Conjugate addition of arylboronic acids to α,β-unsaturated carbonyl compounds in aqueous medium using Pd(II) complexes with dihydroxy-2,2′-bipyridine ligands: homogeneous or heterogeneous nano-catalysis?†
Eder
Tomás-Mendivil
,
Josefina
Díez
and
Victorio
Cadierno
*
Departamento de Química Orgánica e Inorgánica, IUQOEM, University of Oviedo, E-33006 Oviedo, Spain. E-mail: vcm@uniovi.es; Fax: (+34)985103446; Tel: (+34)985103453
First published on 22nd August 2011
Abstract
The conjugate addition of arylboronic acids to α,β-unsaturated carbonyl compounds in water under air has been studied using a series of palladium(II) derivatives containing symmetrically disubstituted-2,2′-bipyridine ligands. Among them, best results were obtained with complex [PdCl2{3,3′-(OH)2-2,2′-bipy}] (2a), which selectively provides the desired addition products in good to high yields under mild conditions. Catalytically active palladium nanoparticles are formed in the course of the reactions. In the presence of sodium dodecyl sulfate (SDS) these nanoparticles can be stabilized and recycled.
Introduction
The conjugate 1,4-addition of organometallic compounds to electron-deficient olefins represents one of the most powerful tools presently available to create new C–C bonds.1 In this context, since the pioneering work of Miyaura and co-workers in 1997,2 metal-catalyzed conjugated addition of arylboronic acids to α,β-unsaturated carbonyl compounds has attracted considerable attention because of the higher stability of these reagents towards air and moisture, as well as their improved functional group tolerance, as compared to the more classical organocopper, -zinc, -zirconium, -aluminum and -mercury species (Scheme 1).3 In fact, the maturity gained by this reaction in such a short period of time is such that, its asymmetric version, is nowadays considered as the best method for the enantioselective introduction of aryl and alkenyl groups at β-position of these electron-deficient olefins.3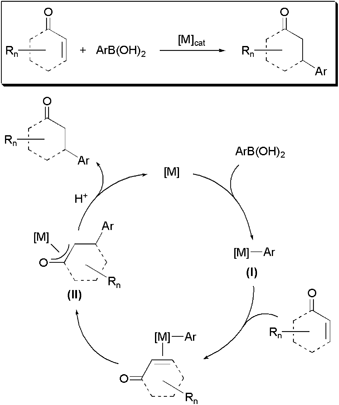 | ||
| Scheme 1 The catalytic 1,4-conjugate addition of arylboronic acids to α,β-unsaturated carbonyl compounds. | ||
From a mechanistic point of view (Scheme 1),3 these catalytic transformations involve the initial transmetallation of the aryl group from the arylboronic acid to the metal, followed by the insertion of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the enone (or enal) into the metal-carbon bond of intermediate I, to give a π-oxa-allyl complex II. Final protonolysis of II liberates the corresponding conjugated addition product with regeneration of the catalytically active species. To date, these catalytic reactions have been dominated by Rh(I)-based complexes.2,3Palladium species are also known to promote the conjugate addition of arylboronic acids to α,β-unsaturated carbonyl compounds.4 However, despite the lower cost associated with this metal, palladium-based catalysts remain comparatively much less studied due to their propensity to promote the competitive formation of Heck-type coupling products via β-hydride elimination pathways (Scheme 2).4,5
C bond of the enone (or enal) into the metal-carbon bond of intermediate I, to give a π-oxa-allyl complex II. Final protonolysis of II liberates the corresponding conjugated addition product with regeneration of the catalytically active species. To date, these catalytic reactions have been dominated by Rh(I)-based complexes.2,3Palladium species are also known to promote the conjugate addition of arylboronic acids to α,β-unsaturated carbonyl compounds.4 However, despite the lower cost associated with this metal, palladium-based catalysts remain comparatively much less studied due to their propensity to promote the competitive formation of Heck-type coupling products via β-hydride elimination pathways (Scheme 2).4,5
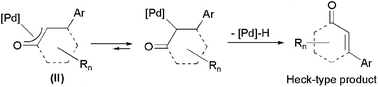 | ||
| Scheme 2 The competitive β-hydride elimination in intermediate II. | ||
Among the different Pd-based systems able to catalyze the conjugate addition of arylboronic acids to α,β-unsaturated carbonyl compounds selectively,6Pd(OAc)2/bipy7 and the dimeric complex [{Pd(μ-OH)(bipy)}2][BF4]28 merit to be highlighted due to their outstanding activity under mild conditions. Apparently, the presence of the π-acceptor 2,2′-bipyridine ligand (bipy) coordinated to the active Pd(II) center inhibits the β-hydride elimination and promotes the protonolysis of intermediate II.9
Remarkably, in the presence of appropriate surfactants, the addition reactions promoted by Pd(OAc)2/bipy could also be conveniently performed in aqueous media.7b This fact represents a significant advance towards Green Chemistry, since one of its principles is to circumvent the use of hazardous organic solvents as they are responsible for a large part of the waste generated by the chemical processes.10 In this sense, water is one of the most appealing candidates to replace organic solvents due its abundance, low-cost, and non-toxic and safe nature.11 Indeed, the use of water as solvent in catalytic and stoichiometric organic synthesis has spread throughout the chemical community at a staggering pace during the last two decades.12
With all these precedents in mind, and as part of our current research work dealing with the development of catalytic transformations in environmentally friendly aqueous media,13 we have investigated the conjugated addition of arylboronic acids to α,β-unsaturated carbonyl compounds in water using well-defined palladium(II) complexes containing symmetrically disubstituted-2,2′-bipyridine ligands functionalized with hydrophilic groups. Our goal was to find palladium catalysts able to promote these addition processes in aqueous medium without the aid of surfactants.14 As the reader will see, in the course of these studies we have discovered a new example of heterogeneous nanocatalysis involving metallic nanoparticles.
Results and discussion
Introduction of hydrophilic ligands in the coordination sphere of a transition-metal is probably the most popular method for the preparation of water-soluble catalysts.15 In this context, although scarcely employed in homogeneous catalysis, a large number of 2,2′-bipyridine ligands substituted with hydrophilic groups are known.16 In particular, our initial efforts focused on the use of the symmetrically substituted dihydroxy-2,2′-bipyridines 1a–d due to their easy access from commercially available starting materials.17 We also believed that the ability of the OH groups to establish hydrogen bonds with water would help the solubilization of the resulting complexes in this medium. Thus, by reacting an stoichiometric amount of these dihydroxylated ligands with PdCl2 in refluxing acetone, we prepared the new complexes 2a–d, which were isolated as air-stable yellow or brown solids in 50–94% yield (Scheme 3). Alternatively, compounds 2a–d can be generated in similar yields starting from the soluble precursor [PdCl2(COD)] (COD = 1,5-cyclooctadiene). In both cases, 2a–d directly precipate in the reaction media.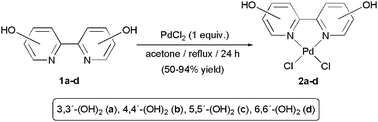 | ||
| Scheme 3 Preparation of complexes 2a–d. | ||
Surprisingly, despite the presence of OH groups in their structure, complexes 2a–d were found to be insoluble in water at room temperature (only in the case of 2a a solubility of 0.5 mg cm−3 could be measured).18 However, we must note that at high temperature (80 °C), or under strongly basic conditions (pH 14),19 we were able to prepare aqueous solutions of these compounds of up to 10 mg cm−3. Characterization of complexes 2a–d was straightforward following their analytical and spectroscopic data, with the 1H and 13C{1H} NMR spectra recorded in DMSO-d6 showing the expected signals for the protons and carbons of the 2,2′-bipyridyl skeletons (details are given in the Experimental Section).20 The presence of the OH units was confirmed by the appearance of a strong absorption band at ca. 3400 cm−1 in the IR spectra of these compounds.
