Ru modified Au catalysts for the selective oxidation of aliphatic alcohols
Laura
Prati
*a,
Francesca
Porta
a,
Di
Wang
b and
Alberto
Villa
a
aDipartimento di Chimica Inorganica Metallorganica e Analitica L.Malatesta, Università degli Studi di Milano, via Venezian 21, 20133 Milano, Italy. E-mail: Laura.Prati@unimi.it; Fax: +39 02503 14405; Tel: +39 02503 14357
bInstitute of Nanotechnology, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 14th September 2011
Abstract
Gold catalysts have been modified by Ru. The sequential deposition of the metals produced very active catalysts for the liquid phase oxidation of aliphatic alcohols which differentiate very much from monometallic counterparts and depends on the order of the addition of the two metals. The typical selectivity showed by Ru catalysed aerobic oxidation of alcohol to aldehyde is maintained but the activity strongly depends on the structure of nanoparticles. Moreover the substrate and the experimental conditions greatly affected the catalyst activity. It appears that in all cases the most active bimetallic system showed a surface enriched by the less active component.
1. Introduction
Selective alcohol oxidation represents an important task from an industrial point of view and many efforts have been made to produce active and selective catalysts. Particularly the aerobic catalytic oxidation has been appreciated in view of environment control and safety reasons despite non-catalytic methods are still in use.1,2Gold and ruthenium have been separately well studied as active catalytic metals for aerobic oxidation, the first mainly for its improved resistance to deactivation, the second for the typical high selectivity to carbonyl compounds instead of carboxylic ones.However, both metals presented some limitations. In fact in the case of gold catalysed alcohol oxidation a basic environment is needed to accelerate the rate determining step (i.e.hydride abstraction). As a consequence carboxylic acids in the form of the corresponding carboxylate are produced. On the other hand ruthenium often shows a low activity which leads to the use of a very low alcohol/metal molar ratio (15–25 mol/mol). In principle the use of large quantities of a catalyst can be acceptable from a practical point of view but only in the case of a highly resistant catalyst, able to be recycled for a long time (high turn over number value).
A lot of catalytic systems have been developed for the aerobic oxidation of alcohols based on monometallic Ru and Au on a variety of supports. Ru on hydroxyapatite (HAP),3,4 on Al2O3,5,6carbon,7Carbon Nano Tubes (CNTs),8zeolite9 and MgO10 as well as Au on CeO2,11 on carbon, on TiO2 and zeolite12,13 have been reported. Bimetallic systems have been used to circumvent the need of a basic environment in the case of gold catalysts: Au–Pd on TiO213,14 and Au–Pd or Pt on activated carbon.13,15,16
A Ru/Au system on SiO2 and MgO has been deeply studied by Schwank et al. since 198117 and some important conclusions have been derived: although the limited miscibility of the two metals,18 an intimate dispersion of Au and Ru can be achieved when the particle diameters are below 5 nm19 as a function of the support.17 In fact it has been reported that Au and Ru on SiO2 formed bimetallic aggregates with a surface composition similar to the bulk one whereas on MgO a Ru enrichment on the surface of the bimetallic particles was observed. Moreover ruthenium chemisorbs O2 at lower temperature than Au and upon increasing the Au/Ru ratio, i.e. increasing Au, the reactivity of the particle decreases for a geometrical dilution effect (ensemble effect).19
More recently AuRu on SiO2 and Al2O3 prepared by incipient wetness impregnation have been tested for the total oxidation of methanol, revealing a high interaction between Au and Ru in the case of silica supported nanoparticles.20 RuAu on Fe2O3 prepared by a deposition-precipitation method have been tested in the water gas shift reaction21 and for H2 production by partial oxidation of methanol22 highlighting the presence of Au–Ru interaction. It was also detected a partial segregation of Au but no segregation of ruthenium.
By using a sequential deposition method, AuRu on activated carbon without any segregation was successfully prepared and tested in the hydrogenolysis of glycerol.23 Using condensed phases, however, it has been shown that the composition of the catalyst was altered during the reaction by the harsh conditions (473 K, 40 bar H2) with gold that appeared to migrate off the ruthenium and agglomerate on carbon. In such a reaction the bimetallic system behaved as a physical mixture of monometallic Au and Ru on a carbon catalyst.
