Methanol-to-hydrocarbon chemistry: The carbon pool (r)evolution
Jeffery L.
White
Department of Chemistry, Oklahoma State University, Stillwater, OK 74078, USA
First published on 19th August 2011
Abstract
Mechanistic investigations related to the conversion of methanol, a single-carbon molecule, to higher hydrocarbons ranging from olefins to fuels, have arguably been one of the most active and controversial areas in heterogeneous catalysis research during the last thirty years. In this Perspectives article, we review the evolution of the discovery of the carbon pool mechanism, which in total proved to be the pivotal mechanistic discovery related to the catalytic conversion of methanol to hydrocarbonsvia solid acid catalysts. In addition to acknowledging the now well-known open literature on the topic, a unique aspect of this article is the inclusion of discoveries described in the patent literature that may be less familiar to practicing scientists in the field, but which contain some of the first discoveries in the carbon-pool area and capture their context relative to the practice of methanol-to-hydrocarbon chemistries. Finally, we discuss the larger implications of the carbon pool mechanism to catalysis in general, and reflect on the current state of methanol-to-hydrocarbon (MTH) commercialization and application worldwide.
Dr Jeffery L. White is currently a Professor of Chemistry at Oklahoma State University in Stillwater, OK. Previously, he was an Assistant and Associate Professor of Chemistry at North Carolina State University in Raleigh, NC. His professional experience in the field of heterogeneous catalysis began as a researcher and group leader in Exxon Chemical's (and later ExxonMobil's) methanol-to-olefins (MTO) project during the late 1990's. During this period, Dr White and his colleague Dr Teng Xu discovered that the structure of the carbon pool was methylated aromatics, resulting in a novel composition of matter patent that for the first time defined a working MTO catalyst as a composite organic/inorganic hybrid composed of methylaromatics proximate to solid acid sites. |
Introduction
Growing worldwide concerns about rising energy consumption fuel a broad and continuously evolving scientific basis for discovery and utilization of hydrocarbons. Obviously this is not a new situation, and it is precisely within this same context that the methanol-to-hydrocarbon (MTH) story began, albeit 35 years prior and primarily for the purpose of making synthetic gasoline.1,2 Advances in natural gas recovery from new and existing (e.g., stranded gas) reservoirs using hydrofracturing3 processes ensure future methanol feedstock supplies, but current interest stems primarily from renewed coal gasification efforts. The heterogeneous catalysis community has demonstrated that methanol is a viable starting point for making an almost infinite array of monomers, polymers, and fuels, as well as feedstocks for fine chemicals. In addition, recent reviews have summarized methanol's advantages as a primary energy carrier and storage option.4–6 Doubtless, methanol's importance in this context will continue to increase, as natural gas to methanol becomes more attractive with increasing natural gas supplies. Current research directions surrounding methanol include (a) increased efficiency/lower cost/lower temperature catalytic routes to methanol synthesis; (b) utilization of carbon dioxide as a reagent in catalytic methanol synthesis; (c) increasing catalyst lifetimes in methanol-to-hydrocarbon conversions; (d) new catalysts for methanol-to-hydrocarbon chemistry that enable tailored selectivities to desired products; (e) methanol in fuel cells.In this Perspectives article, we will discuss the development of the discovery of the key mechanistic step in the methanol-to-hydrocarbon reaction, known as the hydrocarbon pool or carbon pool mechanism. This mechanism transformed our understanding of how methanol actually forms higher hydrocarbons in solid acid catalysts. In addition, the carbon pool mechanism fundamentally redefined the way we view what makes solid acid catalysts actually function as such, which has implications extending well beyond methanol-based chemistries. Finally, the discovery of hydrocarbon pool mechanism, and follow-up work showing its relationship to a variety of catalyst topologies, has provided as clear a working example of transition-state shape selectivity as any reaction in heterogeneous catalysis, and its efficacy might well be summed up by the well-known phrase “location, location, location”. While recent reviews have been published in the MTH area,7–9 many of the original research contributions to the carbon pool mechanism that were reported in the patent literature (many of which predate open literature publications) are less familiar to the catalysis community. Indeed, the overall field of methanol-to-hydrocarbons is deeply rooted in industrial research and practice, with Mobil and UOP being strong early participants in catalyst and process design. Given the historically large amount of activity in the MTH area by researchers and practitioners, the community will benefit from a more integrated view of the open literature and the patent literature. The current increases in worldwide natural gas production through unconventional exploration and production methods indicate that inexpensive methanol may be here to stay, which in turn suggests that large-scale global MTH production may finally be a realized.
