Heterogenization of homogeneous catalytic systems†
Alana E. C.
Collis
and
István T.
Horváth
*
City University of Hong Kong, Kowloon, Hong Kong SAR, China. E-mail: istvan.t.horvath@cityu.edu.hk
First published on 29th July 2011
Abstract
Heterogenization methods of homogeneous catalytic systems to render the catalyst and the product(s) in separable phases to allow facile separation are summarized. The applications of two liquid phases with no, limited, or temperature regulated miscibility of the two phases or a solid phase and a liquid phase with no, limited, or temperature regulated solubility of the constituent(s) of the solid phase in the liquid phase are demonstrated on selected examples.
Introduction
Heterogeneity is the quality or state to express the lack of uniformity among atoms, molecules, materials, phases, catalysts, cells, animals, people, cities, countries, societies, planets, and even universes. Heterogenization of homogeneous catalytic systems can be defined as the modification of homogeneous catalysts to “break” the uniformity among the molecules of the catalyst and the product(s) by rendering them in separable phases to allow their separation. The most frequently used variations involve two liquid phases with no, limited, or temperature regulated miscibility of the two phases1 or a solid phase and a liquid phase with no, limited, or temperature regulated solubility of the constituent(s) of the solid phase in the liquid phase.2Liquid–liquid catalytic systems
A liquid–liquid biphasic catalytic system consists of a catalyst phase containing the dissolved catalyst and a product phase. Water, alcohols, ionic liquids, fluorous media, supercritical fluids, and gas expanded liquids have been used successfully as the catalyst phase. Since the formation of a liquid–liquid biphasic system is due to the sufficiently different intermolecular forces of two liquids,3 the selection of a catalyst phase should be governed by the solvent properties of the product phase at high conversion. The Hildebrand solubility parameter scale (Fig. 1) can be used to assess the solubility and miscibility of the product with various solvents and to identify a well separable catalyst phase.4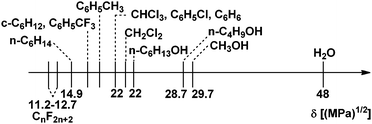 | ||
| Fig. 1 Hildebrand solubility parameters of solvents. | ||
Thus, if the product is apolar the catalyst phase should be polar, and vice versa, if the product is polar the catalyst phase should be apolar. The success of any biphasic system depends on whether the catalyst could be designed to dissolve preferentially in the catalyst phase. Perhaps the most important rule for such design is that the catalyst has to be catalyst phase like, since it has been known for centuries that “similia similibus solvuntur,” or “like dissolves like”. This can be achieved by attaching solubilizing substituents or groups to the catalyst core in appropriate size and number. In order to avoid significant leaching of the catalyst, the solubility properties of all catalytic intermediates should be tuned for high catalyst phase affinity at product separation conditions. It should be emphasized that heterogenized catalysts cannot be evaluated properly without leaching data. Unfortunately, many publications and patents either do not report or perform leaching analysis, which can be time consuming and expensive.
A biphasic reaction could proceed either in the catalyst phase or at the interface of the two phases. When the solubilities of the substrates are very low in the catalyst phase, the chemical reaction may still occur at the interface or phase transfer agents may be added to facilitate the reaction.5 It should be emphasized that a biphase system might become a one-phase system by increasing the temperature (Fig. 2).
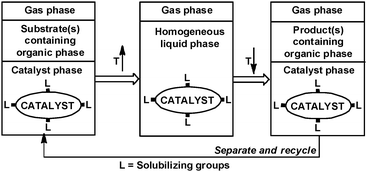 | ||
| Fig. 2 Temperature-dependent liquid–liquid catalyst system. | ||
Thus, a biphasic catalyst could combine the advantages of one-phase chemistry with biphasic product separation by running the reaction at higher temperatures and separating the products at lower temperatures. The first commercial biphasic process, the Shell Higher Olefin Process (SHOP) developed by Keim, uses 1,4-butanediol as the catalyst phase and a nickel catalyst modified with a diol soluble phosphine R2PCH2COOH.6 While ethylene is highly soluble in 1,4-butanediol, the higher olefins readily phase separate from the catalyst phase. Although alcohols could be excellent solvents for various catalytic reactions, their use is limited because of the solvent properties of most products and alcohols are too similar resulting in separation issues.