Activated carbon represents an ideal support for liquid phase application where typically mild temperature conditions (RT–100 °C) are used.24 Indeed carbon has a number of advantages over oxide supports. Carbon supports are stable in both acidic and basic environments. If the supported catalyst deactivates or the precious metal needs to be recycled, the carbon can be burnt off leaving the expensive metal which can easily be collected and reprocessed.
In the present study we investigated the effect of the preparation method on the activity and selectivity of Au–Ru bimetallic systems supported on activated carbon in the liquid phase oxidation of aliphatic alcohols.
2. Experimental
2.1 Catalyst preparation
Activated carbon was obtained from Camel (X40S, AS 900–1100 m2 g−1) and washed with HCl 6 N and water (until pH 6) before using. Commercial Ru/AC (Escat 40, 5 wt%) was obtained from Engelhard whereas Au/AC (1 wt%) was prepared following the procedure reported elsewhere.25 A PVA stabilized sol was prepared by reducing a HAuCl4 solution in the presence of polyvinyl alcohol (PVA) with NaBH4. The sol was then supported on activated carbon (1 wt% loading).Bimetallic systems were prepared following a two-step procedure using two different techniques. Core–shell Ru@Au nanoparticles have been prepared as reported in the literature26 and then supported on activated carbon (1 wt% loading) (i.e. (Ru@Au)/AC). A second preparation was also employed following the same procedure as the one reported for the AuPd system.27Ru(III) was reduced with H2 at 80 °C in the presence of preformed Au/AC. The catalyst was labelled as Ru@(Au/AC).
Finally PVA stabilized AuNPs were deposited on commercial Ru/AC (i.e. Au@(Ru/AC)) following the same procedure as before.
ICP of the filtrate allowed us to determine the metal loading of each sample (Table 1).
2.2 Characterisation
The catalysts were examined in a FEI Titan 80–300 electron microscope equipped with a CEOS image spherical aberration corrector, a Fischione model 3000 HAADF STEM detector and an EDAX SUTW EDX detector. The microscope was operated at an accelerating voltage of 300 kV in TEM mode for HRTEM and in STEM mode for STEM and EDX spectrum imaging. The probe size was about 1 nm as a good compromise of spatial resolution and beam current. The sizes in pixels of the spectrum images were 30 × 40. The pixel size is 1.5 nm. The dwell time per pixel was 2.5 seconds. The element maps were generated by integrating the Au L and Ru K intensities, respectively. The quantification was using Au L and Ru K lines processed in TIA software.The metal content was checked by ICP analysis on a Jobin Yvon JY24.
2.3 Oxidation of alcohols
Reactions were carried out in a 30 mL glass reactor equipped with a thermostat and an electronically controlled magnetic stirrer connected to a 5000 mL reservoir charged with oxygen (300 kPa). The oxygen uptake was followed by a mass-flow controller connected to a PC through an A/D board, plotting a flow time diagram.Glycerol oxidation : glycerol 0.3 M, and the catalyst (substrate/total metal = 1000 mol/mol) were mixed in distilled water (total volume 10 mL) and 4 eq. of NaOH. The reactor was pressurized at 300 kPa of nitrogen and set to 50 °C. Once this temperature was reached, the gas supply was switched to oxygen and the monitoring of the reaction started. The reaction was initiated by stirring. Samples were removed periodically and analyzed by high-performance liquid chromatography (HPLC) using a column (Alltech OA-10308, 300 mm–7.8 mm) with UV and refractive index (RI) detection to analyze the mixture of the samples. Aqueous H3PO4 solution (0.1 wt%) was used as the eluent. Products were identified by comparison with the original samples.
Octanol oxidation : n-Octanol and the catalyst (alcohol/total metal = 100 mol/mol) were mixed in toluene (octanol/toluene 10/90 vol/vol; total volume, 10 ml). The reactor was pressurized at 300 kPa of oxygen and set to 80 °C. The reaction was initiated by stirring. Periodic removal of samples from the reactor was performed. Identification and analysis of the products were done by comparison with the authentic samples by GC using a HP 7820A gas chromatograph equipped with a capillary column (HP-5 30 m × 0.32 mm, 0.25 μm film, by Agilent Technologies) and a TCD detector. Quantification of the reaction products was done by the external calibration method.
