Immobilized Pd complexes over MCM-41 as supported catalysts for effective reformation of hydrocarbons
Gopal S.
Mishra†
*a and
Anil
Kumar
b
aCentro de Química Estrutural, Complexo I, Instituto Superior Técnico, TU Lisbon, Av. Rovisco Pais, 1049-001, Lisboa, Portugal
bDepartment of Chemical Engineering, Indian Institute of Technology, Kanpur-208016, India
First published on 9th August 2011
Abstract
The ethylthio alkane ligands i.e. di(ethylthio)ethane, I, and di(ethylthio)propane, II, have been used for the synthesis of Pd(II) complexes. These complexes were covalently anchored to the modified MCM-41 matrix as supported hybrid catalysts (MCM-41/I as Pd–I and MCM-41/II as Pd–II). Remarkable catalytic effects were observed when these supported catalysts act in n-hexane and n-heptane reformation reactions in the presence of H2. Catalyst Pd–I provided the high TONs of 2325 with 91% selectivity for n-hexane and high TONs of 2145 with 88% selectivity for n-heptane under optimized and relatively mild conditions. A carbenium-based mechanism has been included to explain the product formation. TGA analysis indicates that these catalysts were thermally stable up to the reaction temperature and recyclable for further utilization.
1. Introduction
Palladium complexes have received a lot of interest owing to their potential applications in catalysis, biological activities with industrial significance such as catalytic converters, petroleum cracking or reforming.1,2 In fact, more than 90% of industrial hydrocarbon reformation processes are catalyzed by the Pt, Pd and Ni metals, either as soluble or solid catalysts.3,4 For this purpose, supports like silica, zeolites, clays, etc. have been used for the immobilization of metal complexes.5,6 This is generally achieved by the formation of covalent bonds between the silanol (–OH) of the support matrix and the terminal active groups of metal complexes (e.g. –Cl, –NH2, –CN, –OCH3 and –SH).7,8 Particularly, potentially active di(ethylthio)alkane Pd complexes have been known for a long time to undergo reversible coordination with the –SH moiety of a ligand in solution.9 These immobilized complexes as supported catalysts can be exploited in the reformation of hydrocarbons with the typical advantages of heterogeneous catalysts over the homogenous ones e.g. in terms of easy removal, increased selectivity, low toxicity and reutilization.Petroleum refining is one of the processes employed to provide high-octane-number hydrocarbon blending components for gasoline.10,11 The typical alkanes reformation reaction sometimes competes with reactions like dehydrogenation (gives alkenes and H2), cracking (gives lower-alkanes), cyclization (gives cyclo-alkanes) and dehydrocyclization (gives cycloalkanes and H2).12 Heterogenized Pd and Pt catalysts have been widely used for the alkane reformation.13,14 Mono- or bimetallic Pd and Pt complexes have been immobilized on various types of supports (i.e. ZrO2, Al2O3 and WOx) and used for n-hexane isomerization. Monometallic Pd catalysts have provided considerably higher catalytic activity (conversion range ca. 39–57% at 260 °C).15 Natural zeolites as supports with Pd and Pt catalysts have been used for the n-hexane reformation (conversion ca. 17–80% under various temperatures).16 Several supports such as MgO, ZrO2 and MgO–ZrO2 have been used for Pd catalysts. Pd–MgO was found to be the best catalyst for n-hexane isomerization, favoring mainly the formation of aromatized products (12% conversion at 550 °C, with 90% selectivity).17 A nanocrystalline β zeolite over Pt catalyst has provided 95% selectivity at 300–400 °C.18 We have also obtained significant conversions in the n-hexane reformation (ca. 72–74% at 200 °C) with Co and Zr complexes over modified SiO2 and Al2O3, respectively.19–21
Isomerization of n-heptane on a β type zeolite/Pt catalyst has been used to produce mono- and multi-branched isomers.22 Bifunctional catalysts such as Pt metal on WOx–ZrO2 (PWZ) for different tungsten loadings were tested for n-heptane reactions and branched products were obtained, in addition to C1 to C5 cracking products.23 The MoOx and MoOx supports bearing Ni catalysts were also used for n-heptane isomerization, providing a significant good conversion result at 200 °C.24 Herein, we report the preparation of modified MCM-41 which is covalently anchored by di(ethylthio)ethane ligands of Pd complexes as supported hybrid catalysts. These catalysts have been tested under optimized and relatively mild conditions for the reformation of n-hexane and n-heptane in order to obtain high TONs and high selectivity of corresponding lower isomers of alkanes under optimized and relatively mild conditions.