Water is a very attractive medium for the heterogenization of homogeneous catalytic systems, because it is readily available, nonflammable, non-toxic, cheap, and easy to separate from many organic products. Water is a particularly good medium for ionic reactions because it can readily solvate anions and cations and it could serve as a solvent and/or as a ligand. The water solubility of catalysts can be ensured by either attaching appropriate hydrophilic substituents such as hydroxides, carboxylates, phosphonates, ammonium, etc. or polar solubilizing groups that could involve in hydrogen bonding in the aqueous media. The water-soluble catalyst can have different structures depending on the pH offering another powerful designer tool.
Rhône-Poulenc developed and Rurhchemie commercialized an aqueous biphasic process for the hydroformylation of propylene in the presence of triphenylphosphine trisulfonate (TPPTS) modified rhodium catalyst, HRh(CO)(TPPTS)3.7 The main product is n-butanal and the normal- and iso-butanal or n/i ratio is surprisingly high in comparison to the PPh3 modified rhodium catalyst. The overall reaction mechanism operating in the aqueous and organic phases is very similar.8a Although the water soluble catalyst has similar electronic and steric properties to the organic analogue, it is less active and more selective. It is believed that the coordination of the olefin to the coordinatively unsaturated {HRh(CO)(TPPTS)2} or {HRh(CO)2(TPPTS)} species results in high or low n/i ratio, respectively. Indeed, HRh(CO)2(TPPTS) was not detectable in water because of the higher activation energy of the dissociation of TPPTS from HRh(CO)(TPPTS)3 compared with PPh3 from HRh(CO)(PPh3)3. The lower activity and higher selectivity in the aqueous biphasic system is therefore due to the intramolecular association of sulfonate substituents of the neighboring TPPTS ligands viahydrogen bonding in aqueous medium.8
One of the limitations of water is the low solubility of some gases (H2 and CO) and many organic compounds.9 Accordingly, the potential application of aqueous biphasic hydroformylation is limited by the low solubility of higher olefins (Cn > 6) in water.10 It was reported that supported aqueous phase catalysis could overcome this limitation by immobilizing the aqueous solution of HRh(CO)(TPPTS)3 on a high surface area silica.11 It was later shown that the hydrophilic support loses a significant amount of water and holds the water soluble phosphine by hydrogen bonding of the hydrated sodium-sulfonate groups to the surface of silica (Fig. 3) and the rhodium site actually operates in the organic phase resulting in no solubility limitations.10
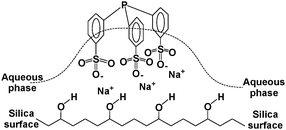 | ||
| Fig. 3 Immobilization of a water soluble phosphine by hydrogen bonding of the hydrated sodium-sulfonate groups to the surface of silica. | ||
The immobilization of various liquids on solid carriers could be an attractive solution to the separation problem of homogeneous catalysts. The supported liquid phase can be the catalyst itself or it may contain a dissolved catalyst. In order to maintain the catalytic performance a supported liquid phase catalyst should not lose its liquid phase, which is therefore stationary, and the catalyst may be called a stationary liquid-phase (SLP) catalyst.‡
Panster has investigated the design of SAPCs of Ru/TPPTS on silica for hydrogenation.12 Upon recycling of the catalyst, the conversion dropped off but this was attributed to poisoning of the catalyst by organic compounds rather than the observed leaching of <0.02% of the metal. These catalysts are limited to non-polar solvents. More success was achieved in the hydrogenation of α,β-unsaturated aldehydes using a supported homogeneous iridium catalyst. No leaching was observed and the catalysts retained high conversions with good chemo- and stereoselectivity. A hybrid SAPC catalyst was used by van Leeuwen et al. using an inorganic silica core with polystyrene attached to it. The polystyrene had phosphite ligands for rhodium-catalyzed hydroformylation.13
Besides water, liquid acids14 and ionic liquids15 have been used in supported liquid phase catalysis. For example, an SLP catalyst was developed for the facile hydrogenation of aromatics by impregnating silica or clay (attapulgite) with the solution of PtCl2(CH3CN)2 in BF3·H2O.14Hydrogenation of arenes occurs rapidly even at 25 °C under 2.76 MPa H2 and the colorless products can be separated from the catalyst by simple filtration.