3. Results and discussion
3.1 Catalyst characterisation
The use of Au seeds as a nucleation centre for the deposition of a second metal has been already reported to be successful in preparing bimetallic nanoparticles even if the method should be adapted depending on the desired results. In fact it has been reported that the sol technique produced Rushell–Aucorenanoparticles using Au seeds but no Aushell–Rucore are produced by the reverse addition.26 On the contrary using Ru supported on carbon as starting material for the subsequential deposition of Au, gold nanoparticles can be successfully deposited onto Ru ones.23 In this study we prepared different kinds of catalysts in order to have Ru or Au as a modifier on the external surface of the nanoparticles.Table 1 shows the catalysts prepared with their characteristics.
Monometallic gold was prepared by pre-formed sol immobilisation whereas ruthenium was commercially available.
(Ru@Au)/AC was prepared following the literature procedure for having Ru deposition on Au nanoparticles. A representative HRTEM image of this catalyst is shown in Fig. 1. STEM-EDX analysis did not show any Ru or Au segregation even if the quantitative analysis of different regions revealed a slightly different composition Ru![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au. We also deposited Ru nanoparticles on preformed Au/AC following the procedure that our group set up for bimetallic AuPd/AC.27,28 The Ru@(Au/AC) showed a better metal dispersion on the support than (Ru@Au)/AC but a similar distribution of the metals. Also in this case STEM-EDX mapping (Fig. 2) showed a slightly varying composition of the nanoparticles with Ru situates on Au particles.
:Au. We also deposited Ru nanoparticles on preformed Au/AC following the procedure that our group set up for bimetallic AuPd/AC.27,28 The Ru@(Au/AC) showed a better metal dispersion on the support than (Ru@Au)/AC but a similar distribution of the metals. Also in this case STEM-EDX mapping (Fig. 2) showed a slightly varying composition of the nanoparticles with Ru situates on Au particles.
 | ||
| Fig. 1 Representative HRTEM image of (Ru@Au)/AC. | ||
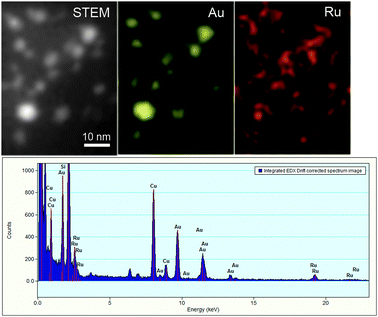 | ||
| Fig. 2 STEM-EDX mapping of Ru@(Au/AC). | ||
A representative EDS line profile is exhibited as an example for the varying composition in one nanoparticle (Fig. 3).
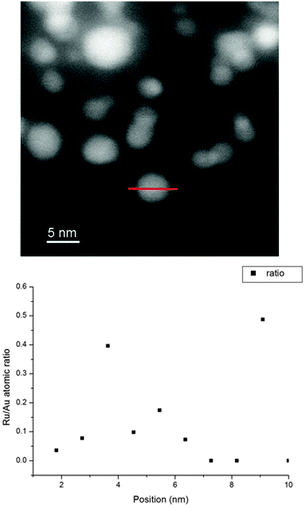 | ||
| Fig. 3 EDS line profile of a representative Ru@(Au/AC) particle. | ||
In the third case Ru/AC was used as seeds for nucleating Au nanoparticles according to the literature.23 In this case, however, we obtained an almost inhomogeneous distribution, with the presence of small particles principally composed by Ru and larger bimetallic particles without any obvious enrichment of a single metal. HAADF STEM and EDS spectra are reported (Fig. 4).
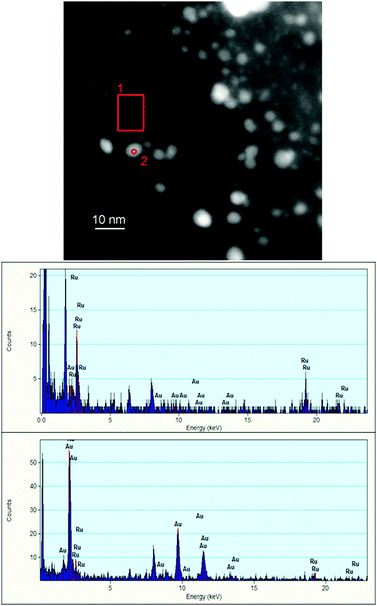 | ||
| Fig. 4 HAADF STEM and EDS spectra from two types of different particles for Au@(Ru/AC). | ||
Au@(Ru/AC) has been reported to be not stable under harsh conditions in the presence of water where migration of Au was observed. Although our conditions appear much more mild we could expect a possible changing of particles composition during the reaction.