Questions and answers
Even though thirty years of targeted research and countless publications had been devoted to MTH work, two key mechanistic questions recently remained unanswered. Firstly, how does methanol, a reagent containing a single carbon atom, form carbon–carbon bonds? In a key 1999 review of the subject, approximately eight to ten different mechanisms were included as possible C–C bond formation routes from methanol.10 Secondly, why does the MTH reaction exhibit an induction period, i.e., an initial time on catalyst where no conversion takes place? As an example, we show a representative result in Fig. 1, taken from our laboratory in the late 1990's, in which a pulsed microreactor is run under conditions of low total conversion (to minimize secondary reactions), and where methanol is introduced into a flowing helium stream that passes through a fixed bed of acidic SAPO-34 catalyst. Clearly, the reaction in SAPO-34 does not begin to occur with any appreciable conversion until the catalyst has been exposed to a significant amount of the total methanol. Although product distributions vary across different catalyst types, an extensive body of literature exists with plots exhibiting an induction time, and involving data collected from a large variety of microcrystalline solid acids, including most notably HZSM-5 but many other small, medium, and large pore zeolites.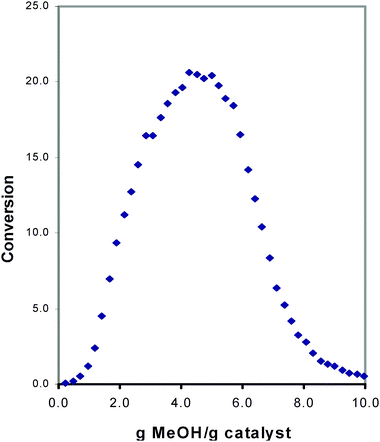 | ||
| Fig. 1 Representative plot showing conversion of methanolversus the amount of methanol contacted with the solid acid catalyst H-SAPO-34. Each data point was generated using a pulsed microreactor with computer-controlled injections of CH3OH into a flowing He stream passing through a tubular fixed bed reactor. | ||
It proves convenient to discuss the resolution to each key question chronologically, i.e., in the order they were discovered. Interestingly, the first question related to initial C–C bond formation, which had received the most emphasis in the literature and which one might presume must be answered in order to understand the induction time mystery, was not the first question experimentally answered. A key factor enabling solution to these mechanistic questions was the growing interest during the early 1990's in UOP's eight-membered ring SAPO-34 catalysts, which were structurally analogous to the chabazite zeolites. As such, product distributions from methanol were much simpler (favoring ethylene and propylene, and formally giving rise to the MTO or Methanol-to-Olefins process) than the traditional ten-membered ring ZSM-5 catalyst. More importantly, SAPO-34 had large cages but small cage openings (0.38–0.40 nm), thereby trapping species that would diffuse out of the larger channel structure of ZSM-5. The first systematic experiments that demonstrated these “trapped” species in SAPO-34 cages were integral to the reaction came from the Kolboe group in the mid 1990's.11–13 In this important set of initial experiments, Dahl and Kolboe used isotopically labeled reagents (a mix of 13C-labeled methanol and unlabeled ethene or propene) to propose a parallel reaction route, which they termed the “carbon pool” mechanism. Experimental results were not consistent with proposed mechanisms for direct methylation of olefins, which was a key experimental finding at that time, given that ethylene was a natural assumption for the first multiple-carbon species formed. Fig. 2 shows the general form of the mechanism proposed by Dahl and Kolboe, even though the exact structure of the (CH2)n “pool” species was not known, in which the adsorbed “pool” reacts with methanol to generate products. As Fig. 2 indicates, the chief products in SAPO-34 were ethylene, propylene, and butene in contrast to the larger hydrocarbons typically produced in methanol/H-ZSM-5 reactions.
