Microwave-assisted, environmentally friendly, one-pot preparation of Pd nanoparticles/graphene nanocomposites and their application in electrocatalytic oxidation of methanol†
Yingwei
Zhang
a,
Guohui
Chang
a,
Sen
Liu
a,
Jingqi
Tian
ab,
Lei
Wang
a,
Wenbo
Lu
a,
Xiaoyun
Qin
a and
Xuping
Sun
*a
aState Key Lab of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022 Jilin, China. E-mail: sunxp@ciac.jl.cn; Fax: +86 431-85262065; Tel: +86 431-85262065
bGraduate School of the Chinese Academy of Sciences, Beijing 100039, China
First published on 21st September 2011
Abstract
In this communication, we demonstrate for the first time that Pd nanoparticles/graphene nanocomposites (PdNPs–G) can be rapidly synthesized through a microwave-assisted, environmentally friendly, one-pot method with the use of tannic acid (TA) as a reducing agent. It is suggested that the loading amount and sizes of PdNPs on a G sheet can be controlled by the ratio of raw materials and microwave irradiation time. It is also found that the electrocatalytic activity and stability of the resultant PdNPs–G nanocomposites are much better than that of commercial Pd/C catalysts towards methanol electrooxidation in alkaline media.
The direct methanol fuel cell (DMFC) has attracted enormous attention as a high-efficiency, low-emission future power source for fuel-cell vehicles.1 A significant challenge in the development of DMFC technology is the need for highly active anode catalysts for the methanol oxidation reaction. Pt-based catalysts are generally identified as the best catalysts for methanol electrooxidation in acid solution and have been widely studied in recent years. However, Pt-based catalysts generally undergo deactivation/poisoning by CO-like intermediates in an acid medium.2 In addition, the high cost and limited resource of Pt also do not allow its efficient commercial usage.3 Therefore, as the replacement of Pt based catalyst, Pd based catalyst is emerging as a good choice due to its abundance and good electrocatalytic activity for methanol in alkaline media.4 Additionally, Pd based catalysts can overcome the CO-poisoning effect and thus present high performance in DMFCs.5 Therefore, Pd-based catalysts have become promising anode catalysts in an alkaline DMFC.
Recently, hybridization provides an effective strategy for enhancing the functionality of materials.6 In order to further maximize the activity of Pd and minimize the use of previous Pd, it is very necessary to load Pd nanostructures with high activity on the surface of supporting nanomaterials with low cost, high surface area, and good electrical conductivity, which not only maximize the availability of nanosized electrocatalyst surface area for electron transfer but also provide better mass transport of reactants to the electrocatalyst. The recent emergence of graphene (G) nanosheets has opened a new avenue for utilizing 2D new carbon material as a support because of its high conductivity, unique graphitized basal plane structure and potential low manufacturing cost.7 Currently, it has been demonstrated that the integration of graphene and certain functional metal particles (such as Pd nanoparticles) can present special features in the new hybrids, useful, for example, in optics, electronics, catalysis, sensors, and so on.8 However, the metal–G nanocomposites are usually obtained from in situ reduction of metallic salts on preformed G sheets or the decoration of G sheets with presynthesized nanoparticles, which increased the complexity of the process. Furthermore, the resulting composites are mostly in the form of aggregation due to π–π stacking interactions between G nanosheets. Therefore, a one-pot route for synthesizing well-dispersed metal–G nanocomposites with high performance is highly required.
Herein, we report on microwave-assisted, environmentally friendly, one-pot preparation of Pd nanoparticles/graphene nanocomposites (PdNPs–G) with the use of tannic acid (TA, Fig. S1, ESI†), a water-soluble, phenolic hydroxyl-rich compound and widely present in woods, as a reducing agent. Scheme 1 presents a scheme (not to scale) to illustrate the preparation process of the PdNPs–G nanocomposites via microwave-assisted reduction of graphene oxide (GO) and H2PdCl4 by TA. The resultant nanocomposites exhibit notable catalytic performance toward methanol oxidation.
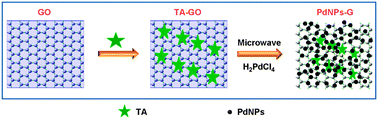 | ||
| Scheme 1 A scheme (not to scale) to illustrate the preparation process of PdNPs–G nanocomposites via microwave-assisted reduction of H2PdCl4 and GO by TA. | ||
TA is a mild reducing agent with a redox potential from −0.57 to −1.05 V (pH = 3.03–6.24).9 Compared with the redox potential of −0.82 to −1.09 V needed for GO,10 the reduction of GO by TA is thermodynamically favoured and there is a successful report recently.11 In our present study, the successful reduction of GO and H2PdCl4 by TA was firstly verified by Raman spectroscopy, as shown in Fig. 1. It is well-known that G obtained by chemical reduction of GO exhibits two characteristic main peaks: the D band at ∼1350 cm−1, arising from a breathing mode of κ-point photons of A1g symmetry; the G band at ∼1575 cm−1, arising from the first order scattering of the E2g phonon of sp2 C atoms. In our present study, it is seen that GO exhibits a D band at 1353 cm−1 and a G band at 1603 cm−1, while the corresponding bands of the products are 1355 and 1599 cm−1, respectively. The shift of the G band of the products from 1603 to 1599 cm−1 is attributed to the recovery of the hexagonal network of carbon atom. It is also found that products show relative higher intensity of D to G band (0.98) than that of GO (0.81), providing another piece of evidence to support the formation of new graphitic domains after 1 min microwave irradiation.12
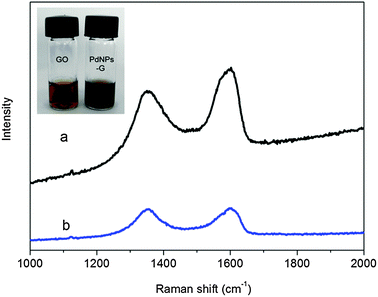 | ||
| Fig. 1 Raman spectra of GO (a) and the products obtained after 1 min microwave irradiation (b). Inset shows photographs of corresponding samples. | ||
It is known that most of the oxygen-containing functional groups in GO exist in the form of either hydroxyl or epoxide groups,7e,13 and the successful removal of epoxy and hydroxyl means the formation of G, which can be confirmed by X-ray photoelectron spectroscopy (XPS). Fig. 2 shows the C 1s XPS spectra of GO and the products, respectively. The C 1s spectra of GO and the products could be deconvoluted into three peaks at 284.5, 286.6, and 288.2 eV, which are associated with C–C, C–O (epoxyl and hydroxyl), and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (carbonyl), respectively. It is seen that the peak intensity of C–O is strong in GO due to the presence of oxygen-containing functional groups (Fig. 2a). In contrast, after microwave irradiation, the peak intensity of C–O in the products (Fig. 2b) tremendously reduced. Note that TA consists of a large amount of –OH and –COOH and thus makes much contribution to the intensity of C–O and CO peaks in products. All the observations suggest that the most oxygen-containing functional groups are successfully removed after microwave irradiation.13 The XPS pattern of the products also shows significant Pd 3d signals corresponding to the binding energy of metal Pd (Fig. 2c),7d indicating that H2PdCl4 has been reduced by TA. Although there is a report about microwave irradiation reduction of GO without the use of additional reducing agents,14 in order to clarify the role of TA, a control experiment under microwave irradiation in the absence of TA was performed. We didn't observe the appearance of Pd nanoparticle precipitation and the reduction of GO did not occur based on the XPS result in Fig. S2 (ESI†). Hence, TA is very important in our experiments as the reductant for the preparation of PdNPs–G nanocomposites.
O (carbonyl), respectively. It is seen that the peak intensity of C–O is strong in GO due to the presence of oxygen-containing functional groups (Fig. 2a). In contrast, after microwave irradiation, the peak intensity of C–O in the products (Fig. 2b) tremendously reduced. Note that TA consists of a large amount of –OH and –COOH and thus makes much contribution to the intensity of C–O and CO peaks in products. All the observations suggest that the most oxygen-containing functional groups are successfully removed after microwave irradiation.13 The XPS pattern of the products also shows significant Pd 3d signals corresponding to the binding energy of metal Pd (Fig. 2c),7d indicating that H2PdCl4 has been reduced by TA. Although there is a report about microwave irradiation reduction of GO without the use of additional reducing agents,14 in order to clarify the role of TA, a control experiment under microwave irradiation in the absence of TA was performed. We didn't observe the appearance of Pd nanoparticle precipitation and the reduction of GO did not occur based on the XPS result in Fig. S2 (ESI†). Hence, TA is very important in our experiments as the reductant for the preparation of PdNPs–G nanocomposites.
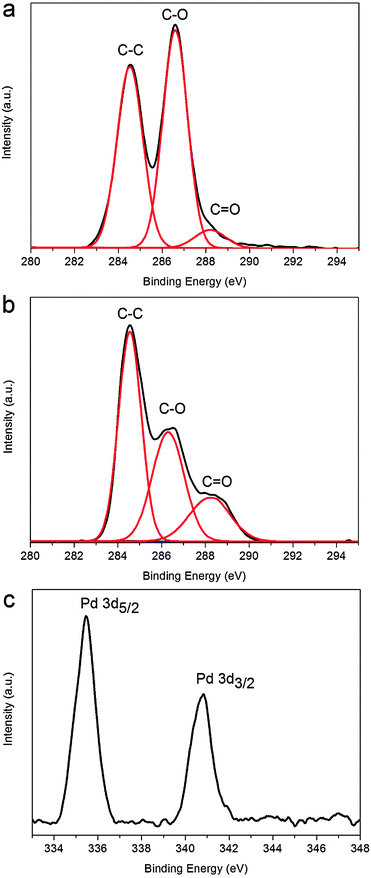 | ||
| Fig. 2 C 1s spectra of GO (a) and the products (b); the Pd 3d XPS spectrum of the products (c). | ||
Fig. 3 shows the TEM images of products thus formed. Fig. 3a shows a TA-functionalized GO sheet in the suspension of TA–GO. After microwave irradiation of the mixture of H2PdCl4 and TA–GO, it is seen that the G sheet has been decorated with a large amount of PdNPs about 10 nm to 20 nm in size (Fig. 3b). When the GO amount was decreased from 0.5 mL to 0.25 mL and 0.125 mL, the loading amount of PdNPs increased with sizes almost unchanged, as shown in Fig. 3c and d. It was also found that increasing irradiation time from 1 min to 2 min leads to an increase of PdNP size from several nanometres to 20 nm, as shown in Fig. 4 (the samples of PdNPs–G obtained by microwave irradiation for 1 min and 2 min are as PdNPs–G1 and PdNPs–G2, respectively). Such observations indicate that the loading amount and sizes of PdNPs on a G sheet can be controlled by the ratio of raw materials and microwave irradiation time.