3.2 Oxidation reactions
Ru catalysts have been shown to be of great interest as oxidation catalysts under aerobic conditions especially for the high selectivity to carbonyl compounds. n-Octanol has been often used as a model for aliphatic alcohol in testing the activity of a catalyst. Up to now however only few cases reported successful results in this field. Ru on HAP3,4,6 or on Al2O3 appeared the most promising system even if the reaction has to be performed using a high amount of catalyst (alcohol/metal molar ratio 15–25). Ru on carbon7 and on carbon nanotubes8 are also reported. In particular Ru/C when used with linear alcohol showed a low selectivity to aldehyde, being carboxylic acid the main product.Gold catalysts under neutral conditions appeared often unreactive12 even in some cases bimetallic systems circumvent such a problem.13 Under basic conditions the selectivity of the reaction is normally shifted toward the corresponding carboxylate.
As expected, when we tested Au/AC in n-octanol oxidation (10% vol in toluene, 80 °C, octanol/Au 100 mol/mol) we did not observe any reaction (3 h) (Table 2). On the contrary, commercial Ru/C showed, under the same conditions, one of the highest activities up to now reported with a conversion of 45% in 3 h and an almost total selectivity to aldehyde.
Checking the bimetallic systems (Ru@Au)/AC and Ru@(Au/AC), it clearly appears that the presence of gold almost completely suppressed the Ru activity. In fact also the catalyst with a higher content of Ru, i.e. Ru@(Au/AC), showed a negligible activity. The activity was not significantly improved even when using a metal/alcohol molar ratio of 10. We also tested a physical mixture: when 10 mol% of ruthenium was substituted by gold (Au:Ru 1:10) we expected to observe a corresponding decrease of activity with respect to the monometallic Ru catalyst. However an unexpected increase of activity was observed (50% for the mixture vs. 45% for the Ru alone). During our previous study on AuPd systems we already observed a similar phenomenon that was ascribed to Pd migration on Au forming a more active alloy.29 A similar migration process can be envisaged also in this case.
Au@(Ru/AC) where small Ru nanoparticles coexist with larger bimetallic particles of random alloy composition showed the highest activity. This supports the envisaged beneficial effect produced by gold on a Ru/AC catalyst but also the importance of the composition of the metallic particles.
According to the literature it appears that when a surface enrichment of Ru is present, Au has a detrimental effect on activity due to a geometric dilution effect (ensemble effect).19 On the contrary in the present case it appeared that when alloyed Ru/Au nanoparticles are present, as in the case of Au@(Ru/AC), a significant increase of the activity was observed without any change in selectivity.
STEM-EDX analyses of used Ru@(Au/AC) (Fig. 5) and also Au@(Ru/AC) (Fig. 6) revealed a progressive segregation of the two metals under the reaction conditions.
 | ||
| Fig. 5 STEM-EDX analyses of used Ru@(Au/AC). | ||
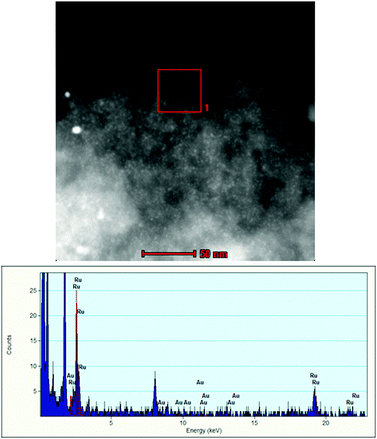 | ||
| Fig. 6 STEM-EDX analyses of used Au@(Ru/AC). | ||
The same catalysts have been tested in glycerol oxidation which is performed in aqueous conditions, but under milder conditions than the previous ones (50 °C instead of 80 °C). Table 3 reports the results we obtained under the usual conditions used for glycerol oxidation (0.3 M in water; 4 eq. of NaOH; metal/alcohol = 1/1000 mol/mol; 300 kPa O2; T = 50 °C). On the contrary of the case of n-octanol, a base was added. Au/AC showed the expected activity/selectivity on the base of literature data.30 On the opposite we observed a negligible activity for Ru/AC, confirmed also using a lower glycerol/Ru ratio (100 mol/mol).
