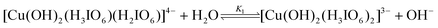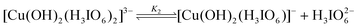Mechanistic aspects of uncatalyzed and ruthenium(III) catalyzed oxidation of 1,4-dioxane by a copper(III) periodate complex in aqueous alkaline medium
Received
31st May 2011
, Accepted 25th June 2011
First published on 3rd August 2011
Abstract
Oxidation of 1,4-dioxane (Dio) by a copper(III) periodate complex (DPC) has been investigated spectrophotometrically both in the absence and presence of a ruthenium(III) catalyst in aqueous alkaline medium at 298 K. Stoichiometry of (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8) was observed in both the reactions and the order of the reaction with respect to [DPC] was unity while the order with respect to [Dio] was <1 over the concentration range studied. Order with respect to [OH−] and [IO4−] had negative order in both cases. Order with respect to [Ru(III)] was unity. The activation parameters (ΔH# = 38 ± 2 kJ mol−1, ΔS# = −164 ± 15 J K−1 mol−1 and ΔG# = 87 ± 4 for uncatalyzed and ΔH# = 34 ± 3 kJ mol−1, ΔS# = −43 ± 2 J K−1 mol−1 and ΔG# = 47 ± 3 for catalyzed reaction) with respect to the slow step of the proposed mechanism were computed and thermodynamic quantities were also calculated. Kinetic studies suggest that [Cu(OH)2(H3IO6)]− is the reactive copper(III) species and [Ru(H2O)5(OH)]2+ is the reactive Ru(III) species.
:8) was observed in both the reactions and the order of the reaction with respect to [DPC] was unity while the order with respect to [Dio] was <1 over the concentration range studied. Order with respect to [OH−] and [IO4−] had negative order in both cases. Order with respect to [Ru(III)] was unity. The activation parameters (ΔH# = 38 ± 2 kJ mol−1, ΔS# = −164 ± 15 J K−1 mol−1 and ΔG# = 87 ± 4 for uncatalyzed and ΔH# = 34 ± 3 kJ mol−1, ΔS# = −43 ± 2 J K−1 mol−1 and ΔG# = 47 ± 3 for catalyzed reaction) with respect to the slow step of the proposed mechanism were computed and thermodynamic quantities were also calculated. Kinetic studies suggest that [Cu(OH)2(H3IO6)]− is the reactive copper(III) species and [Ru(H2O)5(OH)]2+ is the reactive Ru(III) species.
1. Introduction
1,4-Dioxane (Dio) is an important solvent both in the laboratory and in industry. It has a valuable property of being miscible with water in all proportions and also possesses excellent dissolution power for several organic compounds.1 It is a popular solvent that is often used in place of glacial acetic acid. It is also used as a stabilizer1 in chlorinated solvents and in organic syntheses. It is an excellent chelating diether ligand. It reacts with Grignard reagents to precipitate the magnesium dihalide. In this way, dioxane is used to drive the Schlenk equilibrium.2
Transition metals in their higher oxidation states can generally be stabilized by chelation with suitable polydentate ligands. These metal chelates such as diperiodatocuprate(III),3diperiodatoargentate(III)4 and diperiodatonickelate(IV)5 are good oxidants in a medium with an appropriate pH value. Diperiodatocuprate(III) is a versatile one-electron oxidant and the oxidation study of DPC is scarce in view of its limited solubility and stability in aqueous medium. Its use as an analytical reagent is now well recognized.6Copper complexes have played a major role in oxidation chemistry as experimentally copper(III) is involved and relevance is biological.7,8Copper(III) is involved in many biological electron transfer reactions.9 When a copper(III) periodate complex is the oxidant and multiple equilibria between different copper(III) species are involved, it would be interesting to know which of the species is the active oxidant.
Transition metals are known to catalyze many oxidation–reduction reactions since they involve multiple oxidation states. In recent years the use of transition metal ions, such as ruthenium, osmium, palladium, manganese, chromium, iridium, either alone or as binary mixtures, as catalysts in various redox processes has attracted considerable interest.10 The Ru(III) acts as a catalyst in the oxidation of many organic and inorganic substrates.11,12 The catalyzed mechanism can be quite complicated due to formation of different intermediate complexes and different oxidation states of ruthenium(III). Although the mechanism of catalysis depends on the nature of substrates, oxidants and experimental conditions, it has been shown13 that metal ions act as catalysts by one of the different paths such as the formation of complexes with reactants or oxidation of a substrate itself or through the formation of free radicals. In earlier reports,14 it has been observed that Ru(III) forms a complex with the substrate, which gets oxidized by the oxidant to form a Ru(IV)–substrate complex followed by the rapid redox decomposition to regenerate Ru(III). In another report,15 it has been observed that there involves the formation of a Ru(III)–substrate complex with further cleavage in a concerted manner giving rise to a Ru(I) species, which gets rapidly oxidized by the oxidant to regenerate the catalyst. In some other reports,16 it is observed that Ru(III) forms a complex with a substrate and is oxidized by the oxidant with the regeneration of the catalyst. Hence, understanding the role of Ru(III) catalyzed reaction is important. We have observed that Ru(III) catalyzes the oxidation of Dio by DPC in alkaline medium in small amounts.
In earlier reports9 on DPC oxidation, periodate had a retarding effect and the order in [OH−] was found to be less than unity in most of the reactions. However in the present study we have obtained entirely different kinetic observations. A literature survey revealed that there are no reports on the oxidation of 1,4-dioxane with DPC in the absence as well as presence of any catalyst. Hence, the present investigation is aimed at checking the reactivity of dioxane towards DPC in both uncatalyzed and ruthenium(III) catalyzed reactions and to arrive at the plausible mechanisms.
2. Experimental
2.1. Materials and reagents
All chemicals used were of reagent grade and double distilled water was used throughout the work. A solution of 1,4-dioxane (S.D. Fine Chem.) was prepared by dissolving an appropriate amount of purified17 sample in double distilled water. The purity of the Dio sample was checked by comparing its IR spectrum with the literature data and its boiling point (101.5 °C) with literature data (101 °C). The required concentration of Dio was obtained from its stock solution. A standard stock solution of Ru(III) was prepared by dissolving RuCl3 (S.D. Fine Chem.) in 0.20 mol dm−3HCl. The concentration was determined18 by EDTA titration. KNO3 and KOH (BDH) were used to maintain ionic strength and alkalinity of the reaction, respectively.
The copper(III) periodate complex was prepared by a standard procedure.19 Aqueous solution of copper(III) was standardized by iodometric titration and gravimetrically by a thiocyanate method.20 The copper(II) solutions were prepared by dissolving a known amount of copper sulfate (BDH) in distilled water. Periodate solution was prepared by weighing out the required amount of the sample in hot water and used after keeping it for 24 h. Its concentration was ascertained iodometrically21 at neutral pH maintained using phosphate buffer. Since periodate is present in excess in DPC, the possibility of oxidation of Dio by periodate in alkaline medium at 25 °C was tested. The progress of the reaction was followed iodometrically. There was no significant reaction under the experimental conditions employed compared to the DPC oxidation of Dio. The pH of the medium in the solution was measured by ELICO (LI613) pH meter. Solutions of Dio and DPC were always freshly prepared before use.