To prove the catalytic potential of complexes 2a–d, we investigated the conjugate addition of phenylboronic acid (4a) to 2-cyclohexenone (3a) as model reaction. Initial exploratory experiments were performed at 80 °C in pure aqueous medium under air, using a 3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4a molar ratio of 1:3 and a palladium loading of 5 mol%. As shown in Table 1, under these conditions, all the complexes synthesized were able to provide the desired 3-phenylcyclohexanone (5aa) as the major reaction product along with variable amounts of the Heck-type product 6aa and cyclohexanone, the latter resulting from the reduction of the CC bond of 3a (entries 1–4).21 However, marked differences in activity and selectivity were observed depending on the substitution pattern of the 2,2′-bipyridine ligand attached to palladium. Thus, higher activities were observed with complexes 2a and 2c, substituted with the OH groups in 3,3′ and 5,5′ positions, as compared to their 4,4′ and 6,6′-disubstituted counterparts 2b and 2d (entries 1 and 3 vs. 2 and 4). This fact clearly indicates that electronic effects govern the catalytic activity of these complexes, increasing when the electronic density on the metal is lower.22 In particular, the best results were obtained with complex [PdCl2{3,3′-(OH)2-2,2′-bipy}] (2a) which was able to generate selectively the desired ketone 5aa in 98% GC-yield after only 1 h of heating (entry 1). As shown in entry 5, under identical reaction conditions, significantly worse results were obtained using PdCl2 as catalyst, thus confirming the key role played by the dihydroxylated bipy ligand in this catalytic reaction.23 It is also worth of note that no differences in the activity and selectivity of 2a were observed when the catalytic reactions were performed under inert atmosphere (entry 6) or in the presence of an excess of the ligand 1a (entry 7).
:4a molar ratio of 1:3 and a palladium loading of 5 mol%. As shown in Table 1, under these conditions, all the complexes synthesized were able to provide the desired 3-phenylcyclohexanone (5aa) as the major reaction product along with variable amounts of the Heck-type product 6aa and cyclohexanone, the latter resulting from the reduction of the CC bond of 3a (entries 1–4).21 However, marked differences in activity and selectivity were observed depending on the substitution pattern of the 2,2′-bipyridine ligand attached to palladium. Thus, higher activities were observed with complexes 2a and 2c, substituted with the OH groups in 3,3′ and 5,5′ positions, as compared to their 4,4′ and 6,6′-disubstituted counterparts 2b and 2d (entries 1 and 3 vs. 2 and 4). This fact clearly indicates that electronic effects govern the catalytic activity of these complexes, increasing when the electronic density on the metal is lower.22 In particular, the best results were obtained with complex [PdCl2{3,3′-(OH)2-2,2′-bipy}] (2a) which was able to generate selectively the desired ketone 5aa in 98% GC-yield after only 1 h of heating (entry 1). As shown in entry 5, under identical reaction conditions, significantly worse results were obtained using PdCl2 as catalyst, thus confirming the key role played by the dihydroxylated bipy ligand in this catalytic reaction.23 It is also worth of note that no differences in the activity and selectivity of 2a were observed when the catalytic reactions were performed under inert atmosphere (entry 6) or in the presence of an excess of the ligand 1a (entry 7).

| Entry | Catalyst | Time | Conversionb | Yield 5aab | Yield 6aab |
|---|---|---|---|---|---|
| a Reactions performed at 80 °C under air using 0.5 mmol of 3a, 1.5 mmol of 4a, 0.025 mmol of the corresponding Pd(II) complex 2a–d and 1 cm3 of water. Unless otherwise stated, formation of trace amounts of cyclohexanone was in all cases observed (up to 1%). b Determined by GC. c 5% of cyclohexanone was formed. d 14% of cyclohexanone was formed. e 6% of cyclohexanone was formed. f Reaction performed under N2 atmosphere. g Reaction performed in the presence of 15 mol% of 1a. | |||||
| 1 | 2a | 1 h | >99% | 98% | 1% |
| 2 | 2b | 24 h | >99% | 95% | 4% |
| 3c | 2c | 5 h | 99% | 87% | 7% |
| 4d | 2d | 24 h | 35% | 18% | 3% |
| 5e | PdCl2 | 3 h | 51% | 37% | 8% |
| 6f | 2a | 1 h | >99% | 98% | 1% |
| 7g | 2a | 1 h | >99% | 98% | 1% |
A striking feature of all the catalytic reactions listed in Table 1 is the gradual appearance of a black solid suspension. The solid material generated in one of these reactions (entry 1) was isolated by filtration, washed with diethyl ether and acetone, and studied by means of TEM (Transmission Electron Microscopy) and SEM/EDX (Scanning Electron Microscopy/Energy-Dispersive X-ray spectroscopy) techniques. The micrographs obtained (Fig. 1) revealed the formation of aggregates (100–250 nm) of nanoparticles with spherical morphology and a size range of 6–12 nm. SEM/EDX measurements confirmed that the solid was mainly composed of elemental palladium (73.7 wt%).24 The additional presence of carbon (19.2 wt%), oxygen (3.3 wt%) and nitrogen (1.8 wt%) in the sample also evidenced that part of the 3,3′-dihydroxy-2,2′-bipyridine ligand 1a is “retained” by the nanoparticles. At this point, we must note that decomposition of the Pd(II) complex 2a into Pd(0) is closely related to the dissociation of the bipy ligand, since a fluorescence phenomenon was observed when the catalytic reaction mixture was exposed to UV light (Fig. 2). Green light emission when irradiated with 365 nm of ultraviolet light is a hallmark of ligand 1a,17b property that loses when coordinated to a metal center.
 | ||
| Fig. 1 TEM (left) and SEM (right) images of the agglomerated nanoparticles isolated from the catalytic reaction described in entry 1 of Table 1. | ||
 | ||
| Fig. 2 The fluorescence behaviour observed in the presence of UV-light (right; non-irradiated sample on the left). | ||
Formation of palladium nanoparticles starting from 2a–d is rather surprising since in previous studies by Lu and Tsai, using related Pd(II)-bipy systems, such a fact was not evidenced.7,8,14 That's why, in order to determine whether this property is unique of complexes 2a–d, we decided to prepare a series of Pd(II) derivatives with different 2,2′-bipy ligands and explore their behaviour under identical reaction conditions. In particular, in addition to the parent compound [PdCl2(bipy)] (11),25 we synthetized the hydrophilic derivatives 12–14 by refluxing an equimolar mixture of PdCl2 with the appropriate 4,4′-disubstituted-2,2′-bipyridine 8–10 in acetone for 24 h (Scheme 4). These complexes were isolated as air-stable yellow-orange solids in 93–95% yield after a simple filtration of the reaction mixtures since all of them directly precipitate in the medium. Similarly to 2a–d, the dihydroxymethyl-substituted derivative 13 was found to be insoluble in water at room temperature (a solubility of 5 mg cm−3 was observed in basic medium at pH 14).19 In contrast, the Pd(II) complexes 12 and 14, substituted with trimethylammonium and carboxylic acid groups, respectively, dissolve in water at neutral pH (10 and 5 mg cm−3 at 20 °C).