2. Experimental
2.1 General materials and procedure
All the reactions and manipulations were carried out under dinitrogen using standard Schlenk techniques. All chemicals (Sigma) were used as received and solvents purified by standard methods, freshly distilled prior to use.2.2 Preparation of modified MCM-41
The modified MCM-41 has been prepared according to the previously described methods.9,25 A solution of ethyltriethylammonium bromide (2.2 g) in H2O (52 ml) was slowly added at 40 °C into ammonium hydroxide (26 mL) solution. This mixture was stirred at room temperature (RT) for 1 h. Then tetraethyl orthosilicate (10 mL) was slowly added into the solution with continuous stirring for 3 h at RT. The resulting mixture was shifted into an autoclave and heated at 110 °C for 72 h. MCM-41 was obtained by the calcinations in air at 600 °C for 12 h.For the MCM-41 modification, a solution of chloropropyl trimethoxysilane (0.44 mol), 1,2-ethanedithiol (0.2 mol) and/or 1,2-propyldithiol were stirred separately in a Schlenk flask, for 1 h at RT. These colourless oils were again separately dissolved in toluene (20 mL, each) and MCM-41 powder was added and refluxed for 8 h, after that it was filtered and dried. Finally, we obtained two types of modified MCM-41 i.e. di(ethylthio)ethane anchored modified MCM-41 as I and di(ethylthio)propane anchored modified MCM-41 as II (Scheme 1, steps 1 and 2).
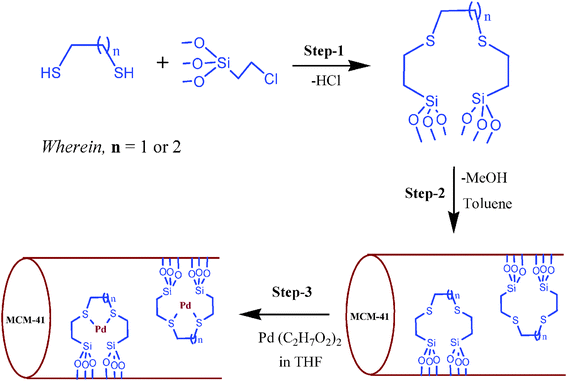 | ||
| Scheme 1 Preparation of di(ethylthio)alkanes Pd(II) complexes immobilized on MCM-41. | ||
2.3 Preparation of Pd–I catalyst
A solution of Pd(C5H7O2)2 (0.70 mmol) in THF (20 mL) was added to the above obtained I (2.0 g). The mixture was then stirred for a 12 h period. It was washed in a Soxhlet extractor with toluene and dried under reduced pressure at 60 °C. A reddish colour modified MCM-41/I as catalyst Pd–I was finally obtained (Scheme 1, step 3).2.4 Preparation of Pd–II catalyst
A solution of Pd(C5H7O2)2 (0.70 mmol) in THF (20 mL) was added to the above obtained II (2.0 g). The mixture was then stirred for a 12 h period. It was washed in a Soxhlet extractor with toluene and dried under reduced pressure at 60 °C. A reddish colour modified MCM-41/II as catalyst Pd–II was finally obtained (Scheme 1, step 3).The identity of Pd metal complexes (Pd–I and Pd–II) on the modified MCM-41 support has been verified by the analytical characterization i.e. SEM-EDX, TGA, ICP, FT-IR and TPD analysis.
2.5 Isomerization procedure and product analysis
A rocking type SS micro-batch reactor (22 cm3 capacity) provided with a gas delivery inlet and a pressure gauge was used for the reformation reactions. The inner temperature of the reactor was monitored by a thermocouple. Initially the reactor was charged with the n-alkane and a catalyst. The reactor was then closed, the air was removed and H2 was introduced (2 atm) for each desired temperature and time. At the end of reaction, the solid catalyst was separated from the liquid product by normal filtration. It was washed several times with acetonitrile and dried at 100 °C for 6 h for further recycling studies. The reaction products were analyzed by gas chromatography (GC) (30 μL of hexanone added as internal standard to 1.0 mL of the filtered final reaction solution). The reaction products were further identified by gas chromatography–mass analysis.2.6 Instruments for characterization
FT-IR spectra (4000–400 cm−1) were recorded on a Unicam Research Series spectrophotometer in transmission mode using KBr pellets (abbreviations: vs = very strong, s = strong, m = medium, w = weak, br = broad). TGA analysis of the catalysts was performed on a TA Instruments Q50. Palladium concentrations were determined by ICP analysis on a Perkin-Elmer 41000ZL spectrometer. Morphology of the solid supported catalysts was analyzed with scanning electron microscopy using FEI Quanta 400, equipped with an EDS detector. GC analysis has been carried out with a FISONS GC 8000 gas chromatograph equipped with a FID detector and a DB-WAX capillary column (length: 30 m; internal diameter: 0.32 mm). The products were further confirmed by GC-MS using a Carlo-Erba Auto/HRGC/MS instrument.3. Results and discussion
3.1 Catalysts synthesis and characterization