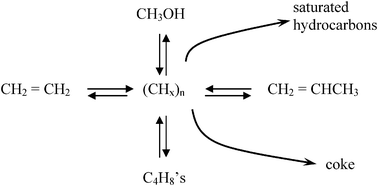 | ||
| Fig. 2 Representation of the carbon pool parallel mechanism originally proposed by Kolboe. (adapted from ref. 12). | ||
At that time, several chemical structures had been discussed in the literature that could represent the carbon pool, including various alkoxy species (which presumably underwent sequential methylations to facilitate chain growth)14 and stable substituted-carbenium ions.15 However, the ExxonMobil group of Xu and White were the first to unambiguously determine the structure of the carbon pool that was active in the MTO reaction in HSAPO-34, the first methanol catalyst system that was fully understood mechanistically. Their work, which began in 1998, culminated in the filing of two patents in early 2000 detailing for the first time that the hydrocarbon pool was methyl-aromatic structures (e.g.toluene, xylenes, methylnaphthalenes, etc.).16,17Solid-state NMR was the key experimental method used to elucidate the carbon pool chemical structure, supported by a variety of isotopic label exchange microreactor experiments and specific molecular synthesis methods. In the first set of results, experiments in which CH3OH was reacted with acidic HSAPO-34 were stopped at various early points along the conversion curve of Fig. 1, and the solid-state NMR spectra were acquired for catalyst aliquots at those specific points without exposure to atmosphere. Fig. 3 shows representative examples of results obtained in this way by White and Xu, in which the time evolution of several hydrocarbons species proceeds to selectively produce methylaromatics as the primary occluded species in the SAPO-34 cages, as particularly evident in the middle Fig. 3spectrum.
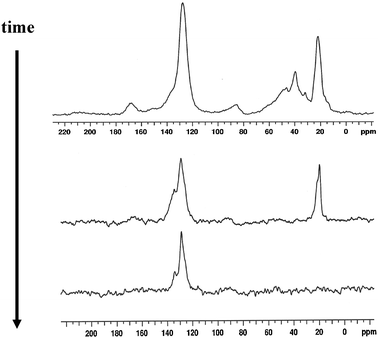 | ||
| Fig. 3 Representative solid-state 125 MHz 13C Bloch decay MAS NMR spectra of trapped organic species following various reaction and He flow times (top = short, bottom = long), showing the cumulative and selective buildup of methylaromatic species indicated by the methyl signal at 19 ppm, the protonated aromatic signal at 128 ppm, and the nonprotonated aromatic signal at 135 ppm. The reactor temperature was 400 °C for this data, but similar results were obtained for temperatures ranging from 250–450 °C. (spectra taken from original U. S. patent filing February 24, 2000; ref. 16) | ||
Based on the findings shown in Fig. 3, Xu and White systematically proved that methylaromatic structures are the reactive carbon pool by synthesizing fully 13C-labeled 1,3,5-trimethylbenzenevia the aldol condensation of triply-labeled acetone-13C inside H-SAPO-34. In this case, each SAPO-34 cage had an active Brønsted acid site in close proximity to the trimethylbenzene molecule; one of the original solid-state 13C NMR results taken from the original patent (with clarifying text in the caption) is shown below in Fig. 4.16,17 Note in particular the similarity in the overall distribution of signal intensities to that shown above in the middle spectrum of Fig. 3.