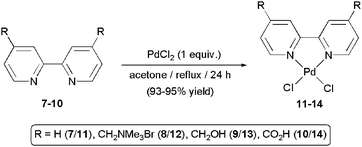 | ||
| Scheme 4 Preparation of complexes 11–14. | ||
Complexes 12–14 were characterized by means of standard spectroscopic techniques (IR and multinuclear NMR) and elemental analyses, all data being fully consistent with the proposed formulations (details are given in the Experimental Section).26 In addition, the structures of 13 and 14 could be unambiguously confirmed by means of X-ray diffraction analyses.‡ X-ray quality crystals were obtained by slow diffusion of methanol into saturated solutions of complexes 13–14 in dimethylsulfoxide. ORTEP views of the molecules are shown in Fig. 3 and 4, respectively (selected bonding parameters are listed in the captions).27 The geometry around the Pd atom is slightly distorted square-planar in both structures, with metal-centered angles between 80.45(10)° and 95.50(8)°. The palladium atom is displaced for only 0.0002(3) (13) and 0.0064(2) (14) Å from the plane defined by the Cl(1), Cl(2), N(1) and N(2) atoms. These data, along with the Pd-N (2.018(3)–2.027(2) Å) and Pd-Cl (2.2752(8)–2.2967(9) Å) bond distances observed, are comparable to those previously reported in the literature for the parent compound [PdCl2(bipy)] (11).28
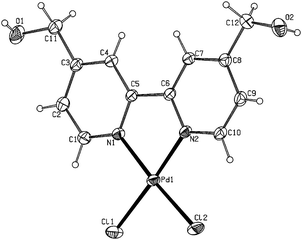 | ||
| Fig. 3 ORTEP-type view of the structure of compound 13 showing the crystallographic labelling scheme. Thermal ellipsoids are drawn at 20% probability level. Selected bond distances (Å): Pd–Cl(1) 2.2967(9); Pd–Cl(2) 2.2869(8); Pd–N(1) 2.022(2); Pd–N(2) 2.018(3); C(11)–O(1) 1.398(5); C(12)–O(2) 1.410(5); Selected bond angles (°): Cl(1)–Pd–Cl(2) 89.33(3); Cl(1)–Pd–N(1) 95.50(8); Cl(1)–Pd–N(2) 175.85(7); Cl(2)–Pd–N(1) 175.10(8); Cl(2)–Pd–N(2) 94.74(7); N(1)–Pd–N(2) 80.45(10); C(3)–C(11)–O(1) 113.8(3); C(8)–C(12)–O(2) 113.9(3). | ||
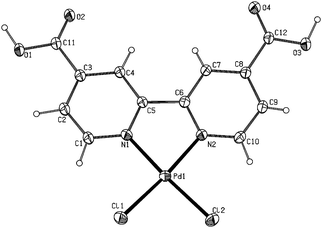 | ||
| Fig. 4 ORTEP-type view of the structure of compound 14 showing the crystallographic labelling scheme. Thermal ellipsoids are drawn at 20% probability level. Selected bond distances (Å): Pd–Cl(1) 2.2752(8); Pd–Cl(2) 2.2771(8); Pd–N(1) 2.027(2); Pd–N(2) 2.023(2); C(11)–O(1) 1.306(3); C(11)–O(2) 1.201(3); C(12)–O(3) 1.312(3); C(12)–O(4) 1.202(4). Selected bond angles (º): Cl(1)–Pd–Cl(2) 90.01(3); Cl(1)–Pd–N(1) 94.98(7); Cl(1)–Pd–N(2) 174.66(7); Cl(2)–Pd–N(1) 174.42(7); Cl(2)–Pd–N(2) 94.41(7); N(1)–Pd–N(2) 80.75(9); C(3)–C(11)–O(1) 113.7(2); C(3)–C(11)–O(2) 122.5(2); O(1)–C(11)–O(2) 123.8(3); C(8)–C(12)–O(3) 113.0(2); C(8)–C(12)–O(4) 123.0(2); O(3)–C(12)–O(4) 124.1(3). | ||
As shown in Table 2, complexes 11–14 were active in the conjugate addition of phenylboronic acid (4a) to 2-cyclohexenone (3a) in water, delivering all of them 3-phenylcyclohexanone (5aa) as the major reaction product. As observed with the dihydroxylated derivatives 2a–d, minor amounts of the Heck-type coupling product 6aa and cyclohexanone were also formed in these reactions and, more importantly, the apparition of a black precipitate of palladium metal was in all cases evidenced during the catalytic events. Instability of the Pd(II)/bipy systems seems to be, therefore, a general trend under the aqueous catalytic conditions employed. The result collected in entry 2 deserves some additional comments since the catalytic behaviour of complex 12, generated in situ from [PdCl2(NH3)2] and the cationic 2,2′-bipyridine ligand 8, has been recently described by Tsai and co-workers.14 In their work, in which the addition reaction of 4a to 3a was studied in the pH range 1–12, there is no feedback on the formation of palladium black and a very poor yield of 5aa was reached when the catalytic reaction was conducted at neutral pH (6% vs. our 72%). In contrast, under extreme acidic conditions (pH 1) almost quantitative formation of 5aa was observed and the catalytic system could be recycled several times. Note that, as shown in entries 5-6 of Table 2, complexes cis-[PdCl2(PTA)2] (15) and cis-[PdCl2(DAPTA)2] (16) containing the water-soluble phosphine ligands 1,3,5-triaza-7-phosphatricyclo[3.3.1.1[3,7]]decane (PTA) and 3,7-diacetyl-1,3,7-triaza-5-phosphabicyclo[3.3.1]nonane (DAPTA) were almost inoperative in this catalytic transformation.
| Entry | Catalyst | Time | Conversionb | Yield 5aab | Yield 6aab |
|---|---|---|---|---|---|
| a Reactions performed at 80 °C under air using 0.5 mmol of 3a, 1.5 mmol of 4a, 0.025 mmol of the corresponding Pd(II) complex 11–14 and 1 cm3 of water. Formation of trace amounts of cyclohexanone was in all cases observed. b Determined by GC. | |||||
| 1 | 11 | 3 h | >99% | 91% | 7% |
| 2 | 12 | 32 h | 79% | 72% | 6% |
| 3 | 13 | 5 h | >99% | 92% | 6% |
| 4 | 14 | 24 h | >99% | 86% | 11% |
| 5 | 15 | 24 h | 4% | 3% | 1% |
| 6 | 16 | 24 h | 12% | 11% | 1% |
From this general catalyst screening, and despite its tendency to decompose into elemental palladium, the dihydroxylated derivative [PdCl2{3,3′-(OH)2-2,2′-bipy}] (2a) emerged as the top choice due to its selectivity and efficiency (entry 1 in Table 1). In this regard, additional studies showed that this complex (5 mol%) is also able to promote efficiently the addition of phenylboronic acid (4a) to 2-cyclohexenone (3a) at room temperature, even when a 1:1 molar ratio of the reactants was employed.29 Under this challenging reaction conditions, ketone 5aa was quantitatively generated after 5 h, and could be isolated in analytically pure form (86% yield) by extraction from the aqueous phase with hexanes and subsequent chromatographic purification (entry 1 in Table 3). It is also worth mentioning that the activity of complex 2a is retained at lower catalyst loadings. Thus, using only 3 mol% of 2a, quantitative formation of 5aa was observed by GC after 8 h of stirring at r.t. (incomplete reactions after 24 h were observed with palladium loadings of 0.5 and 0.005 mol%).