| Catalyst | Conv. (%) | Selectivity | |||
|---|---|---|---|---|---|
| Glyc | Gly | Tartr | Form | ||
| a Glycerol 0.3 M in water; 4 eq. of NaOH; metal/alcohol = 1/1000 mol/mol; 300 kPa O2; T = 50 °C; 1 h reaction. Glyc = glycerate; Gly = glycolate; Tartr = tartronate; Form = formate.b Also using a glycerol/Ru ratio 100 mol/mol. | |||||
| Au/AC | 73 | 68 | 25 | 4 | 2 |
| Ru/ACb | <1 | — | — | — | — |
| (Ru@Au)/AC | 81 | 60 | 20 | 10 | 9 |
| Ru@(Au/AC) | 99 | 57 | 18 | 7 | 18 |
| Au/C + Ru/AC | |||||
| [Au:Ru = 1:10 mol/mol] |
1 | 26 | 3 | — | 30 |
| [Au:Ru = 10:1 mol/mol] |
62 | 45 | 26 | 12 | 14 |
| Au@(Ru/AC) | 26 | 43 | 41 | — | 16 |
On the basis of the activity of monometallics, unexpectedly (Ru@Au)/AC and Ru@(Au/AC) both showed a high activity (higher than monometallic Au) with Ru@(Au/AC) which contain a higher amount of Ru than (Ru@Au)/AC (0.10 wt% vs. 0.04 wt%) being more active. This behaviour could be explained by a possible electronic modification acted by the presence of gold on Ru at the surface.
The selectivity toward C–C bond cleavage products (glycolate and formate) increased by increasing the Ru content especially in the case of formate which increased from 9% to 18% by doubling the Ru content (0.04% vs. 0.10%). This trend has been confirmed by using a physical mixture Au/AC + Ru/AC as the catalyst. In this case the activity is very low and formate became the main product. Using the physically mixed Au/AC and Ru/AC we obtained an activity consistent with the Au content. The only visible effect of the presence of Ru/AC is the increase of the C–C bond cleavage products.
Au@(Ru/AC) showed a lower activity than Au/AC with a strong increase of selectivity to glycolate (41% with respect to 25% of Au/AC) thus with enhancement of C–C bond cleavage. It should be noted that under these conditions the structures of the catalysts appeared to be more stable than in the case of the octanol ones.
Considering the two tests we performed, it can be concluded that the oxidation of n-octanol carried out in toluene and the oxidation of glycerol in water and in the presence of a base profoundly differ. The catalyst that is the most active for octanol oxidationi.e. Au@(Ru/AC) is the less active for glycerol and vice versa the one less active for n-octanoli.e. Ru@(Au/AC) is the most active for glycerol. It should also be noted that in the two reactions Au/AC and Ru/AC catalysts show a negligible activity, respectively, in octanol and glycerol oxidation. Considering the characterisation of the catalyst we could conclude that, differently from the case of octanol, the Ru surface enriched Ru–Au nanoparticles are more active than alloyed ones.
4. Conclusions
Gold and ruthenium catalysts have been studied for a long time as catalysts for the selective oxidation of alcohols. Bimetallic systems prepared by different known methods provided different distributions of the metals within the supported particles. Ru enriched surfaces or alloyed Ru/Au particles similar in size have been thus prepared and tested under different reaction conditions.Aliphatic alcohols, that can be smoothly oxidized to aldehyde in the presence of a monometallic Ru/AC catalyst in toluene, resulted quite unreactive under such conditions in the presence of Au/AC. Bimetallic alloyed AuRu catalysts showed a higher activity with respect to the monometallic counterparts whereas an enrichment of Ru at the particle surface resulted in an almost inactive catalyst. Under the reaction conditions used however we observed a progressive segregation of metals in both catalysts.
Water soluble and highly hydrophilic polyols, such as glycerol, are smoothly oxidized in the presence of Au/AC under basic conditions. Under these conditions Ru/AC didn’t show any appreciable activity. In this case the bimetallic systems that show an enhanced activity with respect to Au/AC resulted the ones where the surface of the nanoparticles is Ru-rich, the opposite to the case of aliphatic alcohols.
It has been reported that Au in MeOH oxidation acts as a geometric diluting agent for Ru thus decreasing the reactivity of this latter for an ensemble effect.19 We can then argue that it can happen also in the case of n-octanol oxidation. However we have shown that when alloyed Au/Ru nanoparticles are used, the activity of the catalyst increased. When glycerol is oxidized in aqueous, basic media, an opposite trend for bimetallic catalysts has been revealed, Ru-surface rich nanoparticles becoming the most active.