Two main types of ethylthio alkanes have been used for the synthesis of modified MCM-41. It was mainly obtained by a grafted reaction method of the silanol (–OH) on the surface of the support matrix (MCM-41) with methoxy (–OCH3) of the di(ethylthio)alkanes ligands (I and II), by the condensation process and liberation of three molecules of CH3OH (Scheme 1). At the final stage of reaction, the Pd(C5H7O2)2 was added into the above mentioned two types of modified MCM-41 supports. Finally two types of potentially active Pd metal containing complexes were produced over MCM-41 as supported hybrid catalysts, i.e. di(ethylthio)ethane Pd(II)/MCM-41 as Pd–I and di(ethylthio)propane Pd(II)/MCM-41 as Pd–II. These ligands of Pd complexes were bonded to the surface of the support matrix by means of covalent attachment. Any residual or excess of Pd complexes were subsequently removed by the toluene in the Soxhlet extractor (thimble filled with the Pd/MCM-41 catalyst).The major aim of the current study is to use these potentially active Pd(II) complexes as reusable and selective catalysts for the n-alkanes reformation under relatively mild conditions.For the catalyst characterization, several analytical techniques have been applied to confirm the identity of Pd complexes on the MCM-41 matrix. In the FT-IR spectra, broad shoulders appeared between br = 3815 cm−1 to 3305 cm−1 and between br = 2765 cm−1 to 1830 cm−1 in both the supported catalysts due to the MCM-41 support. Anchoring of Pd metal complexes on support matrix was confirmed by the C–S bond at m = 800 cm−1. In the near-IR, the Pd–S bond was observed at s = 351 cm−1. These IR values were consistent with the literature.26
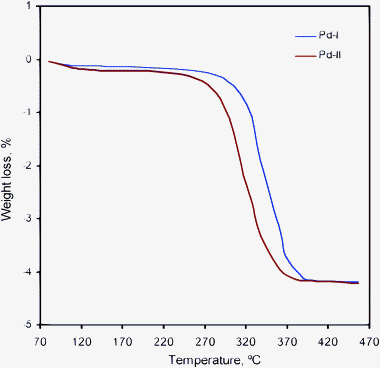
Morphological study of the Pd–I catalyst giving most effective reformation was carried out by scanning electron microscopy (SEM) coupled with an EDS detector. The SEM image of Pd–I (×20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times magnification) is shown in Fig. 1. Some dark regions appeared on the cylinder-shaped surface of the MCM-41 support. It indicated the presence of the Pd complex on the support matrix. The same regions were analyzed with the SEM-EDS detector which confirmed the presence of Pd (0.3% wt) on the MCM-41 support. Thermal stability experiments of both the supported catalysts were performed using the TGA instrument (Fig. 2). Initially small weight losses of these catalysts were observed at ca. 70 to 288 °C, probably due to the absorbed atmospheric moisture and after that decomposition of surface metal species. Beyond that, rapid weight losses were observed for these catalysts (at ca. 290 °C to 400 °C). Absolute decay was observed above 400 °C (ca. 4.3% wt loss). These TGA analysis results point towards that these catalysts were stable over the temperature range of n-alkanes reformation experiments.
000 times magnification) is shown in Fig. 1. Some dark regions appeared on the cylinder-shaped surface of the MCM-41 support. It indicated the presence of the Pd complex on the support matrix. The same regions were analyzed with the SEM-EDS detector which confirmed the presence of Pd (0.3% wt) on the MCM-41 support. Thermal stability experiments of both the supported catalysts were performed using the TGA instrument (Fig. 2). Initially small weight losses of these catalysts were observed at ca. 70 to 288 °C, probably due to the absorbed atmospheric moisture and after that decomposition of surface metal species. Beyond that, rapid weight losses were observed for these catalysts (at ca. 290 °C to 400 °C). Absolute decay was observed above 400 °C (ca. 4.3% wt loss). These TGA analysis results point towards that these catalysts were stable over the temperature range of n-alkanes reformation experiments.
 | ||
| Fig. 1 SEM morphological study of Pd–I catalyst (× 20000 times magnification), and the SEM-EDS detector graph (above crop image). | ||
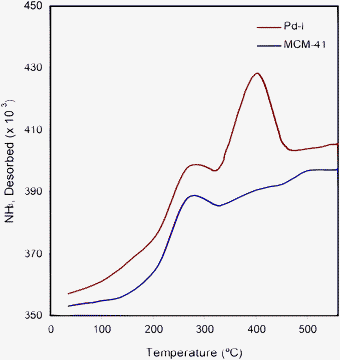
The TPD method with ammonia was used (a basic molecule titrating to weak acid sites) to penetrate into all pores of the solid catalyst and determine the quantity and strength of the acid sites on the MCM-41/I catalyst. We used 10 mg of a fresh Pd–I sample with NH3 (1.0 mL introduced over 10 pulses) under a He gas flow at 150 °C. The graph of TPD shows two main desorption peaks, one at 265 °C (MCM-41 peak) and the other at around 410 °C, due to NH3 adsorption on the surface Pd catalyst (Fig. 3). It was also confirmed by comparing the FT-IR spectra of the catalyst before and after NH3 adsorption (very small additional peaks at 3495 cm−1 most likely due to NH3). This result showed that the Pd–I catalyst was porous and acidic in nature.
3.2 Reformation of hydrocarbons by Pd-catalysts
We have tested modified MCM-41 supported di(ethylthio)ethane ligand I and di(ethylthio)propane ligand II (without Pd metal, Scheme 1, step 1) and their supported Pd metal complex catalysts (Pd–I and Pd–II) under heterogeneous conditions in the reformation (H2) of n-hexane and n-heptane. These catalytic reactions were performed under relatively mild conditions in a rocking type batch reactor without requiring any solvents and additives. At the end of the chemical reformation, the colours of both the substrates were changed from colourless to light yellow and both the catalysts from reddish to brown. These n-alkane reaction mixtures were quantified in the GC and again identified in the GC/MS using the same column. Mainly mono- and di-branched lower-alkane products appeared as their counterparts.Initially, we have examined modified MCM-41 anchored di(ethylthio)ethane ligand, I, and modified MCM-41 anchored di(ethylthio)propane ligand, II, derivatives (25 mg, each) for the reformation of n-alkanes under analogous conditions as given in Table 1. These supported derivatives I and II gave slightly improved overall conversion results (8.9% for n-hexane and 7.7% for n-heptane with I and 10.2% for n-hexane and 8.3% for n-heptane with II) in comparison to blank experiments (less than 4.2%). Some more experiments with normal MCM-41 were separately carried out and less than 5.6% conversion of n-hexane and less than 4.9% of n-heptane were obtained. It seems that the structures of ligands also played a significant role in these catalytic reactions.
| 2-MP | 3-MP | 2,2-DMB | 2,3-DMB | — | ||||
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: catalyst = 25 mg, substrate = 3.0 mL (n-hexane = 140 °C for 12 h and n-heptane = 220 °C for 18 h), p(H2) = 2 atm (at 25 °C). b Turnover number (TONs) = moles of main products formed per mole of the immobilized Pd complex. c Overall %conversion = main products + by-products. | ||||||||
| n-Hexane | Pd–I | 2325 | 30.9 | 15.6 | 7.4 | 2.9 | 2.0 | — |
| Pd–II | 2053 | 24.8 | 12.4 | 6.6 | 1.5 | 1.3 | — |
Surprisingly, high TONs, 1886 to 2325, with good overall conversion results, 21.8 to 30.9%, have been observed when these MCM-41 supported ligand derivatives (I and II) coordinated with Pd i.e. catalysts Pd–I and Pd–II (Table 1) were used. Noticeably, best catalytic activities were obtained with the Pd–I catalyst in contrast to Pd–II. The catalyst Pd–I gave 30.9% conversion for n-hexane and 26.6% conversion for n-heptane. In the case of catalyst Pd–II, we observed 24.8% for n-hexane and 21.8% for n-heptane. Significantly high TONs were also obtained by both these catalysts. Catalyst Pd–I can lead to highest TONs of 2325 for n-hexane and 2145 for n-heptane. The overview of these results shows that the best TONs and overall conversion were only achieved by the catalyst Pd–I for both the n-alkanes. It reflects that not only supported Pd complexes improve the catalyst activity, but some other improving factors like modified MCM-41 and structures of the I and II also play important roles in these n-alkane reactions (see above). These supported Pd catalysts have mainly produced mono- and di-branched products of n-hexane (2-MP, 3-MP, 2,2-DMB and 2,3-DMB) and n-heptane (2-MH, 3-MH, 2,3-DMP and 2,2-DMP). It showed that there were no cracking, no dehydrocyclization or no dehydrogenation reactions taking place. In view of above results, subsequent detailed catalytic studies of n-alkanes for the chemical reformation were centralized on catalyst Pd–I to optimize best reaction conditions.
The effect of reformation reaction time was examined between 4 to 20 h for both the n-alkanes under constant conditions (catalyst = 25 mg, temp = 140 °C, substrate = 3 mL, H2 = 2 atm). The overall conversion and the concentration of the main products for both the n-alkanes increased slowly until 4 h (overall 4.0%) for n-hexane and 6 h (overall 2.8%) for n-heptane. Beyond this period, the overall conversions sharply increased up to 12 h for n-hexane and 18 h for n-heptane, after which they remained practically constant. The conversion of n-hexane and n-heptane into the main branched isomers was 30.9% and 26.6%, respectively. Furthermore, overall conversions of 34.8% for n-hexane and 30.6% for n-heptane increased slowly up to 20 and 30 h, respectively. However, longer reaction time leads to the formation of polymerized dark brown material decreasing the selectivity of the main products. The dependence of the isomer product selectivity on time is shown in Fig. 4 for n-hexane and Fig. 5 for n-heptane. It indicates that the highest selectivity of n-hexane is towards the main single bond shift isomers (i.e. 2-MP, 51%; 3-MP, 24%) and two or bi-bonds shift isomers (i.e. 2,2-DMB, 9%; 2,3-DMB, 7%) which were obtained at 12 h. Whereas, the n-heptane time graph shows that the maximum selectivity is towards the single bond shift isomers (i.e. 2-MH, 36%; 3-MH, 29%) and two or bi-bonds shift isomers (2,2-DMP, 11%; 2,3-DMP, 13%; 3,3-DMP, 5%) which were formed after 18 h reaction time (Fig. 5).
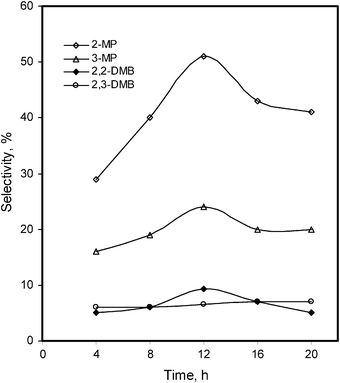 | ||
| Fig. 4 Time effect on the %selectivity of the isomerized products of n-hexane, in the presence of the Pd–I catalyst. | ||
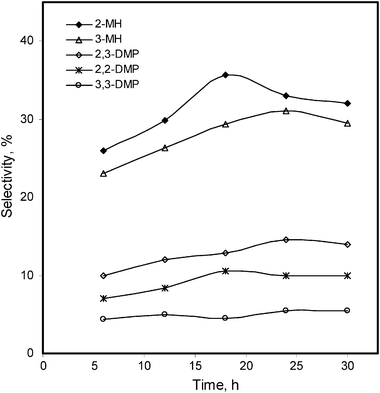 | ||
| Fig. 5 Time effect on the %selectivity of the isomerized products of n-heptane, in the presence of the Pd–I catalyst. | ||
The effect of temperature on the reformation reactions of n-alkanes has been studied. Once again the reactions were carried out under H2, keeping constant the amount of the catalyst (25 mg), substrate = 3 mL and the reaction times (12 h for n-hexane and 18 h for n-heptane). The same trends were observed for both the n-alkanes. Initially overall and main isomers conversions increased slowly at 80 °C (overall 7.3% for n-hexane) and 100 °C (overall 4.6% for n-heptane). Beyond that it increased until 160 °C for n-hexane (overall 36%) and 260 °C for n-heptane (overall 30%). At higher temperatures e.g. 200 °C for n-hexane and 260 °C for n-heptane, the overall conversions did not change significantly. Above these values the main isomers selectivity was decreased. It showed that the highest selectivity towards the main isomers of n-hexane (i.e. 2-MP, 3-MP) was achieved at 140 °C (Fig. 6), while for n-heptane (2-MH and 3-MH) it was attained at 220 °C (Fig. 7).
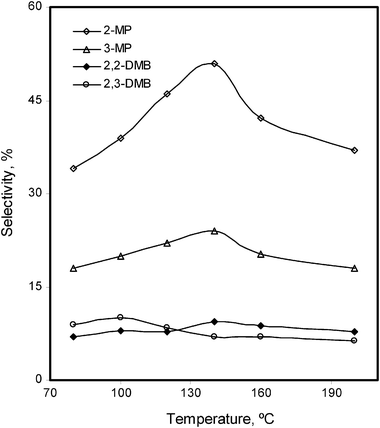 | ||
| Fig. 6 Temperature effect on the %selectivity of the isomerized products of n-hexane, in the presence of Pd–I catalyst. | ||
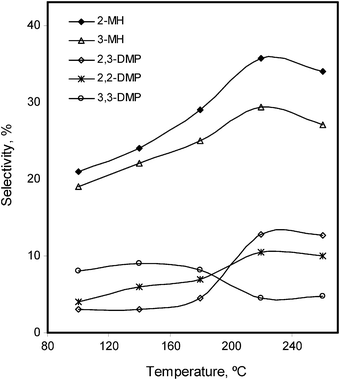 | ||
| Fig. 7 Temperature effect on the %selectivity of the isomerized products of n-heptane, in the presence of Pd–I catalyst. | ||
The effect of Pd–I amounts has been also studied from 5 mg to 45 mg under analogous conditions as given in Table 1. Initially, low overall conversions, 2.7% and 3.2%, were observed with 5 mg of the catalyst for n-hexane and n-heptane, respectively. The catalyst amount was increased slowly up to 45 mg (36.9% for n-hexane and 31.1% for n-heptane). At 45 mg of catalyst, we obtained main mono-branched products i.e. 2-MP, 15%; 3-MP, 7%; 2-MH, 8%; and 3-MH, 6%. On the other hand, much less di-branched products i.e. 2,2-DMB, 3%; 2,3-DMB, 3%; 2,2-DMP, 3%; 2,3-DMP, 4% and 3,3-DMP, 2% were obtained. In the selectivity parameter the main product 2-MP of n-hexane remains higher in comparison to others (Fig. 8). The selectivity towards the main products 2-MH, 3-MH of n-heptane tends to decrease significantly, when a high concentration of the catalyst was used, increasing the other di-branched isomers (Fig. 9).