| Entry | Substrate 3 | ArB(OH)24 | Temperature | Time | Conversionb | Yield of 5b | Yield of 6b |
|---|---|---|---|---|---|---|---|
| a Reactions performed under air using 0.5 mmol of the corresponding α,β-unsaturated carbonyl compound 3a–e and arylboronic acid 4a–f, 0.025 mmol of complex 2a and 1 cm3 of water. b Determined by GC (isolated yields after chromatographic work-up are given in brackets). c Reaction performed using 3 equivalents of the arylboronic acid. | |||||||
| 1 | 2-cyclohexenone (3a) | Ar = Ph (4a) | r.t. | 5 h | >99% | 5aa; > 99% (86%) | 6aa; < 1% |
| 2 | 2-cyclohexenone (3a) | Ar = 4-C6H4Me (4b) | r.t. | 24 h | 98% | 5ab; 97% (78%) | 6ab; 1% |
| 3 | 2-cyclohexenone (3a) | Ar = 4-C6H4OMe (4c) | r.t. | 48 h | 99% | 5ac; 98% (77%) | 6ac; 1% |
| 4 | 2-cyclohexenone (3a) | Ar = 4-C6H4Cl (4d) | r.t. | 24 h | 99% | 5ad; 98% (76%) | 6ad; 1% |
| 5 | 2-cyclohexenone (3a) | Ar = 4-C6H4Br (4e) | 80 °C | 1 h | >99% | 5ae; > 99% (80%) | 6ae;<1% |
| 6c | 2-cyclohexenone (3a) | Ar = 2-C6H4Me (4f) | 80 °C | 15 h | 99% | 5af; 98% (77%) | 6af; 1% |
| 7 | 2-cyclopentenone (3b) | Ar = Ph (4a) | r.t. | 4 h | 97% | 5ba; 96% (80%) | 6ba; 1% |
| 8 | 2-cyclopentenone (3b) | Ar = 4-C6H4Me (4b) | r.t. | 24 h | 93% | 5bb; > 92% (86%) | 6bb;<1% |
| 9c | 2-cyclopentenone (3b) | Ar = 4-C6H4OMe (4c) | r.t. | 48 h | >99% | 5bc; > 98% (90%) | 6bc;<1% |
| 10c | 2-cyclopentenone (3b) | Ar = 4-C6H4Cl (4d) | r.t. | 24 h | >99% | 5bd; > 98% (82%) | 6bd;<1% |
| 11c | 2-cyclopentenone (3b) | Ar = 4-C6H4Br (4e) | 80 °C | 24 h | 83% | 5be; 82% (73%) | 6be; 1% |
| 12c | 2-cyclopentenone (3b) | Ar = 2-C6H4Me (4f) | r.t. | 48 h | 99% | 5bf; 98% (79%) | 6bf; 1% |
| 13 | 2-cycloheptenone (3c) | Ar = Ph (4a) | r.t. | 16 h | >99% | 5ca; > 98% (88%) | 6ca;<1% |
| 14 | 3-buten-2-one (3d) | Ar = Ph (4a) | 80 °C | 1 h | 99% | 5da; 83% (69%) | 6da; 16% |
| 15 | cinnamaldehyde (3e) | Ar = Ph (4a) | 80 °C | 26 h | 75% | 5ea; 75% (60%) | 6ea; not observed |
To further define the scope of this green catalytic system, the addition of a number of other arylboronic acids to 2-cyclohexenone (3a), 2-cyclopentenone (3b) and 2-cycloheptenone (3c), was explored using 5 mol% of 2a. Thus, as shown in Table 3 (entries 2–13), the expected addition products 5ab–5ca could be selectively prepared in good to excellent yields (82–99% GC yields; 73–88% isolated yields), regardless of the electronic nature of the aromatic ring of the boronic acid. Only trace amounts of the corresponding Heck-type products 6ab–6ca were detected by GC in the crude reaction mixtures (≤1%), thus confirming the remarkable selectivity of 2a. Although all these processes were operative at r.t. employing equimolar ratios of the reactants, in some cases the use of high temperature (80 °C) and/or excess of the boronic acid was required to achieve good conversions. The generality of the process was further demonstrated by using the acyclic substrates 3d–e (entries 14–15), which also led to the addition products 5da–5ea in high yields.
Again, in all the reactions listed in Table 3 extensive formation of palladium nanoparticles was observed. This fact raised the question on the real nature of the catalytically active species in these aqueous 1,4-addition reactions.30 Thus, in order to determine whether the process is really homogeneous, the conjugate addition of phenylboronic acid (4a) to 2-cyclohexenone (3a) promoted by [PdCl2{3,3′-(OH)2-2,2′-bipy}] (2a) was performed in the presence of mercury.31 As shown in Fig. 5, although the reaction works in the presence of Hg(0), a remarkably lower conversion was attained after 5 h as compared to that performed in its absence (64% vs. > 99%), thus suggesting that the palladium nanoparticles play a role in this catalytic transformation.
![Catalytic addition of phenylboronic acid (4a) to 2-cyclohexenone (3a) using [PdCl2{3,3′-(OH)2-2,2′-bipy}] (2a) in the absence (a) or presence (b) of mercury. Conditions: Reactions performed at r.t under air using 0.5 mmol of 3a, 0.5 mmol of 4a, 0.025 mmol of 2a and 1 cm3 of water.](/image/article/2011/CY/c1cy00214g/c1cy00214g-f5.gif) | ||
| Fig. 5 Catalytic addition of phenylboronic acid (4a) to 2-cyclohexenone (3a) using [PdCl2{3,3′-(OH)2-2,2′-bipy}] (2a) in the absence (a) or presence (b) of mercury. Conditions: Reactions performed at r.t under air using 0.5 mmol of 3a, 0.5 mmol of 4a, 0.025 mmol of 2a and 1 cm3 of water. | ||
In line with this, an interesting observation was made when the same catalytic reaction was carried out, in the absence of Hg(0), under more dilute conditions (graphs (b), (c) and (d) in Fig. 6). The irregularities in the curves at short times (20–40 min) visually match with the progressive change of color of the aqueous solution from yellow to orange and finally to dark green, a color change that is accompanied by a turbidity. TEM images recorded from a sample collected at this moment (reaction (c)) confirmed the appearance of the first nanoparticles (Fig. 7).