Acknowledgements
Financial support by Fondazione Cariplo and Euminafab is gratefully acknowledged.Notes and references
- W. J. Mijs and C. R. H. de Jonge, Organic Syntheses by Oxidation with Metal Compounds, Plenum, New York, 1986 Search PubMed.
- M. Hudlicky, Oxidations in Organic Chemistry, ACS Monograph 186, ACS, Washington, DC, 1990 Search PubMed.
- K. Yamaguchi, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2000, 122, 7144 CrossRef CAS.
- Z. Opre, J.-D. Grunwaldt, M. Maciejeski, D. Ferri, T. Mallat and A. Baiker, J. Catal., 2005, 230, 406 CrossRef CAS.
- K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2002, 41, 4538 CrossRef CAS.
- N. Zotova, K. Hellgardt, G. H. Kelsall, A. S. Jessiman and K. K. Hii, Green Chem., 2010, 12, 2157 RSC.
- S. Mori, M. Takubo, K. Makida, T. Yanase, S. Aoyagi, T. Maegawa, Y. Monguchi and H. Sajiki, Chem. Commun., 2009, 5159 RSC.
- X. Yang, X. Wang and J. Qiu, Appl. Catal., A, 2010, 382, 131 CrossRef CAS.
- B.-Z. Zhan, M. A. White, T.-K. Sham, J. A. Pinock, R. J. Doucet, K. V. R. Rao, K. N. Robertsin and T. S. Camerson, J. Am. Chem. Soc, 2003, 125, 125 Search PubMed.
- M. L. Kantam, U. Pal, B. Sreedhar, S. Bhargava, Y. Iwasawa, M. Tada and B. M. Choudary, Adv. Synth. Catal., 2008, 350, 1225 CrossRef CAS.
- A. Abad, P. Concepcion, A. Corma and H. García, Angew. Chem., Int. Ed., 2005, 44, 4066 CrossRef CAS.
- L. Prati and F. Porta, Appl. Catal., A, 2005, 291, 199 CrossRef CAS.
- C. Della Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077 RSC.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362 CrossRef CAS.
- N. Dimitratos, A. Villa, D. Wang, F. Porta, D. Su and L. Prati, J. Catal., 2006, 244, 113 CrossRef CAS.
- D. Wang, A. Villa, F. Porta, L. Prati and D. Su, J. Phys. Chem. C, 2008, 112, 8617 CAS.
- H. Okamoto and T. B. Massalski, Bull. Alloy Phase Diagrams, 1984, 5, 388 CrossRef CAS.
- S. Galvagno, J. Schwank, G. Parravano, F. Garabassi, A. Marzi and G. R. Tauszik, J. Catal., 1981, 69, 283 CrossRef CAS.
- A. G. Shastri and J. Schwank, J. Catal., 1985, 95, 271 CrossRef CAS.
- T. Sreethawong, D. Sukjit, P. Ouraipryvan, J. W. Scwank and S. Chavadej, Catal. Lett., 2010, 138, 160 CrossRef CAS.
- A. Venugopal, J. Aluha, D. Mogano and M. S. Scurrell, Appl. Catal., A, 2003, 245, 149 CrossRef CAS.
- F.-W. Chang, L. S. Roselin and T.-C. Ou, Appl. Catal., A, 2008, 334, 147 CrossRef CAS.
- E. P. Maris, W. C. Ketchie, M. Murayama and R. J. Davis, J. Catal., 2007, 251, 281 CrossRef CAS.
- G. J. Hutchings, S. Carrettin, P. Landon, J. Edwards, D. Enache, D. Knight, Y.-J. Xu and A. F. Carley, Top. Catal., 2006, 38(4), 223 CrossRef CAS.
- A. Villa, D. Wang, D. S. Su and L. Prati, ChemCatChem, 2009, 1, 510 CrossRef CAS.
- J. Yang, J. Y. Li and H.-P. Too, Anal. Chim. Acta, 2005, 537, 279 CrossRef CAS.
- D. Wang, A. Villa, F. Porta, D. Su and L. Prati, Chem. Commun., 2006, 1956 RSC.
- A. Villa, D. Wang, D. Su, G. M. Veith and L. Prati, Phys. Chem. Chem. Phys., 2010, 12, 2183 RSC.
- D. Wang, A. Villa, P. Spontoni, D. S. Su and L. Prati, Chem.–Eur. J., 2010, 16, 10007 CrossRef CAS.
- F. Porta and L. Prati, J. Catal., 2004, 224, 397 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |