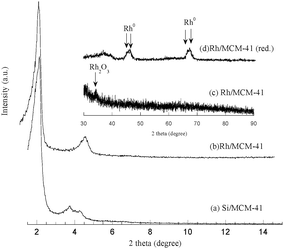 |
| | Fig. 2
X-Ray diffraction pattern of (a) Si/MCM-41, (b) Rh/MCM-41; inset shows higher angle diffraction for (c) Rh/MCM-41 and (d) Rh/MCM-41 (reduced). | |
3.1.2.
Transmission electron microscopy (TEM).
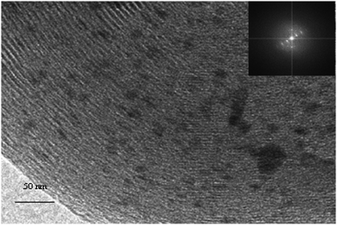
The structure of Rh/MCM-41 is evident from the TEM image of the material (Fig. 3). The ordered array of channels has been confirmed by the FFT pattern shown in the inset of Fig. 3. In addition, a large number of particles can be viewed. From the image it is evident that Rh2O3 particles are almost spherical and randomly distributed. An average particle size of ∼10–15 nm is revealed from the calculation of the particle size distribution. However, few larger particles are also present along with the smaller particles.
3.2. Hydrogenolysis of THFA
Scheme 1 exhibits the probable reaction pathway of the hydrogenolysis of THFA, which is converted to 1,5-pentanediol (1,5-PD) and 1,2-pentanediol (1,2-PD) followed by the formation of 1 or 2-pentanol (POL).
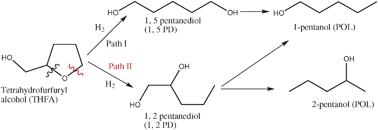 |
| | Scheme 1 Possible reaction pathways of THFA hydrogenolysis. | |
Table 1 Hydrogenolysis of THFA over Rh and Pd catalysts in scCO2
| Entry |
Catalyst
|
Conv. (%) |
Selectivity (%) |
|
1,5-PD
|
1,2-PD
|
POL
|
Othersd |
| Reaction conditions: catalyst = 0.1 g, THFA = 0.4 g, temperature = 80 °C, PCO2 = 14 MPa, PH2 = 4 MPa, time =24 h. Reduced at 300 °C in the presence of H2. Solvent = 2 ml of H2O. Solvent = 2 ml of H2O + 7 MPa of CO2. Others = tetrahydro-2-methylfuran and dimer. |
| 1 |
Rh/MCM-41 |
80.5 |
91.2 |
0.0 |
8.8 |
0.0 |
| 2 |
Rh/Al2O3 |
60.0 |
21.9 |
51.4 |
19.5 |
7.2 |
| 3 |
Rh/C |
34.8 |
29.8 |
42.3 |
20.0 |
7.9 |
| 4 |
Rh/SiO2 |
30.4 |
78.2 |
11.4 |
8.2 |
12.2 |
| 5 |
Pd/MCM-41 |
50.5 |
12.6 |
77.4 |
10.0 |
0.0 |
| 6 |
Pd/Al2O3 |
32.5 |
9.1 |
0.9 |
47.5 |
42.5 |
| 7 |
Pd/C |
48.9 |
99.2 |
0.3 |
0.5 |
0.0 |
| 8a |
Rh/MCM-41 |
5.0 |
12.1 |
82.5 |
5.4 |
0.0 |
| 9b |
Rh/MCM-41 |
20.2 |
35.1 |
0.0 |
4.9 |
60.0 |
| 10c |
Rh/MCM-41 |
42.3 |
62.5 |
0.0 |
0.0 |
37.5 |
For Pd catalysts, conversion of THFA is low and in the range of 30–50% (Table 1; entries 5–7). Moreover, from Table 1, it has been found that POL was the main product of Pd/Al2O3, which suggested the possible conversion of diols to POL within the reaction time of 24 h. On the other hand, the selectivity of 1,5-PD was 99.2 and 12.6% over Pd/C (conversion = 48%) and Pd/MCM-41 (conversion = 50%), respectively (Table 1; entries 5 and 7). Based on the highest conversion and the selectivity of the desired product (1,5-PD), Rh/MCM-41 was chosen for further study to optimize the reaction conditions. Notably, no reaction was proceeding without the catalyst.
3.4. Effect of CO2 pressure
Fig. 4 shows the effect of CO2 pressure from 8 to 18 MPa and a comparison was made under the solvent-less conditions for hydrogenolysis of THFA over Rh/MCM-41 at the fixed temperature of 80 °C and a hydrogen pressure of 4 MPa. As the pressure changed from 8 to 14 MPa, the conversion was increased from <10 to 80% and then remained almost constant. From the phase behaviour (solubility of THFA in CO2), as shown in Fig. 1, it is observed that the solubility of THFA increased with the CO2 pressure and reached the single-phase condition above 12 MPa. Fig. 1a presents the image of only THFA in the view cell. After the introduction of CO2, at the lower pressure (Fig. 1b and c), the system was biphasic. On the other hand, the meniscus between two phases disappeared (Fig. 1d) and the system attained a single phase (substrate–CO2–H2) (Fig. 1e and f) when the CO2 pressure was above 12 MPa. Therefore, hydrogenolysis of THFA required single-phase condition to achieve highest catalytic performance. At the lower pressure region, the lower conversion might be related to the mass-transfer limitation in the gas–liquid interface. On the selectivity of 1,5-PD, no such discrete influence of CO2 pressure was observed.
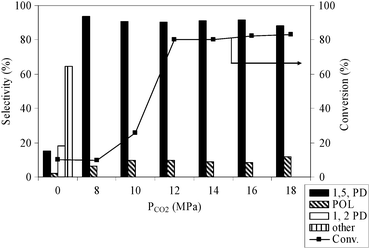 |
| | Fig. 4 Effect of CO2 pressure on the conversion and selectivity of THFA hydrogenolysis; reaction conditions: catalyst = 0.1 g, THFA = 0.4 g, temperature = 80 °C, PH2 = 4 MPa, time = 24 h. | |
Furthermore, lower conversion and a noticeable difference in the product distribution were found under the solvent-less conditions; mainly other by-products are observed along with the formation of 1,2-PD, which suggests a possible role of CO2 in dictating the overall performance of the catalyst.
3.5. Variation of H2 pressure
Fig. 5 presents the effect of the change in H2 pressure from 2–8 MPa, on the hydrogenolysis of THFA using the Rh/MCM-41 catalyst. The temperature and CO2 pressure were kept constant at 80 °C and 14 MPa, respectively, in accordance with the observed highest activity and selectivity of 1,5-PD. As expected, the conversion of THFA was increased from 10.2 to 98% with an increase in H2 pressure from 2 to 8 MPa, but the selectivity of 1,5-PD was reduced due to the formation of POL. It has to be mentioned that at 8 MPa, a negligible amount of 1,2-PD was observed. Thus, according to the maximum selectivity of the 1,5-PD, 4 MPa was chosen as the optimum pressure for THFA hydrogenolysis.
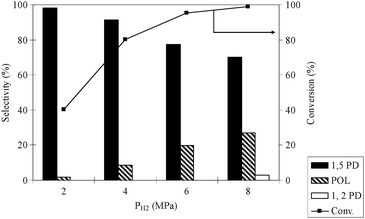 |
| | Fig. 5 Effect of H2 pressure on the conversion and selectivity. Reaction conditions: catalyst = 0.1 g, THFA = 0.4 g, temperature = 80 °C, PCO2 = 14 MPa, time = 24 h. | |
3.6. Effect of temperature
Temperature is an important factor in the hydrogenolysis of THFA. It was observed that higher temperature >60 °C is necessary for the reaction to occur. The effect of temperature on the hydrogenolysis of THFA was studied at 80–120 °C, at the constant pressure of H2 and CO2 of 4 and 14 MPa, respectively (Fig. 6). The conversion of THFA was enhanced as the temperature raised from 80 to 120 °C, accompanied with decreased selectivity of 1,5-PD. The influence of reaction temperature on the conversion and selectivity might be explained by the phase behaviour of the substrate and H2–CO2. In scCO2, the alteration of temperature from 80 to 120 °C changed the density of the medium from 8.73 to 5.83 mol l−1 (NIST, chemistry web book). Hence, there is a possibility of the separation of the single phase into two phases because at higher temperature the CO2 phase becomes less dense. At lower densities, scCO2 is more gas-like and is therefore a poorer solvent. However, increased conversion with temperature may be attributed to the dramatic increase in the reaction rate in the liquid phase and in the gaseous phase, which overcompensates the negative effect of phase separation.21 On the other hand, the selectivity of 1,5-PD was decreased with increasing temperature due to the formation of other by-products. A similar result was also obtained during the hydrogenolysis of tetrahydropyran-2-methanol over the Rh–ReO/C catalyst and suggested that the lower temperature would be better to achieve high selectivity of the corresponding diol.22
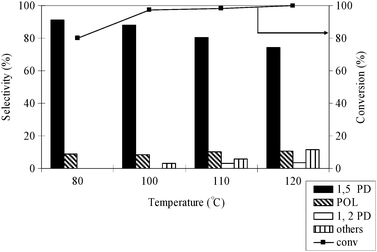 |
| | Fig. 6 Effect of reaction temperature on conversion and selectivity of THFA hydrogenolysis; reaction conditions: catalyst = 0.1 g, THFA = 0.4 g, PCO2 = 14 MPa, PH2 = 4 MPa, time = 24 h. | |
3.7. Effect of reaction time
It is well-known that the product distribution of a reaction is closely related to the working conditions of the catalytic system and the stability of the intermediate species on the catalyst surface. Reaction time is an important parameter, which can provide an idea about the kinetics of the reaction. The effect of reaction time has also been studied over Rh/MCM-41 for hydrogenolysis of THFA. Fig. 7 shows the change in catalytic activity with the change in reaction time from 2 to 36 h. Under the studied reaction conditions, after 2 h conversion of THFA was only 5% suggesting an induction period. However, the conversion increased to 80.2% as the time changes to 24 h. When the reaction was extended to 36 h, conversion was slightly improved, but the selectivity of 1,5-PD decreased. It is evidenced that the rate of the reaction is relatively higher within 2–6 h and then again decreased. To explain this behaviour, the catalyst was characterized ex situ by XRD analysis (Fig. S1 and S2, ESI†). Results revealed that for all the catalysts after 2, 6 and 36 h of reaction, the XRD pattern remained unchanged except for the decreased relative intensities of the peaks. Thus, the crystalline state of the support was intact after the reaction. Regarding the state of Rh, the higher angle XRD pattern (Fig. S2, ESI†; as particles are smaller a broader spectrum was obtained) confirmed the change in state with time. In the beginning, mainly Rh2O3 particles are evident from the XRD pattern of the catalyst after 2 h of reaction. The state of Rh changed into the mixture of Rh0 and Rh2O3 after 6 h and finally mostly Rh0 particles were observed. Therefore, a possible interpretation is that Rh2O3 itself has low activity, and the reduction of Rh2O3 proceeds partially to Rh0, and then the copresence of Rh0 and Rh2O3 promotes the reaction significantly. When all the Rh2O3 was reduced, the activity decreased again. Better conversion of THFA could be achieved within shorter reaction time with the expense of the selectivity of 1,5-PD, if temperature was increased. For example, at 120 °C almost complete conversion of THFA was obtained within 16 h of reaction time, but selectivity of 1,5-PD was dropped to <80% due to the formation of other by-products.
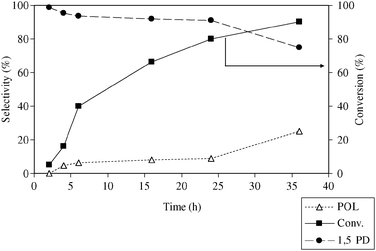 |
| | Fig. 7 Variation of reaction time; reaction conditions: catalyst = 0.1 g, THFA = 0.4 g, temperature = 80 °C, PCO2 = 14 MPa, PH2 = 4 MPa. | |
3.8. Reaction in water and water + CO2 medium
The hydrogenolysis has also been conducted in the presence of water and water + CO2. Under the studied reaction conditions, in the presence of water, conversion was low (20.2%) and the product distribution changed considerably. Instead of diols, high selectivity of by-products (60%) was observed (Table 1; entry 9). Surprisingly, an interesting change in the selectivity was observed when CO2 at a very low pressure of 7 MPa was introduced along with water. With the addition of only 7 MPa of CO2, the selectivity of 1,5-PD was increased to 62.5%, and the conversion became 42.3% (Table 1; entry 10). The detailed study on the promising combination of CO2 and water as the reaction medium for the hydrogenolysis of THFA is currently underway.
4. Conclusion
In conclusion, we have successfully developed an efficient procedure for the hydrogenolysis of THFA in scCO2. The Rh-MCM-41 catalyst demonstrated higher selectivity of 1,5-PD under the mild reaction conditions and without any additive. Alterations of the process variables like CO2 and H2 pressure, temperature and reaction time have a significant effect on the conversion and the selectivity. Under the optimal reaction conditions, the selectivity of 1,5-PD attained 91.2% at 80% conversion of the substrate. In addition, the nature of the metal oxidation state and solvent medium is also an important factor to determine the product selectivity. It has been hypothesized that the presence of Rh2O3 and Rh0 particles on Rh/MCM-41 as confirmed by XRD could be the reason behind the formation of 1,5-PD with high selectivity. An appreciable catalytic performance in terms of conversion and selectivity was also observed for Pd/MCM-41 catalysts. However, the product distribution differs considerably in comparison with the Rh/MCM-41. Although further work is still required to improve the product yield and catalyst recycling, the result achieved provides a simpler way to achieve the formation of 1,5-PD from the oxygenated compounds like THFA using heterogeneous catalysts.
Notes and references
- J. N. Chedha, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164 CrossRef CAS.
- T. Bridgewater, Biomass Bioenergy, 2007, 7, 18 Search PubMed.
- S. Czemik and A. V. Bridgwater, Energy Fuels, 2004, 4, 590 Search PubMed.
- A. Oasmaa, S. Czemik and A. V. Bridgwater, Energy Fuels, 1999, 13, 914 CrossRef CAS.
- A. Pinheiro, D. Hudebine, N. Dupassiex and C. Geantet, Energy Fuels, 2009, 23, 1007 CrossRef CAS.
- L. H. Dao, M. Haniff, A. Houle and D. Lamothe, Pyrolysis Oils and Biomass, ACS Symp. Ser., 1988, 376, 328 CrossRef CAS.
- R. C. D. Brown, Angew. Chem., Int. Ed., 2005, 44, 850 CrossRef CAS.
- M. Schlaf, Dalton Trans., 2006, 4645 RSC.
- C. Zhou, J. N. Beltramini, Y. Fan and G. Q. Lu, Chem. Soc. Rev., 2008, 37, 527 RSC.
- H. Adkins and R. Connor, J. Am. Chem. Soc., 1931, 53, 1091 CrossRef CAS.
- L. E. Schniepp and H. H. Geller, J. Am. Chem. Soc., 1946, 68, 1646 CrossRef CAS.
- S. Koso, I. Furikado, A. Shimao, T. Miyazawa, K. Kunimori and K. Tomishige, Chem. Commun., 2009, 2035 RSC; S. Koso, Y. Nakagawa and K. Tomishige, J. Catal., 2011, 280, 221 Search PubMed.
- S. Koso, N. Ueda, Y. Shinmi, K. Okumura, T. Kizuka and K. Tomishige, J. Catal., 2009, 267, 89 CrossRef CAS.
- W. Xu, H. Wang, X. Liu, J. Ren, Y. Wang and G. Lu, Chem. Commun., 2011, 47, 3924 RSC.
- A. Baiker, Chem. Rev., 1999, 99, 453 CrossRef CAS; A. Fürstner, L. Ackermann, K. Beck, H. Hori, D. Koch, K. Langeman, M. Liebl, C. Six and W. Leitner, J. Am. Chem. Soc., 2001, 123, 9000 CrossRef CAS; K. Wittmann, W. Wisniewski, R. Mynott, W. Leitner, C. L. Kranemann, T. Rische, P. Eilbracht, S. Kluwer, J. M. Ernsting and C. J. Elsevier, Chem.–Eur. J., 2001, 7, 4584 CrossRef CAS; P. Stephenson, B. Kondor, P. Licence, K. Scovell, S. K. Ross and M. Poliakoff, Adv. Synth. Catal., 2006, 348, 1605 CrossRef CAS; J. R. Hyde, B. Walsh, P. Singh and M. Poliakoff, Green Chem., 2005, 7, 357 RSC;
Green Chemistry using Liquid and Supercritical Carbon Dioxide, ed. J. M. DeSimone and W. Tumas, Oxford Press, 2003 Search PubMed;
Modern Solvent in Organic Synthesis, ed. P. Knochel, Springer, 1999 Search PubMed.
- R. Köhn and M. Förba, Catal. Today, 2001, 68, 227 CrossRef CAS; M. Morey, A. Davidson, H. Eckert and G. D. Stucky, Chem. Mater., 1996, 8, 486 CrossRef.
- M. Chatterjee, Y. Ikushima and F. Zhao, New J. Chem., 2003, 27, 510 RSC; M. Chatterjee, F. Y. Zhao and Y. Ikushima, Adv. Synth. Catal., 2004, 346, 459 CrossRef CAS; M. Chatterjee, A. Chatterjee and Y. Ikushima, Green Chem., 2004, 6, 114 RSC; M. Chatterjee, Y. Ikushima, T. Yokoyama and M. Sato, Adv. Synth. Catal., 2008, 350, 624 CrossRef CAS; M. Chatterjee, T. Yokoyama, H. Kawanami, M. Sato and T. Suzuki, Chem. Commun., 2009, 701 RSC.
- R. S. Mulukutla, K. Asakura, T. Kogure, S. Namba and Y. Iwasawa, Phys. Chem. Chem. Phys., 1999, 1, 2027 RSC.
-
H. P. Klug and L. E. Alexander, X-Ray Diffraction Procedures for Polycrystalline Amorphous Materials, Wiley, New York, 2nd edn, 1974 Search PubMed.
- J. Park, Bull. Korean Chem. Soc., 2008, 29, 1787 Search PubMed.
- R. Tschan, R. Wandeler, M. S. Schneider, M. M. Schubert and A. Baiker, J. Catal., 2001, 204, 219 CrossRef CAS.
- K. Chen, S. Koso, T. Kubota, Y. Nakagawa and K. Tomishige, ChemCatChem, 2010, 2, 547 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cy00125f |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.