2.2. Instruments used
(i) For kinetic measurements, a Peltier Accessory (temperature control) attached Varian CARY 50 Bio UV-vis spectrophotometer (Varian, Victoria-3170, Australia) was used.
(ii) For product analysis, a QP-2010S Shimadzu gas chromatograph mass spectrometer and a Nicolet 5700-FT-IR spectrometer (Thermo, U.S.A.), and for pH measurements, an Elico pH meter model LI120 were used.
2.3. Kinetic measurements
Kinetic measurements were performed on a Varian Bio carry-50 spectrophotometer. The reaction was monitored under pseudo first-order conditions where [Dio] > [DPC] in both uncatalyzed and catalyzed reactions at 25.0 ± 0.1 °C, unless specified. In the absence of catalyst the reaction was initiated by mixing DPC with the Dio solution which also contained required concentrations of KNO3, KOH and KIO4. The reaction in the presence of catalyst Ru(III) was initiated by mixing DPC with the Dio solution which also contained the required concentration of KNO3, KOH, KIO4 and Ru(III) catalyst. Progress of the reaction was monitored spectrophotometrically at 415 nm (i.e., decrease in absorbance due to DPC with the molar absorbency index, ‘ε’ to be 6230 ± 100 dm3 mol−1 cm−1 in both catalyzed and uncatalyzed reaction), which is the maximum absorption wavelength of DPC. It was verified that there was almost no interference from other species in the reaction mixture at this wavelength.
The total concentrations of periodate and OH− were calculated by considering the amount present in the DPC solution and that additionally added. Kinetic runs were also carried out in a nitrogen atmosphere in order to understand the effect of dissolved oxygen on the rate of the reaction. No significant difference in the results was obtained under a nitrogen atmosphere and in the presence of air. In view of the ubiquitous contamination of carbonate in the basic medium, the effect of carbonate was also studied. Added carbonate had no effect on the reaction rates.
Pseudo first-order rate constants (kU or kC) in both the cases were determined from the log (absorbance) versus time plots and were the average of duplicate runs. The plots were linear up to 70% completion of reaction under the range of [OH−] used. The orders for various species were determined from the slopes of plots of log (kU or kC) versus respective concentration of species except for [DPC] in which non-variation of ‘kU or kC’ was observed as expected to the reaction conditions. The rate constants were reproducible to within ±5%. Regression analysis of experimental data to obtain regression coefficient r and the standard deviation S, of points from the regression line, was performed with the Microsoft office Excel-2003 programme.
3. Results
3.1. Stoichiometry and product analysis
Different sets of reaction mixtures containing varying ratios of DPC to Dio in the presence of constant amounts of OH−, KIO4, and KNO3 in uncatalyzed reaction and a constant amount of Ru(III) in catalyzed reaction were kept for six hours in a closed vessel under a nitrogen atmosphere. The remaining concentration of DPC was assayed by measuring the absorbance at 415 nm. The results indicated 1:8 stoichiometry for both the reactions as given in eqn (1).| | 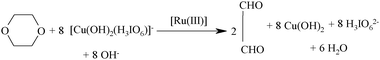 | (1) |
After completion of reaction, the reaction mixture was acidified, concentrated and extracted with ether. The main reaction product was identified as glyoxal. Only one product was obtained as evidenced by a single spot on thin layer chromatography, which was identified by a spot test22 and was characterized by FT-IR, GC-MS spectral studies.
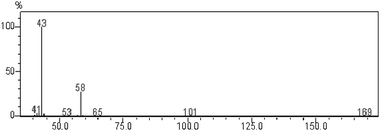
The IR (KBr) spectrum shows a carbonyl stretching of aldehyde at 1636 cm−1 and –CH stretching of aldehyde at 2927 cm−1. The aldehyde was confirmed by preparing its 2,4-DNP derivative.23GC-MS data were obtained on a QP-2010S Shimadzu gas chromatograph mass spectrometer. The mass spectral data showed a molecular ion peak at 58 m/z confirming the presence of glyoxal (Fig. 1). All the peaks observed in GC-MS can be interpreted in accordance with the observed structure of glyoxal. Another product, Cu(II), was identified by UV-vis spectra (Fig. S1). The reaction product did not undergo further oxidation under the present kinetic conditions.
3.2. Reaction orders
As the diperiodatocuprate(III) oxidation of dioxane in alkaline medium proceeds with a measurable rate in the absence of Ru(III), the catalyzed reaction is understood to occur in parallel paths with contributions from both the catalyzed and uncatalyzed paths. Thus the total rate constant (kT) is equal to the sum of the rate constants of the catalyzed (kC) and uncatalyzed (kU) reactions, so kC = kT − kU. Hence the reaction orders have been determined from the slopes of log kCversus log (concentration) plots by varying the concentrations of Dio, alkali, periodate and Ru(III) catalyst in turn while keeping all other concentrations and conditions constant.
3.3. Evaluation of pseudo first-order rate constants
The oxidant DPC concentration was varied in the range of 1.0 × 10−5 to 1.0 × 10−4 mol dm−3 at constant and excess concentrations of [Dio], [KOH] and [KIO4] in both the cases of catalyzed and uncatalyzed reactions. The fairly constant pseudo first-order rate constants, kU and kC, indicate that the order with respect to [DPC] was unity (Fig. 2; Table 1 for uncatalyzed reaction, Table 2 for Ru(III) catalyzed).