![Effect of the concentration in the catalytic addition of phenylboronic acid (4a) to 2-cyclohexenone (3a) using [PdCl2{3,3′-(OH)2-2,2′-bipy}] (2a). Conditions: Reactions performed at r.t. under air using 0.5 mmol of 3a, 0.5 mmol of 4a, 0.025 mmol of 2a and 1 (a), 1.25 (b), 2 (c) or 3.3 (d) cm3 of water.](/image/article/2011/CY/c1cy00214g/c1cy00214g-f6.gif) | ||
| Fig. 6 Effect of the concentration in the catalytic addition of phenylboronic acid (4a) to 2-cyclohexenone (3a) using [PdCl2{3,3′-(OH)2-2,2′-bipy}] (2a). Conditions: Reactions performed at r.t. under air using 0.5 mmol of 3a, 0.5 mmol of 4a, 0.025 mmol of 2a and 1 (a), 1.25 (b), 2 (c) or 3.3 (d) cm3 of water. | ||
 | ||
| Fig. 7 TEM images of the sample taken from the reaction medium when the turbidity begins to appear (the sample corresponds to the reaction denoted as (c) in Fig. 6). | ||
Moreover, as shown in Fig. 8, addition of mercury at this stage almost completely supressed the catalysis. All these facts strongly support that, only at the early stages of the catalytic events, molecular Pd(II) species are the real active species and that, once formed, palladium(0) nanoparticles are the responsible to drive the catalytic reactions to completion.32
![Catalytic addition of phenylboronic acid (4a) to 2-cyclohexenone (3a) using [PdCl2{3,3′-(OH)2-2,2′-bipy}] (2a) without addition of Hg(0) (a) and with addition of Hg(0) after 60 min of reaction (b). Conditions: Reactions performed at r.t. under air using 0.5 mmol of 3a, 0.5 mmol of 4a, 0.025 mmol of 2a and 2 cm3 of water.](/image/article/2011/CY/c1cy00214g/c1cy00214g-f8.gif) | ||
| Fig. 8 Catalytic addition of phenylboronic acid (4a) to 2-cyclohexenone (3a) using [PdCl2{3,3′-(OH)2-2,2′-bipy}] (2a) without addition of Hg(0) (a) and with addition of Hg(0) after 60 min of reaction (b). Conditions: Reactions performed at r.t. under air using 0.5 mmol of 3a, 0.5 mmol of 4a, 0.025 mmol of 2a and 2 cm3 of water. | ||
Finally, recycling of the nanoparticles was also investigated using again the addition of phenylboronic acid (4a) to 2-cyclohexenone (3a) as model reaction. Thus, we observed that the solid material generated from [PdCl2{3,3′-(OH)2-2,2′-bipy}] (2a) shows, after filtration and washing with diethyl ether and acetone (Fig. 1), a very poor activity and a low selectivity towards the formation of the desired 1,4-addition product 5aa. In particular, 40 h of heating at 80 °C were requiered to achieve the complete consumption of 3a, the catalytic reaction leading to 5aa in only 40% GC yield, along with 20% of the Heck product 6aa and 39% of the reduced cyclohexanone. This result is not surprising since agglomeration of metal-nanoparticles is known to have a negative impact on the activity and selectivity of several catalytic transformations.33,34
In order to avoid the agglomeration of the catalytically active Pd nanoparticles, and facilitate their possible recycling, some experiments were performed in the presence of surfactants. This type of additives are known to confer an “electrosteric” stabilization to nanoparticles, thus preventing the formation of bulk metal.35,36 Among the different surfactants employed,37 best results were obtained with sodium dodecyl sulfate (SDS; 0.01 M aqueous solution).38 Thus, as shown in Fig. 9, performing the catalytic addition of phenylboronic acid (4a) to 2-cyclohexenone (3a) with [PdCl2{3,3′-(OH)2-2,2′-bipy}] (2a) in this medium, the aqueous phase containing the stabilized nanoparticles could be effectively reused in at least six consecutive runs after simple extraction of the reaction product 5aa with hexanes. Remarkably, compared to the use of pure water, no differences in activity were observed in SDS(aq) during first cycle. TEM images recorded from the aqueous phase after extraction of 5aa confirmed the formation of micelles (size range 40–150 nm) in which the palladium nanoparticles remain embedded (Fig. 10). Only after the fifth run agglomeration of the nanoparticles was observed. Interestingly, we must also note that the use of SDS-stabilized palladium nanoparticles generated in the absence of the 3,3′-dihydroxy-2,2′-bipyridine ligand 1a led, under the same reaction conditions, to only 9% conversion after 24 h.39 This fact clearly indicates that the ligand 1a “retained” by the nanoparticles plays a key role during the catalytic event.
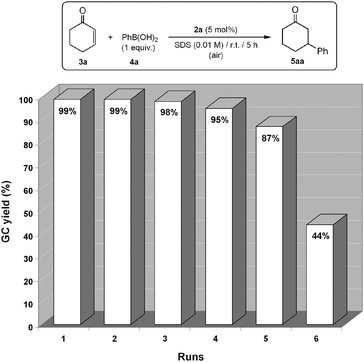 | ||
| Fig. 9 Catalyst recycling by stabilization of the catalytically active nanoparticles with SDS. GC-yields are in all cases given after 5 h of reaction. | ||
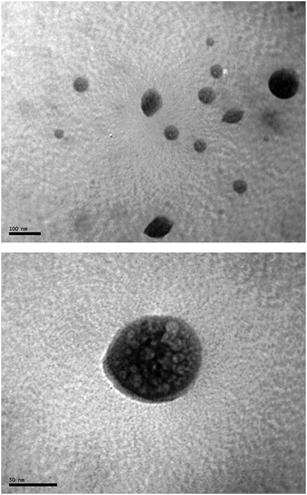 | ||
| Fig. 10 TEM images of the micelles generated in the presence of SDS. Sample taken after the first catalytic cycle. | ||
Conclusions
In summary, we have found that the readily accessible Pd(II) complex [PdCl2{3,3′-(OH)2-2,2′-bipy}] (2a) is an excellent precatalyst for the selective conjugate addition of arylboronic acids to α,β-unsaturated carbonyl compounds under challenging reactions conditions, i.e. in environmentally friendly aqueous media under air. Moreover, we have demonstrated that such a catalytic transformation, which usually proceeds under homogeneous conditions, is in fact a new example of an heterogeneous process involving metallic nanoparticles. Thus, we have evidenced that only at the early stages of the catalytic events are molecular Pd(II) species responsible for the catalysis, with nanoparticles of elemental palladium, generated by decomposition of [PdCl2{3,3′-(OH)2-2,2′-bipy}] (2a), being the true active species at the end of the catalytic reactions. Interestingly, in the presence of the surfactant SDS (sodium docecyl sulfate) these metal nanoparticles can be stabilized by the micelles thus allowing their effective recycling, a key factor for practical applications of this heterogeneous catalytic system.40Experimental section
General methods
All reagents were obtained from commercial suppliers and used as received with the exception of the disubstituted 2,2′-bipyridine ligands 1a–d,178,41942 and 10,43 and the Pd(II) complex 11,251544 and 1644 which were prepared by following the methods reported in the literature. Flash chromatography was performed using Merck silica gel 60 (230-400 mesh). Infrared spectra were recorded on a Perkin-Elmer 1720-XFT spectrometer. NMR spectra were recorded on a Bruker DPX-300 instrument at 300 MHz (1H) or 75.4 MHz (13C). The chemical shift values (δ) are given in parts per million and are referred to the residual peak of the deuterated solvent employed. DEPT experiments have been carried out for all the compounds reported. GC measurements were made on a Hewlett-Packard HP6890 equipment using a Supelco Beta-Dex™ 120 column. GC-MS measurements were performed on a Agilent 6890N equipment coupled to a 5973 mass detector (70eV electron impact ionization) using a HP-1MS column. Elemental analyses were provided by the Analytical Service of the Instituto de Investigaciones Químicas (IIQ-CSIC, Seville) and performed with a Leco-CHNS microanalyzer. TEM (Transmission Electron Microscopy) and SEM/EDX (Scanning Electron Microscopy-Energy/Dispersive X-ray spectroscopy) measurements were provided by the Analytical Service of the University of Oviedo and recorded on MET-JEOL 2000 EX-II and MEB JEOL-6610LV equipments, respectively.| 13 | 14 | |
|---|---|---|
| a R 1 = Σ(|Fo|−|Fc|)/Σ|Fo|; wR2 = {Σ[w(Fo2−Fc2)2]/Σ[w(Fo2)2]}1/2 | ||
| Empirical formula | C12H12Cl2N2O2Pd·2DMSO | C12H8O4Cl2N2Pd·DMSO |
| Formula weight | 549.79 | 499.63 |
| Temperature/K | 293(2) | 293(2) |
| Wavelength/Å | 1.5418 | 1.5418 |
| Crystal system | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
|
P
|
| Crystal size/mm | 0.214 × 0.079 × 0.034 | 0.064 × 0.031 × 0.018 |
| a/Å | 8.4783(3) | 7.6738(3) |
| b/Å | 11.6061(4) | 10.4974(4) |
| c/Å | 13.1535(5) | 12.5545(4) |
| α (°) | 67.125(4) | 112.689(3) |
| β (°) | 89.797(3) | 90.017(3) |
| γ (°) | 68.971(4) | 108.646(3) |
| Z | 2 | 2 |
| Volume/Å3 | 1099.10(7) | 875.20(6) |
| Calculated density/g cm−3 | 1.661 | 1.896 |
| μ/mm−1 | 11.034 | 12.748 |
| F(000) | 556 | 496 |
| θ range/° | 3.69 to 73.88 | 3.85 to 73.97 |
| Index ranges | −9 ≤ h ≤ 10 | −9 ≤ h ≤ 9 |
| −14 ≤ k ≤ 14 | −12 ≤ k ≤ 13 | |
| −16 ≤ l ≤ 16 | −15 ≤ l ≤ 15 | |
| Completeness to θmax | 94.3% | 94.5% |
| No. of reflns. collected | 11501 | 9900 |
| No. of unique reflns. | 4216 (Rint = 0.0217) | 3365 (Rint = 0.0247) |
| No. of parameters/restraints | 349/8 | 292/2 |
| Refinement method | Full-matrix least-squares on F2 | |
| Goodness-of-fit on F2 | 1.060 | 1.039 |
| Weight function (a, b) | 0.0507, 0.7398 | 0.0347, 0.5458 |
| R 1 [I > 2σ(I)]a | 0.0313 | 0.0251 |
| wR2 [I > 2σ(I)]a | 0.0816 | 0.0640 |
| R 1 (all data) | 0.0335 | 0.0288 |
| wR2 (all data) | 0.0839 | 0.0662 |
| Largest diff. peak and hole/e Å−3 | 0.345 and −0.712 | 0.549 and −0.385 |
In both cases data collection was performed on a Oxford Diffraction Xcalibur Nova single crystal diffractometer, using Cu-Kα radiation (λ = 1.5418 Å). Images were collected at a 65 mm fixed crystal-to-detector distance using the oscillation method, with 1° oscillation and a variable exposure time per image of 10–15 s for 13 and 2–6 s for 14. Data collection strategy was calculated with the program CrysAlis Pro CCD.45 Data reduction and cell refinement were performed with the program CrysAlis Pro RED.45 An empirical absorption correction was applied using the SCALE3 ABSPACK algorithm as implemented in the program CrysAlis Pro RED.45
In all cases the software package WINGX was used for space group determination, structure solution and refinement.46 Both structures were solved by direct methods using SIR2004.47 Isotropic least-squares refinement on F2 using SHELXL97 was performed.48 During the final stages of the refinements, all the positional parameters and the anisotropic temperature factors of all the non-H atoms were refined. The H atoms were found from different Fourier maps and included in a refinement with isotropic parameters. In the crystal of 13 two DMSO molecules of solvation per formula unit of the complex were found. One of them is disordered, with the sulphur atom being located in two positions with occupancy of 50%. In the crystal of 14 a DMSO molecule per formula unit of the complex was also found with the sulphur atom equally disordered. The function minimized was [ΣwFo2 − Fc2)/Σw(Fo2)]1/2 where w = 1/[σ2(Fo2) + (aP)2 + bP] (a and b values are collected in Table 4) with σ2(Fo2) from counting statistics and P = (Max (Fo2 + 2Fc2)/3. Atomic scattering factors were taken from the International Tables for X-ray Crystallography.49 Geometrical calculations were made with PARST.50 The crystallographic plots were made with PLATON.51
Acknowledgements
This work was supported by the Spanish Ministry of Science and Innovation (MICINN; projects CTQ2010-14796/BQU and CSD2007-00006). E.T.-M. thanks MICINN and the European Social Fund for the award of a PhD grant (FPU program).Notes and references
- See, for example: (a) P. Perlmutter, in Conjugate Addition Reactions in Organic Synthesis, Pergamon, Oxford, 1992 Search PubMed; (b) B. E. Rossiter and N. M. Swingle, Chem. Rev., 1992, 92, 771 CrossRef CAS; (c) B. H. Lipshutz, in Organometallics in Synthesis: A Manual, ed. M. Schlosser, John Wiley & Sons, New York, 1994, pp. 283–382 Search PubMed; (d) M. P. Sibi and S. Manyen, Tetrahedron, 2000, 56, 8033 CrossRef CAS; (e) N. Krause and A. Hoffmann-Röder, Synthesis, 2001, 171 CrossRef CAS; (f) J. Christoffers, G. Koripelly, A. Rosiak and M. Rössle, Synthesis, 2007, 1279 CrossRef CAS; (g) C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295 RSC.
- M. Sakai, H. Hayashi and N. Miyaura, Organometallics, 1997, 16, 4229 CrossRef CAS.
- For reviews on this topic, see: (a) T. Hayashi, Synlett, 2001, 879 CrossRef CAS; (b) K. Fagnou and M. Lautens, Chem. Rev., 2003, 103, 169 CrossRef CAS; (c) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829 CrossRef CAS; (d) T. Hayashi, Pure Appl. Chem., 2004, 76, 465 CrossRef CAS; (e) T. Hayashi, Bull. Chem. Soc. Jpn., 2004, 77, 13 CrossRef CAS; (f) H. Yoshida and T. Hayashi, in Modern Rhodium-Catalyzed Organic Reactions, ed. P. A. Evans, Wiley-VCH, Weinheim, 2005, pp. 55–77 Search PubMed.
- See, for example: N. Miyaura, Synlett, 2009, 2039 CAS and references cited therein.
- (a) For a specific review on β-hydride elimination processes in palladium complexes, see: X. Lu, Top. Catal., 2005, 35, 73 CrossRef; (b) For a discussion about the competition between conjugate addition and Heck-type reaction with rhodium-based systems, see: G. Zou, Z. Wang, J. Zhu and J. Tang, Chem. Commun., 2003, 2438 RSC.
- For selected examples, see: (a) T. Nashikata, Y. Yamamoto and N. Miyaura, Angew. Chem. Int. Ed., 2003, 42, 2763 Search PubMed; (b) T. Nishikata, Y. Yamamoto and N. Miyaura, Organometallics, 2004, 23, 4317 CrossRef CAS; (c) F. Gini, B. Hessen and A. J. Minnaard, Org. Lett., 2005, 7, 5309 CrossRef CAS; (d) T. Yamamoto, M. Iizuka, T. Ohta and Y. Ito, Chem. Lett., 2006, 35, 198 CrossRef CAS; (e) T. Nishikata, Y. Yamamoto and N. Miyaura, Tetrahedron Lett., 2007, 48, 4007 CrossRef CAS; (f) R. B. Bedford, M. Betham, J. P. H. Charmant, M. F. Haddow, A. G. Orpen, L. T. Pilarski, S. J. Coles and M. B. Hursthouse, Organometallics, 2007, 26, 6346 CrossRef CAS; (g) P. He, Y. Lu, C.-G. Dong and Q.-S. Hu, Org. Lett., 2007, 9, 343 CrossRef CAS; (h) P. He, Y. Lu and Q.-S. Hu, Tetrahedron Lett., 2007, 48, 5283 CrossRef CAS; (i) T. Zhang and M. Shi, Chem.–Eur. J., 2008, 14, 3759 CrossRef CAS; (j) Y. Suzuma, S. Hayashi, T. Yamamoto, Y. Oe, T. Ohta and Y. Ito, Tetrahedron: Asymmetry, 2009, 20, 2751 CrossRef CAS; (k) K. Kikushima, J. C. Holder, M. Gatti and B. M. Stoltz, J. Am. Chem. Soc., 2011, 133, 6902 CrossRef CAS.
- (a) X. Lu and S. Lin, J. Org. Chem., 2005, 70, 9651 CrossRef CAS; (b) S. Lin and X. Lu, Tetrahedron Lett., 2006, 47, 7167 CrossRef CAS; (c) V. Poláčková, V. Bariak, R. Šebesta and Š. Toma, Chem. Pap., 2011, 65, 338 CrossRef.
- S. Lin and X. Lu, Org. Lett., 2010, 12, 2536 CrossRef CAS.
- For recent DFT calculations on the mechanism of these Pd-catalyzed conjugate additions, see: Y. Lan and K. N. Houk, J. Org. Chem., 2011, 76, 4905 CrossRef CAS.
- (a) P. T. Anastas and J. C. Warner, in Green Chemistry Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed; (b) A. S. Matlack, in Introduction to Green Chemistry, Marcel Dekker, New York, 2001 Search PubMed; (c) Handbook of Green Chemistry and Technology, ed. J. H. Clark and D. J. Macquarrie, Blackwell Publishing, Abingdon, 2002 Search PubMed; (d) M. Lancaster, in Green Chemistry: An Introductory Text, RSC Editions, Cambridge, 2002 Search PubMed; (e) M. Poliakoff, J. M. Fitzpatrick, T. R. Farren and P. T. Anastas, Science, 2002, 297, 807 CrossRef CAS.
- See, for example: (a) W. M. Nelson, in Green Solvents for Chemistry: Perspectives and Practice, Oxford University Press, New York, 2003 Search PubMed; (b) J. H. Clark and S. J. Taverner, Org. Process Res. Dev., 2007, 11, 149 CrossRef CAS; (c) F. M. Kerton, in Alternative Solvents for Green Chemistry, RSC Publishing, Cambridge, 2009 Search PubMed.
- For leading references in this field, see: (a) C. J. Li and T. H. Chan, in Comprehensive Organic Reactions in Aqueous Media, John Wiley & Sons, New Jersey, 2007 Search PubMed; (b) Organic Reactions in Water: Principles, Strategies and Applications, ed. U. M. Lindstrom, Blackwell Publishing Ltd., Oxford, 2007 Search PubMed; (c) Aqueous-Phase Organometallic Catalysis Concepts and Applications, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, 1998 Search PubMed; (d) F. Joó, in Aqueous Organometallic Catalysis, Kluver, Dodrecht, 2001 Search PubMed.
- For recent examples, see: (a) V. Cadierno, P. Crochet, J. Francos, S. E. García-Garrido, J. Gimeno and N. Nebra, Green Chem., 2009, 11, 1992 RSC; (b) V. Cadierno, J. Francos and J. Gimeno, Tetrahedron Lett., 2009, 50, 4773 CrossRef CAS; (c) V. Cadierno, J. Francos and J. Gimeno, Green Chem., 2010, 12, 135 RSC; (d) V. Cadierno, J. Francos and J. Gimeno, Chem. Commun., 2010, 46, 4175 RSC; (e) V. Cadierno, J. Díez, J. Francos and J. Gimeno, Chem.–Eur. J., 2010, 16, 9808 CrossRef CAS; (f) R. García-Álvarez, J. Díez, P. Crochet and V. Cadierno, Organometallics, 2010, 29, 3955 CrossRef; (g) J. Francos and V. Cadierno, Green Chem., 2010, 12, 1552 RSC; (h) V. Cadierno, J. Francos, S. E. García-Garrido and J. Gimeno, Green Chem. Lett. Rev., 2011, 4, 55 CrossRef CAS; (i) B. Lastra-Barreira, J. Francos, P. Crochet and V. Cadierno, Green Chem., 2011, 13, 307 RSC; (j) S. E. García-Garrido, J. Francos, V. Cadierno, J.-M. Basset and V. Polshettiwar, Chem. Sus. Chem., 2011, 4, 104 CrossRef; (k) V. Cadierno, J. Francos and J. Gimeno, Organometallics, 2011, 30, 852 CrossRef CAS.
- We must note that, parallel to our work, an article describing the conjugate addition of arylboronic acids to enones in water, without the aid of surfactants and under aerobic conditions, has been described by Tsai and co-workers. Their catalytic system was composed of [PdCl2(NH3)2] and the cationic 2,2′-bipyridine ligand 8 (see discussion): S.-H. Huang, T.-M. Wu and F.-Y. Tsai, Appl. Organomet. Chem., 2010, 24, 619 CrossRef CAS.
- See, for example: K. H. Shaughnessy, Chem. Rev., 2009, 109, 643 CrossRef CAS and references cited therein.
- See, for example: G. R. Newkome, A. K. Patri, E. Holder and U. S. Schubert, Eur. J. Org. Chem., 2004, 235 CrossRef CAS and references cited therein.
- (a) E. V. Dehmlow and A. Sleegers, Liebigs Ann. Chem., 1992, 953 CrossRef CAS; (b) C. Naumann and H. Langhals, Synthesis, 1990, 279 CrossRef CAS; (c) Y. Fukuda, S. Seto, H. Furuta, H. Ebisu, Y. Oomori and S. Terashima, J. Med. Chem., 2001, 44, 1396 CrossRef CAS; (d) Y.-R. Hong and C. B. Gorman, J. Org. Chem., 2003, 68, 9019 CrossRef CAS.
- With the exception of 2a, which is also partially soluble in methanol, dichloromethane and acetone, these complexes only dissolve well in DMSO or DMF.
- Potassium phosphate/KOH buffer solutions were employed for solubility measurements.
- Complex 2d proved to be unstable in DMSO-d6 generating [PdCl2(DMSO-d6)2] and the free ligand 1d. Fortunately, it partially dissolves in CD3NO2 allowing us to run its 1H NMR spectrum in this deuterated solvent (see Experimental Section).
- Formation of variable amounts of biphenyl and phenol was also observed by GC, as the result of self-coupling and hydrolysis processes, respectively, of the phenylboronic acid (4a) employed in excess: M. Moreno-Mañas, M. Pérez and R. Pleixats, J. Org. Chem., 1996, 61, 2346 Search PubMed.
- In the case of complex 2d steric effects could also be responsible of its low activity.
- Contrary to 2a–d, PdCl2 practically does not dissolve in water at 80 °C.
- Pd(II) to Pd(0) reductions in the presence of arylboronic acids have been previously described. In addition, some authors have also observed that such reduction processes are particularly favoured in the presence of water. See, for example, ref. 6f and: (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 Search PubMed and references cited therein; ; (b) D. N. Korolev and N. A. Bumagin, Tetrahedron Lett., 2006, 47, 4225 Search PubMed; (c) Y.-X. Liao, C.-H. Xing, M. Israel and Q.-S. Hu, Tetrahedron Lett., 2011, 52, 3324 Search PubMed; (d) A. N. Marziale, D. Jantke, S. H. Faul, T. Reiner, E. Herdtweck and J. Eppinger, Green Chem., 2011, 13, 169 Search PubMed.
- P. Wehman, G. C. Dol, E. R. Moorman, P. C. J. Kamer, P. W. N. M van Leeuwen, J. Fraanje and K. Goubitz, Organometallics, 1994, 13, 4856 Search PubMed.
- Preparation of complex 14 has been previously described in the literature, but the only characterization data given in the article were IR absorptions: G.-J. ten Brink, I. W. C. E. Arends, M. Hoogenraad, G. Verspui and R. A. Sheldon, Adv. Synth. Catal., 2003, 345, 497 Search PubMed.
- The crystal packing of both molecules is very different. Thus, while in the case of 13 no intermolecular interactions were found in the crystal lattice, a complex hydrogen bonding network is established between the molecules of 14, thus leading to two-dimensional (2D) layers. In the hollows of these layers are retained DMSO molecules which also interact with molecules of 14 through hydrogen bonds. Illustrative figures can be found in the ESI file.
- A. J. Canty, B. W. Skelton, P. R. Traill and A. H. White, Aust. J. Chem., 1992, 45, 417 Search PubMed.
- (a) Excess of the arylboronic acid is systematically employed in the literature with palladium catalysts. See ref. 6–8 and 14; (b) The influence of medium pH was also studied using phosphate buffer solutions. In contrast to the results reported by Tsai and workers, improvement in the catalytic activity of 2a was not observed neither in acid nor basic media. However, we must note that under extreme acidic conditions (pH 1) precipitation of metallic palladium was much lower.
- For a general discussion on the role of nanoparticles and their stabilization in the closely related Heck reaction, see: J. G. de Vries, Dalton Trans., 2006, 421 Search PubMed.
- The Hg(0)-poisoning test is the most direct method to distinguishing homogeneous from heterogeneous catalysis when transition metals able to form an amalgam are employed. See, for example: J. A. Wildegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317 Search PubMed , and references therein.
-
(a) A mechanism related to that proposed by Uemura and co-workers for the Pd(0)-catalyzed conjugated addition of arylboronic acids to α,β-unsaturated carbonyl compounds could be operative at this stage. That is, an initial oxidative addition of the arylboronic acid to palladium atoms on the surface of the nanoparticles to generate transient [ArPdB(OH)2] species, followed by insertion of the CC bond of the enone into the Pd(II)-aryl bond, reduction elimination to generate an O-boronated enol and final hydrolysis: C. S. Cho, S. Motofusa, K. Ohe, S. Uemura and S. C. Shim, J. Org. Chem., 1995, 60, 883 Search PubMed;
(b)
Oxidation of Pd(0) to Pd(II) by air, thus enabling a subsequent transmetallation of the arylboronic acid, can not be totally rouled out. However, we must note that, as indicated in the entry 6 of Table 1, no differences in activity or selectivity were observed when the catalytic addition of phenylboronic acid (4a) to 2-cyclohexenone (3a) promoted by complex 2a was carried out under inert N2 atmosphere.
- See, for example: (a) M. Moreno-Mañas and R. Pleixats, Acc. Chem. Res., 2003, 36, 638 Search PubMed; (b) Nanoparticles and Catalysis, ed. D. Astruc, Wiley-VCH, Weinheim, 2008 Search PubMed; (c) J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, Chem. Sus. Chem., 2009, 2, 18 Search PubMed.
- Best results in terms of selectivity (88%) were obtained performing the catalytic reaction in the presence of 5 mol% of the 3,3′-dihydroxy-2,2′-bipyridine ligand 1a. However, 32 h of heating at 80 °C were still needed to achieve the total consumption of the starting materials.
- For general reviews on nanoparticles stabilization, see: (a) J. D. Aiken III and R. G. Finke, J. Mol. Catal. A: Chem., 1999, 145, 1 Search PubMed; (b) A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757 Search PubMed; (c) D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852 Search PubMed; (d) L. S. Ott and R. G. Finke, Coord. Chem. Rev., 2007, 251, 1075 Search PubMed.
- Representative examples on the use of surfactants to stabilize metal nanoparticles can be found in: (a) Y. Lin and R. Finke, J. Am. Chem. Soc., 1994, 116, 8335 Search PubMed; (b) S. R. Johnson, S. D. Evans and R. Brydson, Langmuir, 1998, 14, 6639 Search PubMed; (c) J. Xu, X. Han, H. Liu and Y. Hu, J. Dispersion Sci. Technol., 2005, 26, 473 Search PubMed; (d) A. Nowicki, V. Le Boulaire and A. Roucoux, Adv. Synth. Catal., 2007, 349, 2326 Search PubMed; (e) J. Duy, L. B. Connell, W. Eck, S. D. Collins and R. L. Smith, J. Nanopart. Res., 2010, 12, 2363 Search PubMed; (f) M. Boutros, G. Shirley, T. Onfroy and F. Launay, Appl. Catal., A, 2011, 394, 158 Search PubMed.
- In the presence of cetyltrimethylammoniumbromide (CTABr; 0.01 M aqueous solution) formation of an agglomerate of palladium nanoparticles was observed during the first catalytic cycle. Using 1 wt.% PTS (polyoxyethanyl-α-tocopheryl sebacate) stabilization of the nanoparticles could be achived, but only during three consecutive runs.
- The critical micelle concentration of SDS is 8 × 10−3 M, the use of 0.01 M solutions of this surfactant assuring the correct formation of micelles: (a) J. H. Fendler and E. J. Fendler, in Catalysis in Micellar and Macromolecular Systems, Academic Press, New York, 1975 Search PubMed; (b) K. Furton and A. Norelus, J. Chem. Educ., 1993, 70, 254 Search PubMed.
- These nanoparticles were generated as described in: W. L. Wang, Y. Y. Wang, C. C. Wan and C. L. Lee, Colloids Surf., A, 2006, 275, 11 Search PubMed.
- Recoverable and Recyclable Catalysts, ed. M. Benaglia, John Wiley & Sons, Chichester, 2009 Search PubMed.
- (a) W.-Y. Wu, S.-N. Chen and F.-Y. Tsai, Tetrahedron Lett., 2006, 47, 9267 Search PubMed; (b) S.-N. Chen, W.-Y. Wu and F.-Y. Tsai, Tetrahedron, 2008, 64, 8164 Search PubMed.
- D. Miyoshi, H. Karimata, Z.-M. Wang, K. Koumoto and N. Sugimoto, J. Am. Chem. Soc., 2007, 129, 5919 Search PubMed.
- O. Schwarz, D. van Loyen, S. Jockusch, N. J. Turro and H. Dürr, J. Photochem. Photobiol., A, 2000, 132, 91 Search PubMed.
- E. Vergara, S. Miranda, F. Mohr, E. Cerrada, E. R. T. Tiekink, P. Romero, A. Mendía and M. Laguna, Eur. J. Inorg. Chem., 2007, 2926 Search PubMed.
- CrysAlisPro CCD & CrysAlisPro RED, Oxford Diffraction Ltd., oxford, UK, 2008 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 Search PubMed.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2005, 38, 381 Search PubMed.
- G. M. Sheldrick, SHELXL97: Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- International Tables for X-Ray Crystallography, Kynoch Press, Birminghan, U.K., 1974, vol. IV. (present distributor: Kluwer Academic Publishers, Dordrecht, The Netherlands) Search PubMed.
- M. Nardelli, Comput. Chem., 1983, 7, 95 Search PubMed.
- A. L. Spek, PLATON A Multipurpose Crystallographic Tool, University of Utrecht, Utrecht, The Netherlands, 2006 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization data of organic compounds 5aa–5ea and figures showing the hydrogen bond network in the crystal packing of complex 14. CCDC reference numbers 828891 and 828892. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1cy00214g |
| ‡ CCDC-828891 (13) and 828892 (14) contain the supplementary crystallographic data for this paper. |
| This journal is © The Royal Society of Chemistry 2011 |