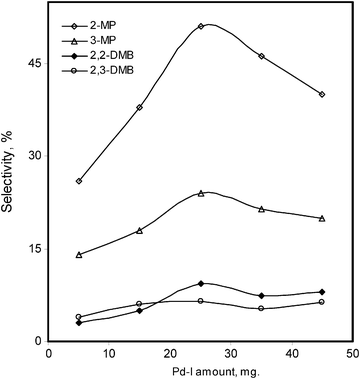 | ||
| Fig. 8 Modified MCM-41 supported Pd–I catalyst amount effect on the %selectivity of the isomerized products of n-hexane. | ||
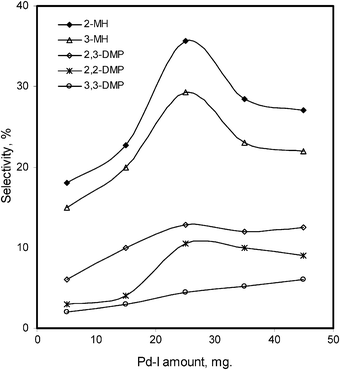 | ||
| Fig. 9 Modified MCM-41 supported Pd–I catalyst amount effect on the %selectivity of the isomerized products of n-heptane. | ||
Recycling and leaching tests have been subsequently carried out with the Pd–I catalyst and it was found that it could be reactivated by washing and drying. Thus, after the first cycle, both the used Pd–I catalysts were filtered off from the reaction mixture, washed with acetone and oven dried at 100 °C for 6 h. These used catalysts were then applied again from the IInd cycle to the Vth cycle under the analogous conditions (Table 1). It was found that the initial overall conversions for n-hexane (after 48 h) and n-heptane (after 90 h) decreased from 27.5% to 12% and from 25.2% to 9.0%, respectively (Fig. 10). The used catalysts were further tested for several more cycles (each time for 12 h for n-hexane and 18 h for n-heptane) and found up to the Vth cycle, the overall conversions slowly dropped. Finally, these catalysts significantly lost their catalytic activity in the VIth cycle (5.6% conversion for n-hexane and 4.4% conversion for n-heptane).
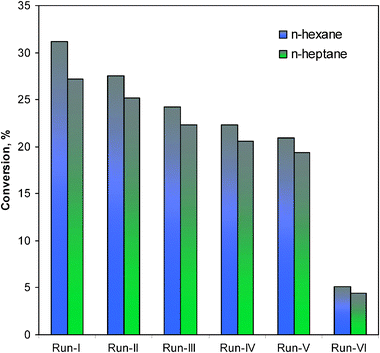
Catalyst metal leaching tests were confirmed with the ICP analysis. The fresh Pd–I catalyst has 0.26 weight% of Pd. After the Ist cycle, 0.24 weight% Pd was obtained in n-hexane reformation and 0.23 weight% of Pd in n-heptane reformation. After the Vth cycle, complete metal leaching was observed. It suggested that the metal loss should not be the main responsible factor for decrease in the catalytic activity. A possible structural change of the supported catalyst (change of catalysts color upon use) or polymerized material deposition at the catalyst surface during deep reformation, which can also act as a pore-blocking agent of the supported catalyst, should possibly account for it.