![First order plots the oxidation of dioxane by diperiodatocuprate(iii) in aqueous alkaline medium at 298 K. (105DPC mol dm−3): (1) 1.0; (2) 3.0; (3) 5.0; (4) 8.0; (5) 10.0. [Dio] = 4 × 10−3, [OH−] = 0.01 and [IO4−] = 1 × 10−5.](/image/article/2011/CY/c1cy00192b/c1cy00192b-f2.gif) |
| | Fig. 2 First order plots the oxidation of dioxane by diperiodatocuprate(III) in aqueous alkaline medium at 298 K. (105DPC mol dm−3): (1) 1.0; (2) 3.0; (3) 5.0; (4) 8.0; (5) 10.0. [Dio] = 4 × 10−3, [OH−] = 0.01 and [IO4−] = 1 × 10−5. | |
Table 1 Effect of [DPC], [Dio], [IO4−] and [OH−] on the oxidation of dioxane by diperiodatocuprate(III) in alkaline medium at 25 °C, I = 0.11 mol dm−3
| [DPC] × 105/mol dm−3 |
[Dio] × 103/mol dm−3 |
[IO4−] × 105/mol dm−3 |
[OH−]/mol dm−3 |
k
U × 103/s−1 |
| Found |
Calculated |
| 1.0 |
4.0 |
1.0 |
0.01 |
1.25 |
1.22 |
| 3.0 |
4.0 |
1.0 |
0.01 |
1.24 |
1.22 |
| 5.0 |
4.0 |
1.0 |
0.01 |
1.22 |
1.22 |
| 8.0 |
4.0 |
1.0 |
0.01 |
1.29 |
1.22 |
| 10.0 |
4.0 |
1.0 |
0.01 |
1.27 |
1.22 |
| |
| 5.0 |
0.8 |
1.0 |
0.01 |
0.34 |
0.34 |
| 5.0 |
2.0 |
1.0 |
0.01 |
0.74 |
0.74 |
| 5.0 |
4.0 |
1.0 |
0.01 |
1.22 |
1.22 |
| 5.0 |
6.0 |
1.0 |
0.01 |
1.57 |
1.56 |
| 5.0 |
8.0 |
1.0 |
0.01 |
1.85 |
1.82 |
| |
| 5.0 |
4.0 |
1.0 |
0.01 |
1.22 |
1.22 |
| 5.0 |
4.0 |
3.0 |
0.01 |
0.95 |
0.94 |
| 5.0 |
4.0 |
5.0 |
0.01 |
0.79 |
0.76 |
| 5.0 |
4.0 |
8.0 |
0.01 |
0.61 |
0.59 |
| 5.0 |
4.0 |
10.0 |
0.01 |
0.53 |
0.51 |
| |
| 5.0 |
4.0 |
1.0 |
0.01 |
1.22 |
1.22 |
| 5.0 |
4.0 |
1.0 |
0.03 |
0.98 |
0.96 |
| 5.0 |
4.0 |
1.0 |
0.05 |
0.78 |
0.79 |
| 5.0 |
4.0 |
1.0 |
0.08 |
0.61 |
0.63 |
| 5.0 |
4.0 |
1.0 |
0.1 |
0.55 |
0.55 |
Table 2 Effect of [DPC], [Dio], [IO4−], [OH−] and [Ru(III)] on the ruthenium(III) catalysed oxidation of dioxane by DPC in alkaline medium at 25 °C, I = 0.11 mol dm3
| [DPC] × 105/mol dm−3 |
[Dio] × 103/mol dm−3 |
[IO4−] × 105/mol dm−3 |
[OH−]/mol dm−3 |
[Ru(III)] × 107/mol dm−3 |
k
T × 103/s−1 |
k
U × 103/s−1 |
k
C × 103/s−1 |
| Found |
Calculated |
| 1.0 |
4.0 |
1.0 |
0.01 |
6.0 |
9.19 |
1.25 |
7.94 |
8.38 |
| 3.0 |
4.0 |
1.0 |
0.01 |
6.0 |
9.21 |
1.24 |
7.97 |
8.38 |
| 5.0 |
4.0 |
1.0 |
0.01 |
6.0 |
9.49 |
1.22 |
8.27 |
8.38 |
| 8.0 |
4.0 |
1.0 |
0.01 |
6.0 |
9.5 |
1.29 |
8.21 |
8.38 |
| 10.0 |
4.0 |
1.0 |
0.01 |
6.0 |
9.3 |
1.27 |
8.03 |
8.38 |
| 5.0 |
0.8 |
1.0 |
0.01 |
6.0 |
2.68 |
0.34 |
2.34 |
2.25 |
| 5.0 |
2.0 |
1.0 |
0.01 |
6.0 |
5.93 |
0.74 |
5.19 |
4.95 |
| 5.0 |
4.0 |
1.0 |
0.01 |
6.0 |
9.49 |
1.22 |
8.27 |
8.38 |
| 5.0 |
6.0 |
1.0 |
0.01 |
6.0 |
12.9 |
1.57 |
11.3 |
10.8 |
| 5.0 |
8.0 |
1.0 |
0.01 |
6.0 |
15.2 |
1.85 |
13.3 |
12.6 |
| 5.0 |
4.0 |
1.0 |
0.01 |
6.0 |
9.49 |
1.22 |
8.27 |
8.38 |
| 5.0 |
4.0 |
3.0 |
0.01 |
6.0 |
8.11 |
0.94 |
7.12 |
7.10 |
| 5.0 |
4.0 |
5.0 |
0.01 |
6.0 |
7.39 |
0.79 |
6.59 |
6.16 |
| 5.0 |
4.0 |
8.0 |
0.01 |
6.0 |
6.49 |
0.61 |
5.87 |
5.14 |
| 5.0 |
4.0 |
10.0 |
0.01 |
6.0 |
5.17 |
0.53 |
4.64 |
4.63 |
| 5.0 |
4.0 |
1.0 |
0.01 |
6.0 |
9.49 |
1.22 |
8.27 |
8.38 |
| 5.0 |
4.0 |
1.0 |
0.03 |
6.0 |
8.26 |
0.98 |
7.28 |
7.34 |
| 5.0 |
4.0 |
1.0 |
0.05 |
6.0 |
7.34 |
0.78 |
6.56 |
6.53 |
| 5.0 |
4.0 |
1.0 |
0.08 |
6.0 |
6.52 |
0.61 |
5.91 |
5.60 |
| 5.0 |
4.0 |
1.0 |
0.1 |
6.0 |
5.49 |
0.55 |
4.93 |
5.12 |
| 5.0 |
4.0 |
1.0 |
0.01 |
1.0 |
2.72 |
1.22 |
1.50 |
1.39 |
| 5.0 |
4.0 |
1.0 |
0.01 |
3.0 |
5.64 |
1.22 |
4.42 |
4.19 |
| 5.0 |
4.0 |
1.0 |
0.01 |
6.0 |
9.49 |
1.22 |
8.27 |
8.38 |
| 5.0 |
4.0 |
1.0 |
0.01 |
8.0 |
12.6 |
1.22 |
11.38 |
11.1 |
| 5.0 |
4.0 |
1.0 |
0.01 |
10.0 |
15.4 |
1.22 |
14.1 |
13.9 |
3.4. Effect of varying [dioxane]
The dioxane concentration was varied in the range of 8.0 × 10−4 to 8.0 × 10−3 mol dm−3 at 25 °C while keeping other reactant concentrations and conditions constant in uncatalyzed and with constant concentration of Ru(III) in catalyzed reaction. In the case of uncatalyzed reaction as well as catalyzed reaction, at constant temperature, the kU and kC values increased with the increase in concentration of dioxane. The order with respect to [Dio] was less than unity (Table 1 for uncatalyzed and Table 2 for Ru(III) catalyzed). This less than unit order in the dioxane was also confirmed by the linear plots of kUversus[Dio]0.74 and kCversus[Dio]0.75 which were linear rather than the direct plots of kUversus [Dio] and kCversus [Dio] (Fig. S2).
3.5. Effect of varying [alkali]
The effect of alkali was studied for both the cases in the range of 0.01 to 0.1 mol dm−3 at constant concentrations of DPC, Dio, IO4− and ionic strength in the uncatalyzed and with constant concentrations of Ru(III) in catalyzed reaction. The rate constants decreased with increase in alkali and orders were found to be less than unity in both the reactions (Tables 1 and 2).
3.6. Effect of varying [periodate]
The effect of [IO4−] was observed by varying the concentration from 1.0 × 10−5 to 1.0 × 10−4 mol dm−3 while keeping all other reactants concentrations and conditions constant as well as excess. It was observed that the rate constants decreased by increasing [IO4−] in both the cases (Tables 1 and 2).
3.7. Effect of [Ru(III)]
Ruthenium(III) concentration was varied from 1.0 × 10−7 to 1.0 × 10−6 mol dm−3 range, at excess and constant concentrations of a copper(III) periodate complex, dioxane, alkali and at constant ionic strength. The order in [Ru(III)] was found to be unity from the linearity of the plot of kCversus [Ru(III)] (Table 2Ru(III) catalyzed; Fig. S3).