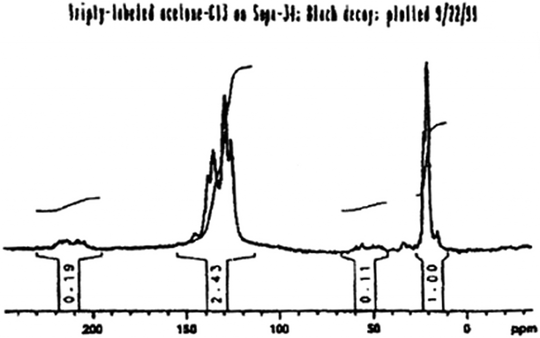 | ||
| Fig. 4 Original solid-state 125 MHz 13C Bloch decay MAS NMR spectrum, taken directly from the initial patent,15 of an acidic SAPO-34 catalyst pre-reacted with triply-labeled acetone-13C to produce one molecule of completely labeled 1,3,5-trimethylbenzene per cage16,17 The spectrum was obtained under quantitative conditions; the caption above the spectrum reads “triply-labeled acetone-C13 on Sapo-34; Bloch decay; plotted 9/22/99”. | ||
Using the catalyst from Fig. 4, and identical catalysts prepared with unlabeled methylaromatic pool molecules, subsequent reactor and NMR experiments showed that the induction period was completely eliminated, and selectivity to ethylene and propylene was increased as shown below in Fig. 5 and originally described in the early patents.16,17
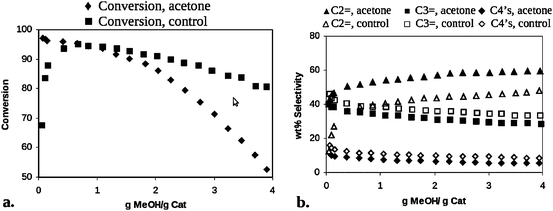 | ||
| Fig. 5 Reactor data demonstrating improved (a) conversion and elimination of induction period, and (b) ethylene (C2=) selectivity of HSAPO-34 catalyst containing hydrocarbon pool (methylaromatics) molecules in each cage formed via a specific synthetic route. | ||
The work from White and Xu unequivocally defined the chemical structure of the hydrocarbon pool and showed that it was an active participant in the reaction, proved that Dahl and Kolboe's parallel “carbon pool” mechanism was correct, and most importantly, showed for the first time that the true composition of a working solid acid catalyst in MTH chemistry is a hybrid organic/inorganic structure, as captured in the composition of matter patent.16 An active hydrocarbon pool molecule could be occluded benzene, toluene, naphthalene, or methyl derivatives thereof (e.g.xylenes, trimethylbenzene, etc.), as long as the molecule was proximate to a Brønsted acid site. Such a view of catalyst chemistry stands in marked contrast to long-held beliefs concerning unreactive and deactivating “coke” molecules, while at the same time clarifying the descriptions of “good coke” and “bad coke” often mentioned in the catalytic literature18 and which were anecdotally pervasive at that time. Label exchange NMR experiments indicate that label scrambling occurs between unlabeled methanol and labeled methylbenzenes, and vice versa. More specifically, solid-state NMR experiments showed that completely-13C labeled carbon pool molecules prepared in SAPO-34 cages (prior to methanol introduction) as described above would react with unlabeled CH3OH, and both aromatic carbons as well as aliphatic carbons from the carbon pool were involved, indicating a ring expansion/contraction/paring reaction step19 that was subsequently discussed in the open literature.20 The key factor that allowed the ExxonMobil group to unambiguously identify the structure of the carbon pool stemmed directly from the fact that the eight-membered ring structure of the HSAPO-34 catalyst cannot allow benzene rings to either enter or leave the reactive cages, in marked contrast to ZSM-5, thereby preserving the structure for parallel NMR studies. Indeed, examination of earlier literature and patents by Mole and coworkers showed that improvements in conversion and changes in selectivity were possible when benzene “promoter” was coadsorbed with methanol in HZSM-5.21–23 Even though these results were early hints to the carbon pool's structure, mechanistic details were difficult to ascertain in that work since the aromatic rings easily escaped the ten-membered ring channels of the ZSM-5 catalyst structure. Indeed, the relationship between the critical diameter of various aromatic molecules that can act as the carbon pool and the pore or channel diameter of the molecular sieve catalyst defines whether a persistent “carbon pool” exists versus transient promoters.24–26 Further, the time dependence of the exact structure of the carbon pool in cage-containing versuschannel-containing catalysts reflects the steric constraints for aromatic side-chain alkylation and growth, which in turn governs overall selectivity to product molecules formed in the dealkylation steps.27,28 That the active carbon pool mechanism is general to all catalyst topologies has recently been demonstrated via a comparative study of several different large-pore catalyst structures.29
Subsequently, and independently, extensive NMR, gas chromatograph, and reactor experiments published by Haw and coworkers in the open literature supported the ExxonMobil findings, and in addition, clarified the long-standing question of how carbon–carbon bonds, and necessarily the hydrocarbon pool, initially formed.27,30–32Fig. 6 shows a result from Song, Fu, and Haw in which the time-dependent concentration of methyl groups in the methylbenzene pool structure is quantified versus time at reaction temperature in flowing helium,31 in agreement with the earlier patent data shown previously in Fig. 3.