 | ||
| Fig. 3 TEM images of TA/GO (a), PdNPs–G nanocomposites obtained by 2 min microwave irradiation with the use of 0.5 mL (b), 0.25 mL (c), and 0.125 mL (d) of GO. | ||
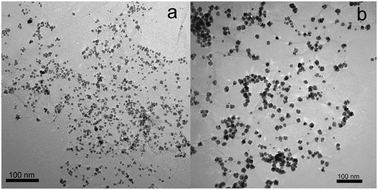 | ||
| Fig. 4 TEM images of PdNPs–G obtained by microwave irradiation for 1 min (a) and 2 min (b), with the use of 0.125 mL GO. | ||
The smaller size and good dispersion of PdNPs are favorable for the oxidation of methanol. Next, we investigated the performance of methanol oxidation with the use of these two samples as catalysts, as shown in Fig. 5. The electrochemically active surface area (ECSA) can provide important information on the available active sites of catalysts, hence, prior to evaluation of the electrocatalytic activity of the as-prepared catalysts toward methanol oxidation, the cyclic voltammograms (CVs) of PdNPs–G1 (curve a) and PdNPs–G2 (curve b) and commercial Pd/C (curve c) electrodes were measured in N2-saturated 0.5 M H2SO4 solution to estimate ECSA, as shown in Fig. 5a. The CVs show typical potential regions for hydrogen adsorption/desorption and the formation/reduction of surfaces of Pd oxide, Pt–OHad or Pd(OH)2.5d It is interesting to note that the reduction peak potential of Pd(OH)2 also shifted positively on the PdNPs–G1 and PdNPs–G2 (0.43 V and 0.45 V), relative to that on the commercial Pd/C catalysts (0.37 V). The positive shift of the reduction potential of Pd(OH)2 indicates the reduced oxophilicity and weakened chemisorption with oxygen-containing species, which can benefit the organic-fuel oxidation reactions in fuel cells.15 The reduction charge of the Pd(OH)2 was employed to estimate electrochemical active area (ECSA) values for the Pd catalysts based on the charge density for the formation of a Pd(OH)2 monolayer (430 μC cm−2).16 The ECSA values for PdNPs–G1 and PdNPs–G2 are 51.0 and 38.5 m2 g−1, which are higher than that of the Pd/C electrode (30.4 m2 g−1). This indicates that the PdNPs–G electrode has a high ECSA, which will probably enhance the active sites for the electrooxidation reaction of methanol.
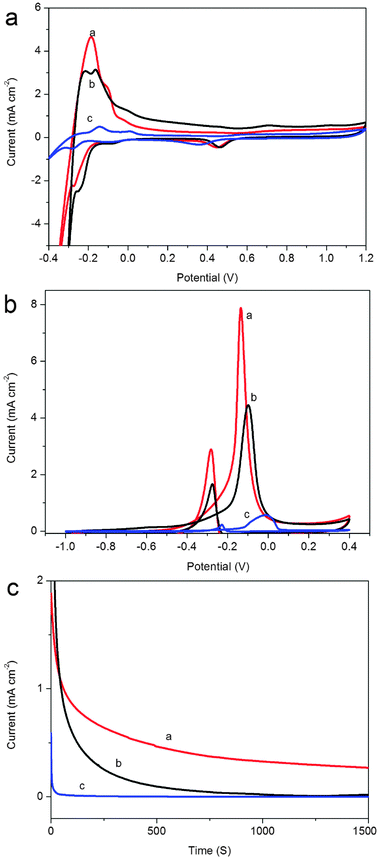 | ||
| Fig. 5 (a) CVs of different catalysts: PdNPs–G1 (curve a), PdNPs–G2 (curve b) and commercial Pd/C (curve c) in N2 saturated 0.5 M H2SO4 solution at a scan rate of 50 mV s−1; (b) CVs of methanol oxidation on corresponding catalysts in 0.5 M NaOH + 1 M CH3OH solution at a scan rate of 50 mV s−1; (c) chronoamperometry of PdNPs–G1 (curve a), PdNPs–G2 (curve b) and Pd/C (curve c) in 0.5 M NaOH + 1 M CH3OH solution at an operation potential of −0.1 V at 25 °C. | ||
The CVs of methanol oxidation on PdNPs–G1, PdNPs–G2 and Pd/C are displayed in Fig. 5b, which were recorded in 0.5 M NaOH + 1 M methanol at a scan rate of 50 mV s−1. Two peaks of methanol oxidation under anodic condition can be clearly observed: the oxidation peak in the forward scan was corresponding to the oxidation of freshly chemisorbed species coming from methanol adsorption; the reverse oxidation peak was primarily associated with removal of carbonaceous species not completely oxidized in the forward scan, rather than caused by freshly chemisorbed species. The magnitude of the peak current on the forward scan indicated the electrocatalytic activity of the electrocatalysts for methanol oxidation. It is clearly seen that PdNPs–G1 catalyst exhibits an especially high current density compared with that of PdNPs–G2 and Pd/C. This result indicates that the PdNPs–G1 catalyst exhibits better electrooxidation activity towards methanol than that of PdNPs–G2 and Pd/C, which can be attributed to the high ECSA from the smaller size and excellent dispersion of PdNPs on a G sheet. It is well known that the stability of catalysts is important for the application of fuel cells. However, redox reactions on the surface of catalysts often cause inactivation of catalytic activity. To evaluate the electrocatalytic stability of the catalysts under continuous operating conditions, long-term chronoamperometric experiments were carried out in a 0.5 M NaOH + 1 M methanol solution.17Fig. 5c shows the curves of current densities versus time recorded at −0.1 V for 1400 s. The rapid current decay showed the poisoning of the electrocatalysts, likely due to the formation of intermediate and some poisoning species during the methanol oxidation reaction.4b,18 It is found that the current decay for the reaction on the PdNPs–G1 (curve a) is significantly slower than those on the PdNPs–G2 (curve b) and commercial Pd/C (curve c), further demonstrating a significantly enhanced electrochemical stability of the PdNPs–G electrode for methanol electrooxidation in alkaline media. This result suggests that the G sheet plays a critical role in promoting the methanol oxidation, which could be attributed to the intrinsic electron transfer characteristic of G sheet and good dispersion of PdNPs on the G sheet.19
In conclusion, microwave irradiation of H2PdCl4 and GO in the presence of TA has been proven to be an effective method for the rapid preparation of PdNPs–G nanocomposites. The resultant nanocomposites exhibit significantly enhanced catalytic activity and stability than that of commercial Pd/C catalysts towards methanol electrooxidation, and therefore, hold great promise as effective electrocatalysts for DMFCs. Our present finding is important because it provides us an environmentally friendly method for rapid preparation of metal nanoparticles–G nanocomposites on a large scale for applications.
Acknowledgements
This work was supported by National Basic Research Program of China (No. 2011CB935800).Notes and references
- (a) M. Jafarian, M. G. Mahjani, H. Heli, F. Gobal, H. Khajehsharifi and M. H. Hamedi, Electrochim. Acta, 2003, 48, 3423 CrossRef CAS; (b) B. C. Norris, W. Li, E. J. Lee, A. Manthiram and C. W. Bielawski, Polymer, 2010, 51, 5352 CAS.
- M. Jafarian, R. B. Moghaddam, M. G. Mahjani and F. Gobal, J. Appl. Electrochem., 2006, 36, 913 CrossRef CAS.
- H. Heli, M. Jafarian, M. G. Mahjani and F. Gobal, Electrochim. Acta, 2004, 49, 4999 CrossRef CAS.
- (a) C. W. Xu, P. K. Shen and Y. L. Liu, J. Power Sources, 2007, 164, 527 CrossRef CAS; (b) Y. C. Zhao, L. Zhan, J. N. Tian, S. L. Nie and Z. Ning, Electrochim. Acta, 2011, 56, 1967 CrossRef CAS.
- (a) S. Ha, R. Larsen and R. I. Masel, J. Power Sources, 2005, 144, 28 CrossRef CAS; (b) P. K. Babu, H. S. Kim, J. H. Chung, E. Oldfield and A. Wieckowski, J. Phys. Chem. B, 2004, 108, 20228 CrossRef CAS; (c) M. Arenz, V. Stamenkovic, T. J. Schmidt, K. Wandelt, P. N. Ross and N. M. Markovic, Phys. Chem. Chem. Phys., 2003, 5, 4242 RSC; (d) S. J. Guo, S. J. Dong and E. K. Wang, Energy Environ. Sci., 2010, 3, 1307 RSC.
- K. Jasuja and V. Berry, ACS Nano, 2009, 3, 2358 CrossRef CAS.