3.8. Effect of initially added products
Initially added products, copper(II) (CuSO4) and glyoxal, did not have any significant effect on the rate of reaction in both the cases.
3.9. Effect of ionic strength (I) and dielectric constant of the medium (D)
Up on addition of KNO3, at constant [DPC], [Dio], [Ru(III)], [OH−] and [IO4−], it was found that increasing ionic strength had no significant effect on the rate of reaction.
The dielectric constant of the medium, ‘D’, was varied by varying the tert-butyl alcohol and water percentage. The D values were calculated from the equation D = DwVw + DBVB, where Dw and DB are dielectric constants of pure water and tert-butyl alcohol, respectively, and Vw and VB are the volume fractions of components, water and tert-butyl alcohol, respectively, in the total mixture. There was no effect of the dielectric constant on the rate of both the cases of catalyzed and uncatalyzed reactions.
Thus from the observed experimental results:
The rate law for uncatalyzed reaction is given as follows:
| Rate = kU [Dio]0.74 [DPC]1.0 [OH−]−0.34 [IO4−]−0.35 |
The rate law for Ru(III) catalyzed reaction is given as follows:
| Rate = kC [Dio]0.75 [DPC]1.0 [OH−]−0.20 [Ru(III)]1.0[IO4−]−0.22 |
3.10. Test for free radicals (polymerisation study)
For both uncatalyzed and catalyzed reactions the possibility of free radicals was tested as follows. The reaction mixture, to which a known quantity of acrylonitrile (scavenger) had been added initially, was kept in an inert atmosphere for six hours. On diluting the reaction mixture with methanol, a white precipitate was formed due to the polymerization of an acrylonitrile monomer, indicating the intervention of free radicals in the reactions. The blank experiments of either DPC or dioxane alone with acrylonitrile did not induce any polymerization under the same conditions as those induced for the reaction mixture. Initially, added acrylonitrile decreases the rate of reaction indicating free radical intervention, which is the case in earlier work.24
3.11. Effect of temperature (T)
The kinetics was studied at five different temperatures under varying concentrations of Dio, alkali, and periodate, keeping other conditions constant. The rate constants were found to increase with increase in temperature. The rate constants of the slow step (k1) of Scheme 1 were obtained from the slopes and intercepts of 1/kobsversus 1/[Dio], 1/kobsversus [H3IO62−], and 1/kobsversus [OH−] plots at five different temperatures and were used to calculate the activation parameters for the uncatalyzed reaction. The activation parameters corresponding to these constants were evaluated from the Eyring plot of (Rlnk1/T × h/kB) (r ≥ 0.9896, S ≤ 0.00035) (h = Planck's constant, kB = Boltzmann constant) versus 1/T and are tabulated in Table 3. For the catalyzed reaction the influence of temperature on the rate of reaction was studied at 15, 20, 25, 30 and 35 °C. The rate constants, k2, of the slow steps in the mechanisms were obtained from the slopes and intercepts of the plots [Ru(III)]/kCversus 1/[Dio], Ru(III)]/kCversus [H3IO62−], and Ru(III)]/kCversus [OH−] at five different temperatures for the catalyzed reaction. The activation parameters corresponding to these constants were evaluated from the Eyring plot of (Rlnk2/T × h/kB) (r ≥ 0.9817, S ≤ 0.4041) (h = Planck's constant, kB = Boltzmann constant) versus 1/T and are tabulated in Table 3.
 |
| | Scheme 1 | |
Table 3 Effect of temperature on k1, k2 and KC for the uncatalyzed and Ru(III) catalyzed oxidation of dioxane by diperiodatocuprate(III) in aqueous alkaline medium
| (A) Effect of temperature |
| Temperature |
k
1 × 103/s−1 |
k
2 × 10−5/dm3 mol−1s−1 |
K
C × 10−3 |
| 288 |
1.87 |
0.24 |
8.8 |
| 293 |
2.60 |
0.35 |
10.5 |
| 298 |
3.54 |
0.43 |
13.8 |
| 303 |
4.69 |
0.51 |
19.3 |
| 308 |
5.49 |
0.68 |
23.0 |
| (B) Thermodynamic activation parameters for the uncatalyzed and Ru(III) catalyzed oxidation of dioxane by diperiodatocuprate(III) in aqueous alkaline medium |
| Parameters |
Values (from k1) |
Values (from k2) |
Values (from KC) |
|
ΔH#/kJ mol−1 |
38 ± 2 |
34 ± 3 |
35 ± 3 |
|
ΔS#/J K−1 mol−1 |
−164 ± 15 |
−43 ± 2 |
−49 ± 2 |
| ΔG#298 K/kJ mol−1 |
87 ± 4 |
47 ± 3 |
49 ± 2 |
| log A |
4.6 ± 0.3 |
10.9 ± 2 |
10.7 ± 1 |
3.12.
Catalytic activity
It has been pointed out by Moelwyn-Hughes25 that, in the presence of the catalyst, the uncatalysed and catalysed reactions proceed simultaneously, so that
Here kT is the observed pseudo first order rate constant in the presence of Ru(III) catalyst, kU is the pseudo first order rate constant for the uncatalysed reaction, KC is the catalytic constant and ‘x’ is the order of the reaction with respect to [Ru(III)]. In the present investigations, x values for the standard run were found to be unity for Ru(III). Then the value of KC can be calculated using eqn (2).| | 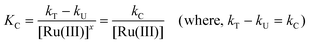 | (2) |
The values of KC were evaluated for the catalyzed reaction at different temperatures and found to vary at different temperatures. Further, plots of log KCversus 1/T were linear and the values of energy of activation and other activation parameters with reference to the catalyst were computed. These results are summarized in Table 3. The value of KC is 1.37 × 10−4 at 298 K.
4. Discussion
As a powerful oxidant, the copper(III) periodate complex in alkaline medium is relatively stable compared to other copper(III) complexes. Malaprade,26 Rozovski et al.,27 and Jensovsky and Milos Rabe28 have prepared a copper(III) periodate complex in pure form.
Water soluble DPC in alkaline medium shows an UV-vis spectrum28 with two strong bands at 413 and 265 nm, one fairly strong band at 334 nm, and a weak one at 560 nm. When solid K3H3[Cu(IO6)2]·4H2O is dissolved in water it undergoes dissociation to give a complex ion.29 In alkaline solution it may exist as [Cu(HL)2]x, [CuL(HL)]y and [Cu(HL)]z, where HL contains an indefinite number of protons and L denotes the unprotonated periodate molecule in the complex.
However, it is well known that periodate exists30 as H5IO6, H4IO6−, H3IO6−, H2IO63− (eqn (3)–(5)) and dimeric forms, depending on the pH of the medium and total concentration of periodate. In the present study, the [OH−] was varied from 0.01 to 0.10 mol dm−3. Thus, the periodate species should be in the form of H3IO62− and H2IO63−. The other species are expected to occur in acidic medium. The existence of dimeric forms can be ruled out as they occur only at higher [IO4−]. Consequently, the active forms of IO4− can be considered as H3IO63− and H2IO62−. Thus, as an alternative to the notation HL and L, respectively, and on incorporating the same into the [Cu(HL)2x], [CuL(HL)y] and [Cu(HL)z] formulae, leads to the structures [Cu(H3IO6)2−], [Cu(H2IO6)(H3IO6)2−] and [Cu(H3IO6)(H2O)+]. These structures retain the dsp2 configuration with square-planar structures. The bidentate ligands H3IO62− and H2IO63− satisfy the coordination number of Cu3+. Similar structures of periodate complexes have been deduced for the Ni(IV) and Ag(III) periodate complexes.31,32
| | | H3IO62− ⇌ H2IO63− + H+ | (5) |
4.1. Mechanism for uncatalyzed reaction
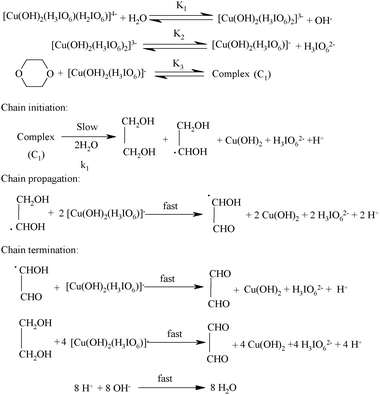
The reaction between DPC and dioxane in alkaline medium presents [DPC]/[Dio] = 1:8 stoichiometry. Since the reaction was retarded by [OH−], added periodate also retarded the rate and first order dependency in [DPC] and fractional order in [Dio] and [OH−], a plausible reaction mechanism has been proposed which also explains all other experimental observations as shown in Scheme 1.
In most of the reports33,34 on DPC oxidation, periodate retards and OH− increases the rate of the reaction. However in the present kinetic study, entirely different kinetic observations have been obtained. In this study OH− retards the rate of reaction and periodate also shows retarding effect on the rate. The result of decrease in rate of reaction with increase in alkalinity (Table 1) can be explained in terms of a prevailing equilibrium of formation of [Cu(OH)2(H3IO6)2)]3− from [Cu(OH)2(H3IO6)(H2IO6)]4−hydrolysis as given in Scheme 1.
Decrease in rate with increase in [H
3IO
62−] (
Table 1) suggests the equilibrium of a copper(
III) periodate complex to form monoperiodatocuprate(
III) (MPC) species as given in
Scheme 1.
The inverse fractional order in [IO
4−] might be due to this equilibrium. The less than unit order in [Dio] presumably results from formation of a complex (C
1) between the DPC species and
dioxane prior to the formation of the products. This complex (C
1) further undergoes
hydrolysis in a slow step to give the free radical species of Dio,
ethane-1,2-diol, periodate and Cu(
II). Further this free radical species of Dio reacts with one molecule of MPC species in a fast step to form
hydroxy acetaldehyde. This
hydroxy acetaldehyde then reacts with two more moles of MPC in a further fast step to form products such as
glyoxal, Cu(
II) and periodate. In the next step the ethane-1,2-diol reacts with four more moles of MPC in a further fast step to form products such as
glyoxal. All these results may be interpreted in the form of
Scheme 1.
Since Scheme 1 is in accordance with the generally well-accepted principle of non-complementary oxidations taking place in a sequence of one-electron steps, the reaction between the substrate and the oxidant would afford a radical intermediate. The free radical scavenging experiment revealed such a possibility (see infra). This type of radical intermediate has also been observed in earlier work.35
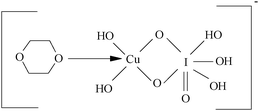
The probable structure of complex (C1) is
Spectroscopic evidence for the complex formation between the oxidant and the substrate was obtained from UV-vis spectra of Dio = (4.0 × 10−3), DPC = (5.0 × 10−5), [OH−] = 0.01 (mol dm−3) and a mixture of both. A bathochromic shift of about 4 nm from 257 nm of DPC to 261 nm of a mixture of DPC and Dio and a hyperchromicity at 261 nm were observed (Fig. 3). A Lineweaver–Burk plot also indicated the complex formation between DPC and Dio, which explains the less than unit order dependence on [Dio]. Such a type of complex between a substrate and an oxidant has been observed in other studies.36Scheme 1 leads to the rate law (eqn (6)).
| | 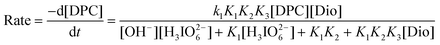 | (6) |
which explains all the observed kinetic orders of different species.
| | 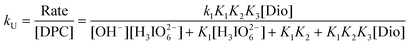 | (7) |
The rate law of
eqn (7) above can be rearranged into the following form which is suitable for verification:
| | 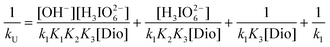 | (8) |
According to
eqn (8) above, other conditions being constant, plots of 1/
kUversus 1/[Dio], 1/
kUversus [OH
−] and 1/
kUversus [H
3IO
62−] should be linear and are found to be so at different temperatures (
Fig. 4). The slopes and intercepts of such plots lead to the values of
K1,
K2,
K3 and
k (
Tables 3 and 4). The values of
K1 and
K2 are in good agreement with the literature.
37 Using these constants, the rate constants were calculated over different experimental conditions, and there is a reasonable agreement between the calculated and the experimental values (
Table 1), which fortifies the proposed mechanism (
Scheme 1). The equilibrium constant
K1 is far greater than
K2 which may be attributed to the greater tendency of DPC to undergo
hydrolysis compared to the dissociation of hydrolysed species in alkaline medium.