Wasserscheid et al. have later reported the use of a rhodium SILP system using silica for the hydroformylation of various alkenes (Fig. 4).16
![Sulphoxantphos ligand and [bmim][n-C8H17OSO3] (bmim = 1-n-butyl-3-methylimidazolium).](/image/article/2011/CY/c1cy00174d/c1cy00174d-f4.gif) | ||
| Fig. 4 Sulphoxantphos ligand and [bmim][n-C8H17OSO3] (bmim = 1-n-butyl-3-methylimidazolium). | ||
Similarly, the carbonylation of methanol using a rhodium iodide Monsanto-type catalyst system on silica, [BMIM][Rh(CO)2I2]-[BMIM]I-SiO2, has also been studied.17
The miscibility of perfluorinated alkanes, dialkylethers, and trialkylamines with common organic solvents such as toluene, THF, acetone, and alcohols is low at room temperature,3,18 thus these materials could form fluorous biphase systems.19,20 The term fluorous was introduced, as the analogue to the term aqueous, to emphasize the fact that one of the phases of a biphase system is richer in fluorocarbons than the other. A fluorous catalyst system consists of a fluorous phase containing a preferentially fluorous soluble catalyst19–21 and a second product phase, which may be any organic or non-organic solvent with limited solubility in the fluorous phase.19 Conventional homogeneous catalysts can be made fluorous soluble by incorporating fluorocarbon moieties to their structure in appropriate size and number. The most effective fluorocarbon moieties are linear or branched perfluoroalkyl chains with high carbon number that may contain other heteroatoms (the “fluorous ponytails”). Fluorous catalysts are best suited for converting apolar substrates to products of higher polarity, as the partition coefficients of the substrates and products will be higher and lower, respectively, in the fluorous phase. The net results are little or no solubility limitation on the substrates and easy separation of the products. Furthermore, as the conversion level increases the amount of polar products increases further enhancing separation. One of the most important advantages of the fluorous biphase catalyst concept is that many well established hydrocarbon soluble catalysts could be converted to fluorous soluble versions. In general, fluorous catalysts have similar structures and spectroscopic properties to the parent compounds. The major difference arises from the presence of the fluorous ponytails, which provide a fluorous blanket around the hydrocarbon domain of the catalyst. If the electron withdrawing effect of the fluorous ponytails on the ligands is not mitigated by insulating groups,22 the reactivity of the organometallic catalysts could be significantly different. Accordingly, most fluorous analogues of hydrocarbon soluble catalysts have been prepared by incorporating insulating groups and shown to have comparable catalytic performance with the additional benefit of facile catalyst recycling.19
The excellent performance of fluorous catalysis was first demonstrated by using the P[CH2CH2(CF2)5CF3]3 modified rhodium catalyst for the hydroformylation of 1-decene at mild conditions in a 50/50 vol% toluene/C6F11CF3 solvent mixture, which forms a homogeneous liquid phase above 100 °C.18 The fluorous soluble P[CH2CH2(CF2)5CF3]3 was selected on the basis of a semiempirical calculation of the electronic properties of P[(CH2)x(CF2)yCF3]3 (x = 0–5; y = 2, 4). It was shown that the structure of HRh(CO){P[CH2CH2(CF2)5CF3]3}3 in C6F11CF3 is similar to that of HRh(CO)(PPh3)3 in toluene and HRh(CO){P(m-C6H4SO3Na)3}3 in water. The catalyst recovery was tested in a semicontinuous hydroformylation of 1-decene using the Rh/P[CH2CH2(CF2)5CF3]3 system. During nine consecutive reaction–separation cycles, a total turnover of more than 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 was achieved with a loss of 1.18 ppm of Rh/mol of undecanals. The fluorous-soluble Rh/P[CH2CH2(CF2)5CF3]3 catalyst was also used for the continuous hydroformylation of ethylene. The long-term stability of the Rh/P[CH2CH2(CF2)5CF3]3 catalyst was better than the Rh/PPh3 catalyst.22 Later work by Cole-Hamilton et al. further showed that using a phosphite were also successful and gave higher l/b ratios.23 Other fluorous-tagged ligands include F-dppp (fluorous-tagged 1,3-bis(diphenylphosphino)propane).24