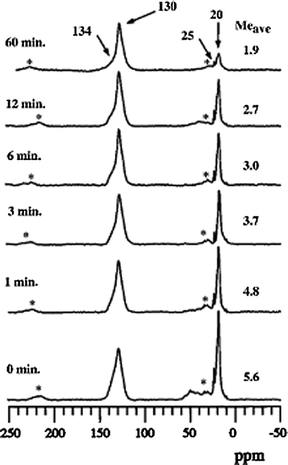 | ||
| Fig. 6 90 MHz 13C CP/MAS NMR spectra showing the loss of methyl groups as a function of time from methylbenzenes trapped in HSAPO-34 at 400 °C (reproduced from ref. 31). | ||
While significant efforts were devoted to cryogenic pulse-quench/NMR experiments and other in situNMR methods (using HZSM-5 as a catalyst), results from these studies did not identify methylbenzenes as the key reaction centers.33 However, in a most important and surprising result, Haw and coworkers showed for the first time that the initial carbon–carbon bonds do not come from methanol directly, but rather from trace impurities (aldehydes, ketones, higher alcohols, etc.) which are extremely difficult to eliminate from either the methanol or catalyst source.32 Based on the extremely careful and methodical experiments by the Haw group involving methanol reagent/catalyst purity, alternative routes to creation of the hydrocarbon pool, e.g., reactive surface methoxy groups as direct methylation sources (as implicated in some continuous and stopped flow NMR experiments) must be excluded as relevant to MTO/MTG, even though multiple side reactions take place following carbon pool formation.34–36 What was clear from the ExxonMobil results is that the methylated aromatic hydrocarbon pool molecules must be in the same SAPO-34 cage as an active Brønsted proton to allow the concerted side-chain alkylation/dealkylation mechanism to occur, and as demonstrated by Haw and coworkers, that those carbon pool molecules are not created via bond formation reactions involving methanol directly but are slowly built up based on reactions of carbon–carbon bond containing impurities in the methanol.
Outlook and future implications
Interest in all aspects of MTH chemistry, ranging from fundamental catalytic science to large-scale process development, has increased during the last decade37–39 and should continue to do so owing to the extremely high flexibility of MTH chemistry as a means to upgrade any gasifiable source of carbon, including coal and biomass. Commercialization of MTH and its various derivatives is underway in China and other developing economies; the Lurgi MTP® process is one strategy that has been used as well as the DMTO (Dalian Institute of Chemical Physics MTO) process.40–42 Further, China is aggressively pursuing coal-to-chemicals/fuelsvia syn gas and methanol intermediates, i.e., the coupling of coal gasification and MTH processes.43,44 Even though the discovery of the carbon pool mechanism occurred primarily in the context of MTO (methanol-to-olefin) chemistry using SAPO-34 catalysts, as mentioned earlier we now know it is general to all MTH processes, e.g.MTG or MTP (methanol to propene, which is MTO with increased propene selectivity), as long as the carbon pool exists in a catalyst that does not sterically restrict formation of the necessary transition states. By virtue of the radically different mechanistic understanding afforded by the discovery of the carbon pool, improvements in catalyst selectivities have already been realized, as well as clarification of alternative methanol reaction pathways in catalysts whose critical internal volumes are too small to allow the bulky carbon-pool mechanism to occur.45 A logical extension of the known transition-state shape selective details surrounding the carbon pool mechanism suggests that designing structure directing templates that mimic the critical dimensions of the desired form of the carbon pool (e.g., templates similar to monosubstituted single-ring aromatics versus multisubstituted single-ring aromatics versus substituted multiring aromatics, etc.) for catalyst synthesis might result in catalysts with selectivity-optimized topologies for very specific and narrow product distributions. Increasing trends toward