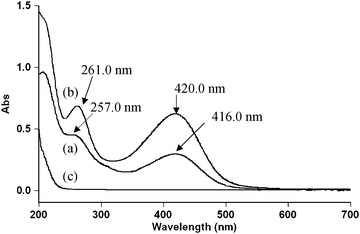 |
| | Fig. 3 Spectroscopic evidence for the complex formation between DPC (5 × 10−5) and Dio (4 × 10−3). (a) UV-vis spectra of a DPC complex (416 and 257 nm), (b) UV-vis spectra of a mixture of DPC and Dio and (c) UV-vis spectra of Dio. | |
![Verification of rate law (eqn (7)) for the oxidation of 1,4-dioxane by diperiodatocuprate(iii). Plots of (A) 1/kUvs. 1/[Dio], (B) 1/kUvs. [H3IO62−] and (C) 1/kUvs. [OH−], at five different temperatures.](/image/article/2011/CY/c1cy00192b/c1cy00192b-f4.gif) |
| | Fig. 4 Verification of rate law (eqn (7)) for the oxidation of 1,4-dioxane by diperiodatocuprate(III). Plots of (A) 1/kUvs. 1/[Dio], (B) 1/kUvs. [H3IO62−] and (C) 1/kUvs. [OH−], at five different temperatures. | |
Table 4
| (A) Effect of temperature on equilibrium constants of Scheme 1 for the oxidation of dioxane by diperiodatocuprate (III) in aqueous alkaline medium |
| Temperature/K |
K
1 × 104/mol dm−3 |
K
2 × 104/mol dm−3 |
K
3 × 10−2/dm3 mol−1 |
| 288 |
2.38 |
4.42 |
1.27 |
| 293 |
6.31 |
3.78 |
1.53 |
| 298 |
11.6 |
3.13 |
1.72 |
| 303 |
16.5 |
2.64 |
2.10 |
| 308 |
22.7 |
1.98 |
2.33 |
| (B) Thermodynamic quantities with respect to K1, K2 and K3 |
| Thermodynamic quantities |
Values from K1 |
Values from K2 |
Values from K3 |
| ΔH/kJ mol−1 |
81 |
−26 |
23 |
| ΔS/J K−1 mol−1 |
214 |
−154 |
119 |
| ΔG298 K/kJ mol−1 |
17 |
20 |
−13 |
The negligible effect of ionic strength and the dielectric constant of medium on the rate explains qualitatively the reaction between neutral and negatively charged ions, as seen in Scheme 1. The thermodynamic quantities for the different equilibrium steps in Scheme 1 can be evaluated as follows. The [Dio], [OH−] and [H3IO62−] (Table 1) were varied at five different temperatures. The plots of 1/kUversus 1/[Dio], 1/kUversus [OH−] and 1/kUversus [H3IO62−] should be linear and are found to be so. From the slopes and intercepts, the values of K1, K2 and K3 were calculated at different temperatures. A vant Hoff's plot was made for the variation of K1, K2 and K3 with temperature (log K1vs. 1/T, log K2vs. 1/T and log K3vs. 1/T). The values of enthalpy of reaction ΔH, entropy of reaction ΔS and free energy of reaction ΔG were calculated for the first, second and third equilibrium steps. These values are given in Table 4. A comparison of the ΔH value (81 kJ mol−1) from K1 with that of the ΔH# value (38 kJ mol−1) of the rate-limiting step supports that the reaction before the rate determining step is fairly slow as it involves low activation energy.38 In the first equilibrium step, only active species of DPC are involved whereas in the slow step, the activated complex is involved, hence this step is taken as the rate determining step. A high negative value of ΔS# (−163.8 J K−1 mol−1) suggests that the intermediate complex (C1) is more ordered than the reactants.39
4.2. Mechanism of ruthenium(III) catalysis
Ruthenium(III) chloride has been extensively used as a catalyst in many redox reactions involving one or two equivalent oxidants40,41 both in acid as well as in alkali. Ru(III) has d5 electronic configuration6 and shows extensive coordination chemistry like forming stable cationic, neutral and anionic, six coordinated complexes etc. which are kinetically42 inert. Aqueous solution of RuCl3 exists in its chloride complexes viz., [Ru(H2O)5Cl]2+, [Ru(H2O)4Cl2]+, [Ru(H2O)2Cl4]− and [RuCl6]3− in addition to its aqueous species as [Ru(H2O)6]3+. The existence of Ru(III) in its chloride complexes is possible only when on heating43 or aging in the presence of higher concentration of HCl. In the present study, the fresh solutions were used and solutions prepared in low concentration of HCl. Hence, such chloride complexes are negligible. Nevertheless, the alternative form of ruthenium(III) may be considered as [Ru(H2O)6]3+. As evidence, the added Cl− did not alter the rate of reaction and its UV-visible spectrum was identical with a reported study.42
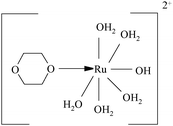
However, in alkaline medium, ruthenium(III) is known to exist44 as its hydroxylated species with general formula [Ru(OH)x(H2O)6−x]3−x where x < 6 and variable, which is depending upon the [OH−] used. Under the present experimental conditions where [OH−] > [Ru(III)], it is mainly existed as [Ru(H2O)5OH]2+.
In the present investigation, the reaction was retarded by [OH−], added periodate also retarded the rate, first order dependency in [DPC] and catalyst [Ru(III)] and fractional order in [Dio] was observed. To explain the observed orders Scheme 2 has been proposed for ruthenium(III) catalysed reaction of dioxane in alkaline medium.
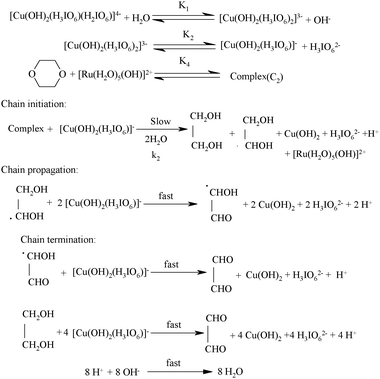 |
| | Scheme 2 | |
In the title reaction the negative less than unit order in periodate on rate of reaction, monoperiodatocuprate(III) (MPC) is considered to be the active species of a copper(III) periodate complex. The results of observed less than order in alkali concentration and negative less than order in periodate concentration suggest that equilibria of different copper(III) periodate complexes are possible as already shown in the above mechanism. The dioxane reacts with ruthenium(III) active species to form a complex (C2) which further undergoes hydrolysis in a slow step to give the free radical species of Dio, ethane-1,2-diol, periodate, Cu(II) with regeneration of the catalyst, ruthenium(III). Further this free radical species of Dio reacts with one molecule of MPC species in a fast step to form hydroxy acetaldehyde. This hydroxy acetaldehyde then reacts with two more moles of MPC in a further fast step to form products such as glyoxal, Cu(II) and periodate. In the next step the ethane-1,2-diol reacts with four more moles of MPC in a further fast step to form products such as glyoxal. All these results may be interpreted in the form of Scheme 2.
The probable structure of complex (C2) is
Spectroscopic evidence for the complex formation between ruthenium(III) and dioxane was obtained from UV–vis spectra of [dioxane] (4.0 × 10−3), [Ru(III)] (6.0 × 10−7), [OH−] = (0.01 mol dm−3) and a mixture of both. A bathochromic shift of about 5 nm from 279 to 284 nm of a mixture of Ru(III) and Dio and a hyperchromicity at 284 nm were observed (Fig. 5). However, the Michaelis–Menten plot proved the complex formation between the catalyst and the reductant, which explains the less than unit order dependence on [Dio]. Such a complex between a substrate and a catalyst has been observed in other studies.16
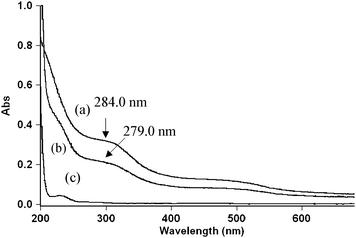 |
| | Fig. 5 Spectroscopic evidence for the complex formation between Ru(III) and Dio. (a) UV-vis spectra of a Ru(III) complex (279 nm), (b) UV-vis spectra of a mixture of Ru(III) and Dio and (c) UV-vis spectra of Dio. | |
From Scheme 2, the rate law (9) can be derived by neglecting some terms in view of the values:
| | 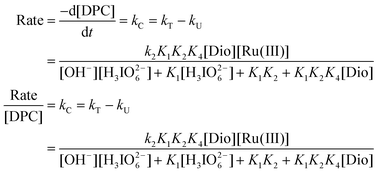 | (9) |
The rate law from
eqn (9) above can be rearranged into the following form which is suitable for verification:
| | 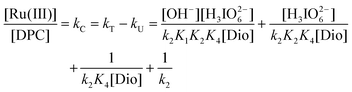 | (10) |
According to
eqn (10), other conditions being constant, plots of [Ru(
III)]/
kCversus [H
3IO
62−], [Ru(
III)]/
kCversus [OH
−] and [Ru(
III)]/
kCversus 1/[Dio] were linear with an intercept supporting the Ru(
III) and
dioxane complex, as is verified at different temperatures in
Fig. 6. From the intercepts and slopes of such plots, the reaction constants
K1,
K2,
K4 and
k2 were obtained (
Tables 3 and 5). The value of
K2 obtained is in good agreement with previously reported value.