000 was achieved with a loss of 1.18 ppm of Rh/mol of undecanals. The fluorous-soluble Rh/P[CH2CH2(CF2)5CF3]3 catalyst was also used for the continuous hydroformylation of ethylene. The long-term stability of the Rh/P[CH2CH2(CF2)5CF3]3 catalyst was better than the Rh/PPh3 catalyst.22 Later work by Cole-Hamilton et al. further showed that using a phosphite were also successful and gave higher l/b ratios.23 Other fluorous-tagged ligands include F-dppp (fluorous-tagged 1,3-bis(diphenylphosphino)propane).24
Solid–liquid catalytic systems
The simplest solid–liquid immobilization method is the use of the solid form of a homogeneous catalyst, if it can be dissolved in the reaction medium at higher temperature and separated from the product after the reaction at lower temperature (Fig. 5).25,26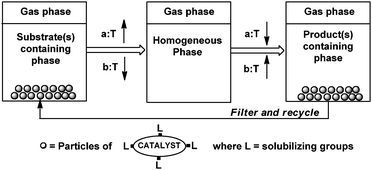 | ||
| Fig. 5 Temperature-dependent solubilisation of solid catalysts. | ||
The temperature dependent solubility of P[CH2CH2(CF2)5CF3]3 in octane was used to catalyze the addition of various alcohols to methyl propiolate (Fig. 6).25a The reaction readily proceeded at 65 °C and the fluorous phoshine catalyst was precipitated at −30 °C allowing the decantation of the product.
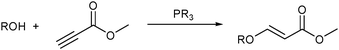 | ||
| Fig. 6 Phosphine catalyzed addition of alcohol to methyl propiolate. | ||
The thermomorphic nature of a fluorous rhodium hydrosilylation catalyst was used to demonstrate the efficient release and capture of the catalyst by a Teflon tape as a function of temperature (Fig. 7).26 The fluorous catalyst, which has limited solubility in the reaction mixture at room temperature, is released from the Teflon tape to the reaction mixture at higher temperature, where it acts as a homogeneous catalyst. When the reaction is completed, the reaction mixture is cooled back to room temperature during which the fluorous catalyst “returns” to the Teflon tape.
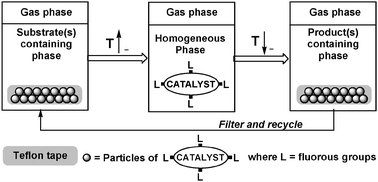 | ||
| Fig. 7 Temperature-dependent release and catch of a fluorous catalyst by a Teflon tape. | ||
The temperature dependent solubility can be used in the opposite manner, when a solid catalyst precursor can be dissolved in the reaction medium at lower temperature and separated from the product by increasing the temperature.27 This approach could particularly be attractive to control highly exothermic reactions as run-away reaction would be shut down by the heterogenization of the catalyst resulting in much lower activity.
Macromolecular dendrimer catalysts give another option for recycling a homogeneous catalyst.28 A relatively inefficient way of isolating dendrimeric catalysts is by precipitation after the addition of an other solvent, leading to a down-stream solvent separation issue.29 More attractively, the products could be separated from the dendrimer catalysts by membrane filtration.
There are many examples of where trace levels of a catalyst can cause undesirable discoloration of a product e.g.polymers and paints. DuPont reported a process by which porphyrin ligands are supported on polyethylene oligomers.30 The thermomorphic catalyst was removed by filtration or centrifugation after cooling. Octadecyl groups have been used to covalently immobilize Cu-catalysts for atom transfer radical polymerization and click-chemistry.31,32 The polymerization of methyl methacrylate was successfully performed by using a Cu(I)Br species with the tetraethyldiethylenetriamine ligand attached to polyethylene using a polyethylene-glycol (PEG) spacer (Fig. 8).33 The catalyst was removed by centrifugation and analysis showed less than 2–3% of the catalyst present in the organic layer. Slight loss in activity was observed with catalyst re-use but this was attributed to the formation of Cu(II) species retarding the rate of reaction.