3.3 Mechanism consideration
In the literature several types of mechanisms have been reported for hydrocarbons reformation.19,27–29 There were two main important mechanisms which involve Pt loaded on solid supports. In these mechanisms, carbocations (carbenium ions) were mainly considered to be responsible for the skeletal type rearrangements. Hence, the most important reformation reaction step was the formation of secondary (2°) carbenium ions and their rearrangement on the heterogeneous catalyst surface in step 1 (where, n = 1 or 2). Once it is formed, the alkylcarbenium ion goes through simple bond shift reactions as well as cyclization of hydrocarbons. In the initial mechanisms, steps 2 and 3, the acidic nature of catalyst, nature of support, hydrogen and supported metal have also played a key role in this process.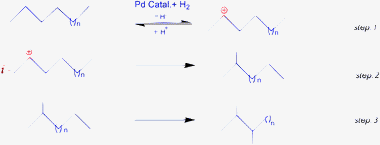
For that reason, one can consider that the catalytic reformation of n-hexane and n-heptane proceeds via formation of alkylcarbenium ions initially formed by reaction of the n-alkanes and promoted by H2 in the presence of MCM-41 supported Pd(II) catalysts. The acidic nature of Pd(II) catalysts (confirmed by TPD) has also taken part in the formation of alkylcarbenium. These formed alkylcarbenium ions easily undergo the bond shift reactions or some other competitive reactions. In this regard, the formation of α, α′, γ-adsorbed species and other adsorbed species was expected to support the preferred branched isomers.30,31
Consequently, the mono-branched isomer species i.e. 2-MP, 2-MH, 3-MP and 3-MH were formed by a single bond shift of the backbone of n-alkane chains. Moreover, the species of di-branched isomers i.e. 2,2-DMB, 2,2-DMP, 3,3-DMP, 2,2-DMB and 2,3-DMP were obtained by two bonds cleavage. The mono-branched species appear as the primary and dominating product with high selectivity of n-alkanes, while di-branched isomers were formed in lesser amount. For this reason, the primary mono-branched isomers were formed (more amount) by a single bond shift from the parent n-alkanes. On the other hand, di-branched isomers were obtained by the bond cleavage of primary mono-branched isomers. Therefore, we obtained lesser amount of di-branched isomers in comparison to mono-branched isomers. Such a type of mechanism was also observed previously in the Pd catalyzed hydrocarbons reformation.31
4. Conclusions
In the current study we have prepared potentially active modified MCM-41 supported di(ethylthio)ethane ligand I and di(ethylthio)propane ligand II and their supported Pd complex catalysts i.e.Pd–I and Pd–II. These hybrid catalysts Pd–I and Pd–II and their modified derivatives I and II were tested under a heterogeneous phase and relatively mild conditions in the reformation (H2) of n-hexane and n-heptane. Catalysts Pd–I and Pd–II appear to exhibit extremely high TONs from 1886 to 2325. The catalyst Pd–I showed best catalytic activity, leading to best overall conversions of 30.9% with a high overall selectivity of 91% and 26.6% with a high overall selectivity of 88% for n-hexane and n-heptane, respectively. Selectivity toward single bond shift of alkane isomers was much higher i.e. 74.4% (2-MP, 3-MP) and 64.9% (2-MH, 3-MH), in comparison to double bond shift isomers i.e. 15.9% (2,2-DMB, 2,3-DMB) and 27.8% (2,3-DMP, 2,2-DMP and 3,3-DMP).The detailed study of the effect of a variety of parameters, i.e. temperature, time, amount of catalyst, indicates that the most adequate operating conditions are 140 °C, 12 h, 25 mg of catalyst (for n-hexane) and 220 °C, 18 h, 25 mg of catalyst (for n-heptane). The reaction should proceed via carbocations in the presence of a catalyst and H2. Catalyst recycling and leaching tests indicated that the Pd does not leach out extensively during the normal reaction conditions, which allows its reutilization. Thermal stability tests confirm that these catalysts were stable during the maximum reaction temperature. In conclusion this is a simple and remarkable catalytic process for the production of branched alkanes from their linear counterparts and an expeditious way of improving the burning efficiency of fuel.
Acknowledgements
One of the authors (GSM) expresses his gratitude to Foundation of Science and Technology, Portugal, for “CIÊNCIA-2007” program and project: PTDC/EQU-ERQ/110825/2009. We also thank Dr P.F. Santos (CQ-VR, UTAD) for the valuable discussions and Analytical Laboratory, IST, Lisbon, for some analytical analysis.References
- V. M. Akhmedov and S. H. A. Khowaiter, Catal. Rev. Sci. Eng., 2007, 49, 33–139 CrossRef CAS.
- S. Raseev and G. D. Suciu, Thermal and Catalytic Processes in Petroleum Refining, Marcel Dekker Inc., New York, 2003, pp. 275–415, ch. 6 Search PubMed.
- D. G. Barton, S. L. Soled, G. D. Meitzner, G. A. Fuentes and E. Iglesia, J. Catal., 1999, 181, 57–72 CrossRef CAS.
- A. S. Goldman, A. H. Roy, Z. Huang, R. Ahuja, W. Schinski and M. Brookhart, Science, 2006, 312, 257–261 CrossRef CAS.
- K. Mukhopadhyay, B. R. Sarkar and R. V. Chaudhari, J. Am. Chem. Soc., 2002, 124, 9692–9693 CrossRef CAS.
- P. C. L'Argentière, E. A. Cagnola, D. A. Liprandi, M. C. Román-Martnez and C. Salinas-Martínez de Lecea, Appl. Catal., A, 1998, 172, 41–48 CrossRef CAS.