total carbon management require optimized selectivities for reduction of unwanted products and greenhouse gases. Further, necessary improvements to catalyst lifetimes, long the weakest link in the MTH technology and especially problematic in MTO/SAPO-34 processes, may be achieved by reducing potential deactivation pathways through the design of catalysts that maintain the nascent form of the active carbon pool. Heretofore this idea has been approached via post-synthetic modifications of known catalyst structures to reduce the space available for longer alkyl branches to form on the host aromatic ring, thereby eliminating the cyclization and dehydrogenation steps necessary for multiring aromatic formation.46 Finally, we note that induction periods are not unique to MTH reactions; alkylation reactions involving olefinic and paraffinic reagents carried out in solid acids also exhibit induction periods prior to the onset of appreciable conversion, as reported by many different investigators using a variety of experimental approaches. The degree to which adsorbed “pre-requisite” structures, i.e. a carbon pool, must exist within the zeolite and within some minimum distance of the active sites prior to appreciable conversion is still unclear.MTH science, technology, and applications continue to benefit from their parallel evolution in both the academic and industrial arenas.47,48 However, the overall impact of new insights generated based on the “carbon-pool revolution” extend far beyond the MTH problem, as it fundamentally redefines the nature of the active site in many solid-acid catalyzed reactions. Re-evaluating the complex diversity of possible reactions in organic transformations via solid acids in view of how active hydrocarbon species contribute to overall reaction mechanisms and the nature of catalyst activity/kinetics should yield new and valuable information for other heterogeneous catalysis chemistries. The discovery of the carbon pool mechanism exemplifies the significance of fundamental understanding of catalytic reaction mechanisms, continues to generate a new perspective on total carbon management in catalytic processes, and demonstrates the cumulative benefits arising from different approaches by multiple investigators to the same problem. This latter point is best revealed by a critical examination of both the open and patent literatures. Indeed, the breadth of length scales involved for MTH chemistry ranges from the truly microscopic (size of atoms) to the mesoscopic (catalyst particle morphology) to the macroscopic (reactor design) to the megascopic (working industrial plants). In practice even the most elegant chemistries must be economically viable, and it now appears that coal-based (and potentially natural gas depending upon economics) MTH processes play central roles in future chemicals and fuels production, particularly with continuing improvements in catalyst lifetimes and total catalyst costs.
References
- C. D. Chang and A. J. Silvestri, J. Catal., 1977, 47, 249 CrossRef CAS
.
-
J. C. W. Kuo, U. S. Patent 3,931,349, Mobil Corporation, 1976 Search PubMed
- R. A. Kerr, Science, 2010, 328, 1624 CrossRef CAS
- G. A. Olah, Chem. Eng. News, 2003, 81, 42 CAS
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636 CrossRef CAS
-
G. A. Olah, A. Goeppert and G. K. Surya Prakash, Beyond Oil and Gas: The Methanol Economy, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed
- U. Olsbye, M. Bjorgen, S. Svelle, K. Lillerud and S. Kolboe, Catal. Today, 2005, 106, 108 CrossRef CAS
- J. F. Haw and D. M. Marcus, Top. Catal., 2005, 34, 41 CrossRef CAS
- M. Hunger, Microporous Mesoporous Mater., 2005, 82, 241 CrossRef CAS
- M. Stocker, Microporous Mesoporous Mater., 1999, 29, 3 CrossRef CAS
- I. Dahl and S. Kolboe, Catal. Lett., 1993, 20, 329 CrossRef CAS