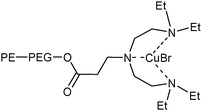 | ||
| Fig. 8 Immobilized CuBr tetraethyldiethylenetriamine complex. | ||
The water soluble poly(N-isopropylacrylamide) (PNIPAM) supported catalyst, Rh(Cl)(CO)2(NH2-polymer) was reported for the hydrogenation of methyl methacrylate under aqueous conditions at 500 psi H2 and 26 °C, though no specific leaching data was reported.34 The formation of hydrogen bonding causes dissolution of the polymer at the ambient temperatures. In this case, warming of the reaction mixture to 40 °C facilitated the precipitation of the catalyst which was isolated by simple filtration. The catalyst could be reused without loss of catalytic activity and no catalytic activity was observed in the filtrate.35
An octadecyl ponytail tag was added to a BINAP ligand to heterogenize a Rh(II) (S)-5,5-diamino BINAP complex.36 This was soluble in hot EtOH–1,4-dioxane solution at 60 °C but then precipitated out at 0 °C. The catalyst was effective at asymmetric hydrogenations of substituted acetoacetates with excellent enantioselectivity in the cases of β-ketoesters and no loss of activity over four cycles. Analysis of the filtrate did show leaching of <2% Ru by the fourth cycle.
In a liquid reaction medium, leaching of a catalyst from a support is often one of the biggest challenges faced in a process. Carbonylation of methanol using [RhI2(CO)2]−catalyst over 50% of the catalyst was leached after one run in MeOH. Cole-Hamilton et al. identified the leaching of Rh species from a polyvinylpyrrolidinone (PVP) support as a problem and utilized supercritical CO2 as a suitable solvent for the reaction; dissolving the reagents but since it is a poor solvent for ionic species there was reduced leaching of the catalyst.37 Above 130 °C in MeOH, the reaction mixture becomes monophasic and less than 1% of the catalyst was leached.
Solid-phase peptide synthesis using polystyrene cross-linked with divinyl benzene (Merrifield resin) was the original model for the permanent heterogenization of homogeneous catalysts. This could be also achieved by attractive interactions between the catalyst and the surface of solids or by the attachment of homogeneous catalysts to surface atoms of organic, inorganic, or organometallic solids by covalent bond(s) (Fig. 9).38 Some supports may be organic functionalized materials e.g. silicas or chemically inert organic supports such as polymers or copolymers of styrene, acrylates, vinyl chlorideetc.39
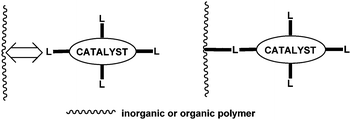 | ||
| Fig. 9 Binding of homogeneous catalysts to support. | ||
Houghten described the use of a tea bag for the solid-phase synthesis of peptides,40 which was further advanced by Seebach. A polymer-bound Ti-TADDOL (Fig. 10) was contained within a polypropylene tea bag.41 A small decrease in enantioselectivity was observed after several runs. After storing the tea bag under Ar for two weeks a decrease in conversion was observed, though there was no specific data for leaching. The tea bag could be rinsed and easily re-used with a different substrates.
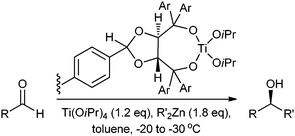
Grubbs has used LiPPh2 to substitute a chloromethylated PS-DVB polymer.42 The immobilized phosphines were then equilibrated with tris(triphenylphosphine)chlororhodium(I) to form a catalyst that was successfully used to hydrogenate olefins. Collman et al. used a triphenylphosphine substituted PS-DVB resin to form resin-bound Rh(I) and Ir(I) complexes for hydrogenation but no leaching data was reported.43 More recently Grubbs has used the same cross-linked polystyrene to hereogenize a ruthenium vinylcarbenene olefin metathesis catalyst.44 The catalyst could be re-used but a 20% drop in activity was observed after each cycle. Slower metathesis rates are caused by diffusion into the polymer but the heterogenization also slows the decomposition of the carbene.