37 These constants were used to calculate the rate constants over different experimental conditions, when compared with the experimental
kC values. There was a reasonable agreement between the two (
Table 2), which fortifies the proposed mechanism (
Scheme 2).
![Verification of rate law (9) for the Ru(iii) catalyzed oxidation of 1,4-dioxane by diperiodatocuprate(iii). Plots of (A) [Ru(iii)/kCvs. 1/[Dio], (B) [Ru(iii)/kCvs. [H3IO62−] and (C) [Ru(iii)/kCvs. [OH−], at five different temperatures.](/image/article/2011/CY/c1cy00192b/c1cy00192b-f6.gif) |
| | Fig. 6 Verification of rate law (9) for the Ru(III) catalyzed oxidation of 1,4-dioxane by diperiodatocuprate(III). Plots of (A) [Ru(III)/kCvs. 1/[Dio], (B) [Ru(III)/kCvs. [H3IO62−] and (C) [Ru(III)/kCvs. [OH−], at five different temperatures. | |
Table 5
| (A) Effect of temperature on equilibrium constants of Scheme 2 for the Ru(III) catalyzed oxidation of dioxane by diperiodatocuprate (III) in aqueous alkaline medium |
| Temperature/K |
K
1 × 103/mol dm−3 |
K
2 × 104/mol dm−3 |
K
4 × 10−2/dm3 mol−1 |
| 288 |
1.14 |
8.78 |
0.87 |
| 293 |
2.06 |
8.07 |
1.09 |
| 298 |
2.69 |
3.09 |
1.35 |
| 303 |
7.26 |
5.23 |
1.53 |
| 308 |
9.24 |
4.14 |
1.91 |
| (B) Thermodynamic quantities with respect to K1, K2 and K4 |
| Thermodynamic quantities |
Values from K1 |
Values from K2 |
Values from K4 |
| ΔH/kJ mol−1 |
80 |
−29 |
28 |
| ΔS/J K−1 mol−1 |
222 |
−159 |
135 |
| ΔG298 K/kJ mol−1 |
14 |
19 |
−12 |
The negligible effect of ionic strength and the dielectric constant of the medium on the rate explains qualitatively the reaction between neutral species, as seen in Scheme 2. Thermodynamic quantities for the different equilibrium steps in Scheme 2 can be evaluated as follows. Dioxane, periodate and hydroxide ion concentrations (Table 2) were varied at five different temperatures. The plots of [Ru(III)]/kCversus 1/[Dio] (r ≥ 0.9995, S.D. ≤ 0.000022), and [Ru(III)]/kCversus [H3IO62−] (r ≥ 0.9918, S.D. ≤ 0.0000046) and [Ru(III)]/kCversus [OH−] (r ≥ 0.981, S.D. ≤ 0.0000043) should be linear, as shown in Fig. 6. From the slopes and intercepts, the values of K1, K2 and K4 were calculated at five different temperatures. A vant Hoff's plot was made for the variation of K1, K2 and K4 with temperature (i.e., log K1versus 1/T, log K2versus 1/T and log K4versus 1/T) and the values of the enthalpy of reaction ΔH, entropy of reaction ΔS and free energy of reaction ΔG were calculated. In the same manner K1, K2 and K4 values were calculated at different temperatures and the corresponding values of the thermodynamic quantities are given in Table 5. A comparison of the ΔH value (80 kJ mol−1) from K1 with that of the ΔH# value (34 kJ mol−1) of a rate-limiting step supports that the reaction before the rate determining step is fairly slow as it involves low activation energy. In the first equilibrium step, only active species of DPC are involved whereas in the slow step, the activated complex is involved, hence this step is taken as the rate determining step.45 The negative value of ΔS# suggests that the intermediate complex is more ordered than the reactants.39 The observed modest enthalpy of activation and a higher rate constant for the slow step indicate that the oxidation presumably occurs via an inner-sphere mechanism. This conclusion is supported by earlier observation.46 The activation parameters evaluated for the catalyzed and uncatalyzed reactions explain the catalytic effect on the reaction. The catalyst Ru(III) forms the complex (C2) with a substrate, which enhances the reducing property of the substrate than that without catalyst. Further, the catalyst Ru(III) modifies the reaction path by lowering the activation energy.
It is also interesting to note that the transient species involved in both the uncatalyzed and Ru(III) catalyzed reactions is different but leads to the formation of the same product. The uncatalyzed reaction in alkaline medium has been shown to proceed via a MPC-Dio complex which decomposes slowly in a rate determining step to give the product via a free radical in the further fast steps, whereas in the catalyzed reaction, it has been shown to proceed via a Ru(III)–Dio complex which further reacts with four moles of MPC in the rate determining step to give the product via a free radical in the further fast steps. Since in both the cases MPC and dioxane were involved, the product obtained were same.
5. Conclusions
A comparative study of uncatalyzed and Ru(III) catalyzed oxidation of dioxane by diperiodatocuprate(III) was conducted. Oxidation products identified were same in both uncatalyzed and catalyzed reactions. Among various species of Cu(III) in alkaline medium, monoperiodatocuprate(III) ([Cu(OH)2(H3IO6))]−) is considered to be the active species for the title reaction. The active species of Ru(III) is found to be [Ru(H2O)5OH]2+. The reaction rates revealed that Ru(III) catalyzed reaction is about eight-fold faster than the uncatalyzed reaction. It becomes apparent that, in carrying out this reaction, the role of reaction medium is crucial. Activation parameters were evaluated for both catalyzed and uncatalyzed reactions. Catalytic constants and the activation parameters with reference to the catalyst were also computed. The overall mechanistic sequence described here is consistent with all the experimental evidences including the product, spectral, mechanistic and kinetic studies.
Appendix A
Derivation of rate law for uncatalyzed reaction
According to Scheme 1:
| | 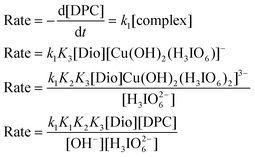 | (A.1) |
The total concentration of [DPC]
T is given by
| [DPC]T = [DPC]f + [Cu(OH)2(H3IO6)23−] + [Cu(OH)2(H3IO6)]− + complex |
where T and f refer to total and free concentrations
| |  | (A.2) |
Similarly, the concentration of OH
− is
| | 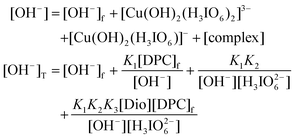 | (A.3) |
In view of low concentration of DPC used, the second, third and fourth terms in the above equation are neglected.
Therefore,
Similarly,
Substituting equations
eqn (A.2), (A.4) and (A.5) in equation
eqn (A.1) we get,
| | 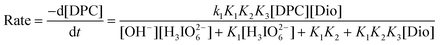 | (A.6) |
The rate law for the ruthenium(
III) catalyzed reaction was derived similarly.
References
-
M. L. Richardson and S. Gangolli, The Dictionary of Substances and their Effects, vol. 4, The Royal Society of Chemistry, UK, 1994, p. 91 Search PubMed;
R. Q. Brewster and W. E. Mcewn,Organic Chemistry, 3rd edn, Prentice-Hall of India private Limited, New Delhi, 1971, p. 172 Search PubMed.