- I. Dahl and S. Kolboe, J. Catal., 1994, 149, 458 CrossRef CAS
- I. Dahl and S. Kolboe, J. Catal., 1996, 161, 304 CrossRef CAS
- T. R. Forester and R. F. Howe, J. Am. Chem. Soc., 1987, 109, 5076 CrossRef CAS
- J. F. Haw, J. Nicholas, T. Xu, L. Beck and D. Ferguson, Acc. Chem. Res., 1996, 29, 259 CrossRef CAS
-
T. Xu and J. L. White, U. S. Patent 6,734,330, 2004, priority filing and PCT published February 2000 Search PubMed
-
T. Xu and J. L. White, U. S. Patent 6,743,747, 2004, priority filing and PCT published February 2000 Search PubMed
-
Handbook of Zeolite Science and Technology, ed. S. Auerbach, K. Carrado and P. Dukka, Marcel Dekker, New York, NY, 2003 Search PubMed
- R. Sullivan, C. Egan, G. Langlois and R. Sieg, J. Am. Chem. Soc., 1961, 83, 1156 CrossRef CAS
- M. Bjørgen, U. Olsbye, D. Petersen and S. Kolboe, J. Catal., 2004, 221(1), 1 CrossRef
- T. Mole, J. A. Whiteside and D. Seddon, J. Catal., 1983, 82, 261 CrossRef CAS
- T. Mole, G. Bett and D. Seddon, J. Catal., 1983, 84, 435 CrossRef CAS
-
D. Seddon, T. Mole and J. Whiteside, U. S. Patent 4,499,314, 1985 Search PubMed
-
S. Kaiser, U. S. Patent 4,677,242, 1987 Search PubMed
-
S. Brown, L. Green, M. Mathias, D. Olson, R. Ware and W. Weber, U. S. Patent 6,046,372, 2000, filing date April 1998 Search PubMed
-
T. Xu, N. Coute, K. Clem and K. Kuechler, U. S. Patent 7,057,083, 2006, filing date November, 2003 Search PubMed
- J. F. Haw, W. Song, D. M. Marcus and J. B. Nicholas, Acc. Chem. Res., 2003, 36, 317 CrossRef CAS
- Z. Cui, Q. Liu, Wei-Guo Song and L. Wan, Angew. Chem., Int. Ed., 2006, 45, 6512 CrossRef CAS
- M. Bjørgen, S. Akyalcin, U. Olsbye, S. Benard, S. Kolboe and S. Svelle, J. Catal., 2010, 275, 170 CrossRef
- W. Song, J. F. Haw, J. B. Nicholas and C. S. Heneghan, J. Am. Chem. Soc., 2000, 122, 10726 CrossRef CAS
- W. Song, H. Fu and J. F. Haw, J. Am. Chem. Soc., 2001, 123, 4749 CrossRef CAS
- W. Song, D. M. Marcus, H. Fu, J. O. Ehresmann and J. F. Haw, J. Am. Chem. Soc., 2002, 124, 3844 CrossRef CAS
- P. W. Goguen, T. Xu, D. H. Barich, T. W. Skloss, W. Song, Z. Wang, J. B. Nicholas and J. F. Haw, J. Am. Chem. Soc., 1998, 120, 2650 CrossRef CAS
- W. Wang, Y. Jiang and M. Hunger, Catal. Today, 2006, 113, 102 CrossRef CAS
- Y. Jiang, M. Hunger and W. Wang, J. Am. Chem. Soc., 2006, 128, 11679 CrossRef CAS
- W. Wang, A. Buchholz, M. Seiler and M. Hunger, J. Am. Chem. Soc., 2003, 125, 15260 CrossRef CAS
-
K. Kuechler, M. J. G. Jansenn, S. Vaughn, S. Fung, N. Coute and J. L. White, U. S. Patent 6,437,208, 2002, filing date January 2000 Search PubMed
-
A. Bozzano, S. Bradley, R. Castillo and J. Chen, U. S. Patent, 7,763,766, 2010, filing date December 2005 Search PubMed
-
S. Zones and A. Burton, U. S. Patent, 7,750,196, 2010, filing date June 2007 Search PubMed
-
G. Belussi and P. Pollesol, Stud. Surf. Sci. Catal., ed. J. Cejka, N. Zilkova and P. Nachtigall, Elsevier, Amsterdam, 2005, vol. 158, p. 1201 Search PubMed
- Chem. Eng. News, 2007, 85, 37.
-
G. Burke, H. Koempel, W. Liebner and H. Bach, U. S. Patent, 7,923,591, 2011, filing date October 27, 2005 Search PubMed
- G. Ondrey, Chemical Engineering, 2011, 118, 16 Search PubMed
- Methanol-to-Olefins unit starts up in China, Oil & Gas Journal, 108, 2010, September 30.
- Z. Cui, Q. Liu, S. Bain, Z. Ma and Wei-Guo Song, J. Catal., 2008, 258 Search PubMed
-
K. Wang, G. Cao, M. Brennan, R. Hall and K. Strohmaier, U S. Patent 7,119,242, 2006, filing date June 1, 2004 Search PubMed
-
M. Stocker, Zeolites and Catalysis, ed. J. Cejka, A. Corma and S. Zones, Wiley-VCH, Weinheim, 2010, vol. 2, p. 687 Search PubMed
-
S. Kvisle, T. Fuglerud, S. Kolboe, U. Olsbye, K. Lillerud and B. Vora, Handbook of Heterogeneous Catalysis, ed. E. Ertl, H. Knozinger, F. Schuth and J. Weitkamp, Wiley-VCH, Weinheim, 2nd edn, 2008, p. 2950 Search PubMed
| This journal is © The Royal Society of Chemistry 2011 |