- M. H. Valkenberg and W. F. Hölderich, Catal. Rev. Sci. Eng., 2002, 44, 321–374 CrossRef CAS.
- D. Karthikeyan, N. Lingappan, B. Sivasankar and N. J. Jabarathinam, Ind. Eng. Chem. Res., 2008, 47, 6538–6546 CrossRef CAS.
- M. A. Hashimi, A. Qazi, A. C. Sullivan and J. R. H. Wilson, J. Mol. Catal. A: Chem., 2007, 278, 160–164 CrossRef.
- G. A. Olah and A. Molnar, Hydrocarbon Chemistry, John Wiley, New York, 3rd edn, 1995, pp. 10–27 Search PubMed.
- H. L. Needleman, Environ. Res., 2000, 84, 20–35 CrossRef CAS.
- (a) G. A. Ali, L. I. Ali, S. M. A. Fotouh and A. K. A. Gheit, Appl. Catal., A, 2001, 215, 161–173 CrossRef; (b) L. I. Ali, A. G. A. Ali, S. M. A. Fotouh and A. K. A. Gheit, Appl. Catal., A, 2001, 205, 129–146 CrossRef CAS.
- N. A. Zakarina, L. D. Volkova, A. K. Akurpekova and L. V. Komashko, Pet. Chem., 2008, 48, 187–193.
- J. N. Condo, S. Yang, Q. Zhu, S. Inagaki and K. Domen, J. Catal., 2007, 248, 53–59 CrossRef CAS.
- A. Barrera, J. A. Montoya, M. Viniegra, J. Navarrete, G. Espinosa, A. Vargas, P. D. Angel and G. Pérez, Appl. Catal., A, 2005, 290, 97–109 CrossRef CAS.
- K. I. Patrylak, F. M. Bobonych, Y. G. Voloshyna, M. M. Levchuk, V. M. Solomakha, L. K. Patrylak, I. A. Manza and O. M. Taranookha, Catal. Today, 2001, 65, 129–135 CrossRef CAS.
- T. Yashima, Z. B. Wang, A. Kamo, T. Yoneda and T. Komatsu, Catal. Today, 1996, 29, 279–283 CrossRef CAS.
- B. Modhera, M. Chakraborty, H. C. Bajaj and P. A. Parikh, React. Kinet. Mech. Catal., 2010, 99, 421–429 Search PubMed.
- G. S. Mishra, A. Kumar Jr and A. Kumar, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2004, 43, 1487–1492.
- M. J. L. Kishore, G. S. Mishra and A. Kumar, J. Mol. Catal. A: Chem., 2004, 216, 157–163 CrossRef CAS.
- K. S. Anisia, G. S. Mishra and A. Kumar, J. Mol. Catal. A: Chem., 2004, 215, 121–128 CrossRef CAS.
- Z. B. Wang, A. Kamo, T. Yoneda, T. Komatsu and T. Yashima, Appl. Catal., A, 1997, 159, 119–132 CrossRef CAS.
- A. M. G. Pedrosa, M. J. B. Souza, B. A. Marinkovic, D. M. A. Melo and A. S. Arauja, Appl. Catal., A, 2008, 342, 56–62 CrossRef.
- X. Wang, C. Li, Y. Wang and T. X. Cai, Catal. Today, 2004, 93, 135–140 CrossRef.
- W. Lin, Q. Cai, W. Pang, Y. Yue and B. Zou, Microporous Mesoporous Mater., 1999, 33, 187–196 CrossRef CAS.
- E. S. Kahn, A. I. Rheingold and S. I. Shupack, J. Crystallogr. Spectrosc. Res., 1993, 23, 697–710 CrossRef CAS.
- C. Costa, I. P. Dzsikh, J. M. Lopes, F. Lemos and F. R. Riberio, J. Mol. Catal. A: Chem., 2001, 154, 193–201.
- J. F. Allain, P. Magnoux, Ph. Schulz and M. Guisnet, Appl. Catal., A, 1997, 152, 221–235 CrossRef CAS.
- Y. Ono, Catal. Today, 2003, 81, 3–16 CrossRef CAS.
- H. Matsuhashi, H. Shibata, H. Nakamura and K. Arata, Appl. Catal., A, 1999, 187, 99–106 CrossRef CAS.
- E. Bolmsma, J. A. Martens and P. A. Jacobs, J. Catal., 1996, 159, 323–331 CrossRef.
Footnote |
| † Present and Correspondence address: Laboratory of Materials, Chemistry Center, Universidade de Trás-os Montes e Alto Douro (UTAD), Vila Real-5001801, Portugal. E-mail: E-mail: mishrags@utad.pt; Fax: +351 259350480 |
| This journal is © The Royal Society of Chemistry 2011 |