Basset has used oxide surfaces (typically silica, alumina or titana) as ligands to carry out surface organometallic chemistry and catalysis. In the case of olefin metathesis, such surface organometallic catalysts have been shown to be successful. For example, a successful tungsten-based heterogeneous alkane metathesis catalyst grafted onto alumina proved successful as well as other transition metals on surfaces (Fig. 11).45
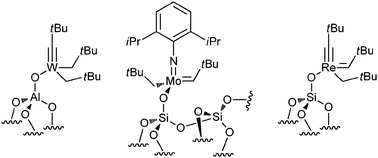 | ||
| Fig. 11 Tungsten alkane metathesis catalyst grafted onto alumina. | ||
Some of the most studied catalysts for heterogenization are those using salen-type ligands, bis(oxazoline) ligands and BINAP systems.46 Inorganic supports widely used include alumina, silica, zeolites and clay. Significant work has been made into the use of clays (natural and synthetic), zeolites and mesoporous materials immobilization by cation exchange.47Leaching is common due to weak physisorption of the catalyst.
The immobilization of Mn(salen) complexes using zeolites, clays and various Al-, Ga-, and Fe-substituted mesoporous silicates for asymmetric catalysis has been studied (Fig. 12). Some metal organic frameworks,48 and polymer supports49 and a sulfoalkyl modified zirconium poly(styrene-isopropenyl phosphonate)-phosphate50 have also been investigated.
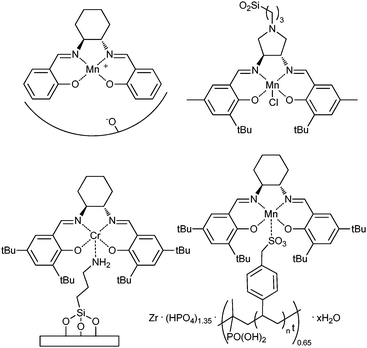 | ||
| Fig. 12 Examples of heterogenized metal(salen) catalysts. | ||
As yet, these immobilized catalysts are generally less active and do not seem able to compete with the enantioselectivities achieved by the homogeneous catalysts. Careful preparation to remove any complex from the surface of the zeolite appears to prevent any leaching of the metal is not observed but there are some reports of ligand leaching.
Mn(salen) catalysts can be supported on bentonite, laponite and clays. Although the activity was comparable with Mn(salen) homogeneous epoxidation reactions, the enantioselectivity was lower and a reduction in activity with catalyst recycling was observed. This was not attributed to the small amount of leaching but more to ligand modification.51
Bis(oxazoline) cupric complexes have been heterogenized with laponite clay, Nafion and a Nafion–silica composite (Fig. 13).52Copper leaching observed using laponite was not observed on the sulfonic Nafion supports. After two runs, the amount of copper (mmol g−1) is lower for the different supported catalysts but this is attributed to adsorption of by-products onto the surface and no catalytic activity is observed in the filtrate.
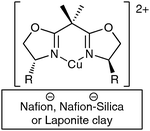 | ||
| Fig. 13 Heterogenization of bis(oxazoline) Cu(II) complexes. | ||
Immobilized bis(oxazoline) on MCM-41 or MCM-48 did not exhibit leaching in the cyclopropanation of styrene, though lower cis/trans selectivities were observed in comparison to the homogeneous catalyst (Fig. 14).53 Similar ligands attached to styrene were copolymerized with styrene and various cross-linkers. The subsequent cyclopropanation reactions gave low yields as was also the case with the Merrifield resin.54 Covalent immobilization over silica led to similar cis/trans ratios in comparison to homogeneous catalysis, but the enantioselectivity was lower.
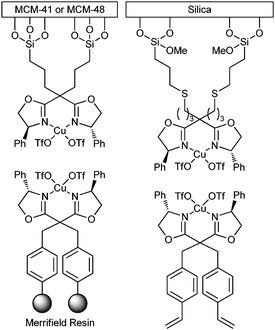 | ||
| Fig. 14 Examples of different immobilized bis(oxazoline) catalysts. | ||
A bis(oxazoline) tethered to a polyurethane and used in a Diels–Alder reaction. The first three cycles of the catalyst, the yields were quantitative with excellent endo selectivity and reasonable enantiomeric excess.55 However, on the fourth cycle there was a complete loss of enantioselectivity. More recently, the same ligand was covalently attached to silica and an improved enantiomeric excess were observed.56
De Vos and Jacobs developed some heterogenized epoxidation catalysts. Different tungsten species were prepared by being exchanged onto Amberlite IRA-900.57 MCM-41 based catalysts were also prepared by silylation then the grafting of quaternary ammonium groups including a phosphoramide group. The active catalyst was then prepared by adding tungstic acid and H2O2. In the presence of the bulky cis-cyclooctene, the Amberlite-based catalysts had varying degrees of conversion in the oxidation reaction. The best catalyst was the Amberlite/{PO4[WO(O2)2]4}3− with 85% conversion and no detected leaching at 16 h. The W-phosphoramide-grafted MCM-41 catalyzed epoxidation of the same substrate gave a 70% conversion and ∼2 ppm of leaching was measured.