-
S. Kenneth, Surprenant “Dioxane” in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2002 Search PubMed.
- K. B. Reddy, B. Sethuram and T. Navaneeth Rao, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 1984, 23, 593–595 Search PubMed.
- A. Kumar, P. Kumar and P. Ramamurthy, Polyhedron, 1999, 18, 773–780 CrossRef CAS.
- R. S. Shettar and S. T. Nandibewoor, J. Mol. Catal. A: Chem., 2005, 234, 137–143 CrossRef CAS.
-
B. Sethuram, Some Aspects of Electron Transfer Reactions Involving Organic Molecules, Allied Publishers (P) Ltd., New Delhi, 2003, p. 78 Search PubMed.
- J. L. Piere, Chem. Soc. Rev., 2000, 29, 251–257 RSC.
- E. I. Solomon, P. Chen, M. Metz, S. K. Lee and A. E. Palmer, Angew. Chem., Int. Ed., 2001, 40, 4570–4590 CrossRef CAS.
-
J. Peisach, P. Alsen and W. E. Bloomberg, The Biochemistry of Copper, Academic Press, New York, 1966, p. 49 Search PubMed.
- A. K. Das, Coord. Chem. Rev., 2001, 213, 307–325 CrossRef CAS.
- K. N. Shivananda, B. Lakshmi, R. V. Jagdeesh, Puttaswamy and K. N. Mahendra, Appl. Catal., A, 2007, 326, 202–212 CrossRef CAS.
- Puttaswamy, J. P. Shubha and R. V. Jagadish, Transition Met. Chem., 2007, 32, 991–999 CrossRef CAS.
- A. K. Singh, S. Srivastava, J. Srivastava, R. Srivastava and P. Singh, J. Mol. Catal. A: Chem., 2007, 278, 72–81 CrossRef CAS.
- S. Sondu, B. Sethuram and T. Navaneeth Rao, J. Indian Chem. Soc., 1983, 60, 198–200 CAS.
- H. P. Panda and B. D. Sahu, Indian J. Chem., 1977, 15, 1066–1069 Search PubMed.
- V. Tegginamath, C. V. Hiremath and S. T. Nandibewoor, J. Phys. Org. Chem., 2007, 20, 55–64 CrossRef CAS.
-
B. S. Furniss, S. Brian, A. J. Hannaford, J. Antony, P. W. G. Smith, W. G. Peter, A. R. Tatchell and R. Austin, Vogel's Textbook of Practical Organic Chemistry, 5th edn, 2007, p. 407 Search PubMed.
- C. S. Reddy and T. Vijaya Kumar, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 1995, 34, 615–620 Search PubMed.
- P. K. Jaiswal and K. L. Yadava, Indian J. Chem., 1973, 11, 837–839 CAS; L. Bai, Y. Liu, M. Run and K. Deng, Iran. Polym. J., 2006, 15(7), 577–582 Search PubMed.
-
G. H. Jeffery, J. Bassett, J. Mendham and R. C. Denney, Vogel's Textbook of Quantitative Chemical Analysis, 5th edn, ELBS, Longman, Essex, UK, 1996, vol. 679, p. 455 Search PubMed.
- G. P. Panigrahi and P. K. Misro, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 1978, 16, 201–204 Search PubMed.
-
F. Feigl, Spot Tests in Organic Analysis, Elsevier, New York, 1975, p. 441 Search PubMed.
-
A. I. Vogel, A Text Book of Practical Organic Chemistry Including Quantitative Organic Analysis, 3rd edn, Longmann, 1973, p. 332 Search PubMed.
- N. P. Shetti, R. N. Hedge and S. T. Nandibewoor, J. Mol. Struct., 2009, 930, 180–186 CrossRef CAS.
-
E. A. Moelwyn-Hughes, Kinetics of reactions in solutions, Oxford University Press, London, 1947, p. 297 Search PubMed.
- L. Malaprade, C. R. Acad. Sci., Paris, 1937, 204, 979–980 CAS.
- G. I. Rozovski, A. Yu. Prokopchik and R. Jankauskas, Liet. TSR Mokslu Akad. Darb., Ser. B, 1968, 1, 25–34 Search PubMed.
- L. Jensovsky and K. Milos Rabe, Z. Anorg. Allg. Chem., 1962, 314, 76–79 CrossRef CAS.
- C. P. Murthy, B. Sethuram and T. Z. Navaneetha Rao, Phys. Chem., 1981, 262, 336–340 CAS.
- C. E. Crouthumel, H. V. Meck, D. S. Martin and C. V. Banks, J. Am. Chem. Soc., 1949, 71, 3031–3035 CrossRef.
- C. E. Crouthumel, A. M. Hayes and B. S. Martin, J. Am. Chem. Soc., 1951, 73, 82–87 CrossRef.
- S. M. Tuwar, S. T. Nandibewoor and J. R. Raju, J. Indian Chem. Soc., 1992, 69, 651–653 CAS.
- T. S. Kiran, D. C. Hiremath and S. T. Nandibewoor, Z. Phys. Chem., 2007, 221, 501–517 CrossRef CAS.
- K. Bal Reddy, Transition Met. Chem., 1990, 15, 9–11 CrossRef.
- M. Jaky, M. Szeverenyi and L. I. Simandi, Inorg. Chim. Acta, 1991, 186, 33–37 CrossRef CAS.
- B. Thimmegowda and J. Ishwarabhat, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 1989, 28, 43–46 Search PubMed.
- N. P. Shetti and S. T. Nandibewoor, Z. Phys. Chem., 2009, 223, 299–317 CrossRef CAS.
- K. T. Sirsalmath, C. V. Hiremath and S. T. Nandibewoor, Appl. Catal., A, 2006, 305, 79–89 CrossRef CAS.
-
Investigations of Rates and Mechenism of Reactions in Techniques of Chemistry, ed. A. Weissberger and E. S. Lewis, Wiley, New York, 1974, vol. 4, p. 421 Search PubMed.
- T. P. Jose, M. A. Angadi, M. S. Salunke and S. M. Tuwar, J. Mol. Struct., 2008, 892, 121–124 CrossRef CAS.
- H. S. Singh, R. K. Singh, S. M. Singh and A. K. Sisodia, J. Phys. Chem., 1977, 81, 1044–1049 CrossRef CAS.
-
F. A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry, Wiley Eastern, New York, 1996, p. 153 CrossRef CAS; R. E. Connick and D. A. Fine, J. Am. Chem. Soc., 1960, 82, 4187–4191 CrossRef CAS.
- H. H. Cady and R. E. Conick, J. Am. Chem. Soc., 1958, 80, 2646–2650 CrossRef CAS.
- V. Uma, B. Sethuram and T. Navaneeth Rao, React. Kinet. Catal. Lett., 1981, 18, 283–288 CrossRef CAS.
- K. S. Rangappa, M. P. Raghavendra, D. S. Mahadevappa and D. J. Channegouda, J. Org. Chem., 1998, 63, 531–536 CrossRef CAS.
- N. Sutin, Annu. Rev. Phys. Chem., 1966, 17, 119–171 CrossRef CAS; S. A. Farokhi and S. T. Nandibewoor, Tetrahedron, 2003, 59, 7595–7602 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.