- (a) A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS; (b) J. S. Bunch, A. M. Van Der Zande, S. S. Verbridge, I. W. Frank, D. M. Tanenbaum, J. M. Parpia, H. G. Craighead and P. L. McEuen, Science, 2007, 315, 490 CrossRef CAS; (c) S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217 CrossRef CAS; (d) S. J. Guo, S. J. Dong and E. K. Wang, ACS Nano, 2010, 4, 547 CrossRef CAS; (e) D. R. Dreyer, S. J. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228 RSC; (f) D. R. Dreyer, R. S. Ruoff and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 9336 CrossRef CAS.
- (a) Z. Wen, S. Yang, Y. Liang, W. He, H. Tong, L. Hao, X. Zhang and Q. Song, Electrochim. Acta, 2010, 56, 139 CrossRef CAS; (b) H. Zhao, J. Yang, L. Wang, C. Tian, B. Jiang and H. Fu, Chem. Commun., 2011, 47, 2014 RSC; (c) J. Yang, C. Tian, L. Wang and H. Fu, J. Mater. Chem., 2011, 21, 3384 RSC; (d) J. J. Shi and J. J. Zhu, Electrochim. Acta, 2011, 56, 6008 CrossRef CAS; (e) J. S. Cheng, G. C. Zhang, J. Du, L. H. Tang, J. Y. Xu and J. H. Li, J. Mater. Chem., 2011, 21, 3485 RSC.
- A. E. Hagerman, K. M. Riedl, G. A. Jones, K. N. Sovik, N. T. Ritchard, P. W. Hartzfeld and T. L. Riechel, J. Agric. Food Chem., 1998, 46, 1887 CrossRef CAS.
- (a) M. Zhou, Y. L. Wang, Y. M. Zhai, J. F. Zhai, W. Ren, F. A. Wang and S. J. Dong, Chem.–Eur. J., 2009, 15, 6116 CrossRef CAS; (b) Z. J. Wang, X. Z. Zhou, J. Zhang, F. Boey and H. Zhang, J. Phys. Chem. C, 2009, 113, 14071 CrossRef CAS.
- Y. D. Lei, Z. H. Tang, R. J. Liao and B. C. Guo, Green Chem., 2011, 13, 1655 RSC.
- V. C. Tung, M. J. Allen, Y. Yang and R. B. Kaner, Nat. Nanotechnol., 2008, 4, 25 CrossRef.
- (a) S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS; (b) Y. Guo, S. Guo, J. Ren, Y. Zhai, S. Dong and E. Wang, ACS Nano, 2010, 4, 4001 CrossRef CAS.
- Y. W. Zhu, S. Murali, M. D. Stoller, A. Velamakanni, R. D. Piner and R. S. Ruoff, Carbon, 2010, 48, 2106 CrossRef.
- S. Wang, S. P. Jiang, T. J. White, J. Guo and X. Wang, J. Phys. Chem. C, 2009, 113, 18935 CAS.
- L. Xiao, L. Zhuang, Y. Liu, J. T. Lu and H. D. Abruna, J. Am. Chem. Soc., 2009, 131, 602 CrossRef CAS.
- Z. Y. Zhou, Z. Z. Huang, D. J. Chen, Q. Wang, N. Tian and S. G. Sun, Angew. Chem., Int. Ed., 2010, 49, 411 CAS.
- (a) Y. H. Qin, H. H. Yang, X. S. Zhang, P. Li and C. A. Ma, Int. J. Hydrogen Energy, 2010, 35, 7667 CrossRef CAS; (b) R. N. Singh, A. Singh and Anindita, Int. J. Hydrogen Energy, 2009, 34, 2052 CrossRef CAS; (c) R. N. Singh, A. Singh and Anindita, Carbon, 2009, 47, 271 CrossRef CAS; (d) L. F. Dong, R. R. S. Gari, Z. Li, M. M. Craig and S. F. Hou, Carbon, 2010, 48, 781 CrossRef CAS.
- J. J. Shi, G. H. Yang and J. J. Zhu, J. Mater. Chem., 2011, 21, 7343 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and supplementary figures. See DOI: 10.1039/c1cy00296a |
| This journal is © The Royal Society of Chemistry 2011 |