In addition to immobilization on silica, Curran recently used a fluorous reverse-phase (FRP) silica gel.58 This enables separation of “light” fluorous compounds (<40 wt% fluorine) from fluorous solvents and other non-fluorinated compounds. Hope et al. used the FRP silica gel to heterogenize BINAP ligands as a ruthenium complex for asymmetric hydrogenation.59
Entrapment or encapsulation is another method to maintain the performance of the homogeneous catalyst and ensure facile separation of the product from the catalyst.60 Although there may be no covalent attachment in all cases, encapsulation can alter the geometry of the structure and therefore affect the catalytic activity of the complex. In general, inorganic encapsulation materials with a high aluminium content are avoided since these sites can act as ligands and reduce catalytic activity. Aluminosilicate and magnesiumsilicate clays act as supports for electrostatic interactions involving cation exchange or complex formation.
In the case of the porous inorganic materials, it is essential that the density of the catalyst is not too high. This would lead to blocking of pores, preventing diffusion of reactants and products in and out of the pores. While pores must be open and accessible to substrates, it is also important that there is sufficient steric confinement to prevent the metal complex being leached out of the structure. Retaining the active metal complex within the zeolite cage is aided by good metal complex stability where there is no ligand substitution or degradation.39 Although non-essential, electrostatic forces aid catalyst retention but there is often significant leaching observed. Many examples are reported in the in the literature of heterogenizing Lewis acid oxidation catalysts but these seem to be susceptible to significant leaching.61
De Vos et al. prepared different Pd-zeolites as heterogeneous catalysts in the Heck reaction to obtain trans-arylated acrylate esters with high yield (Fig. 15).62 A study of the reaction profile then led to a change from a batch reactor process to a continuous flow. Using Pd(NH3)42+-Y (0.4 wt% Pd, 500–800 μm particles), at 130 °C the optimal conversion in the reaction of butyl acrylate and 4-bromoacetophenone was achieved after 3 h. After 103 h only 0.22% Pd had been leached. Leaching was found to be associated with the presence of Pd(II) species.
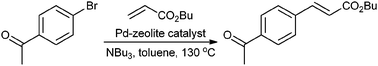 | ||
| Fig. 15 Heck reaction using Pd-zeolite catalysts. | ||
There has been considerable work on the development of polyurea encapsulated microcapsules.63–65 The encapsulation is achieved by the reaction of isocyanate groups of polymethylene polyphenylene diisocyanate with the amines resulting from isocyanate groups that are hydrolysed at the oil–water interface (Fig. 16). The urea backbone is ideal for retaining the Pd species due to ligation of the Pd(OAc)2. Isolated yields from the Suzuki cross-coupling reaction were over 80% and the microcapsules only exhibited 0.2% leaching in a reaction suggesting that the ligating properties of the polyurea scavenged any metal species that may escape.66 These catalysts also did not require the phosphine ligands usually associated with this genre of cross-coupling reaction and could also be applied to reactions in scCO2, carbonylation and Heck reactions.63 In the case of the carbonylation reactions and the Heck reactions, a greater degree of leaching was measured than in the Suzuki reactions.
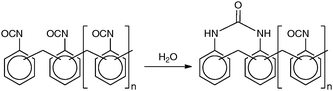 | ||
| Fig. 16 Polyurea microcapsule structure. | ||
Metallocene catalysts are homogeneous catalysts that have many applications and are industrially very significant in olefin polymerization. As a homogeneous catalyst metallocenes are not efficient in slurry and gas phase reactors due to fouling so heterogeneous catalysts have been developed for industrial application. Despite being heterogeneous, these catalysts are not recovered and recycled. Silica, alumina and MgCl2 are widely used for Ziegler–Natta and Phillips catalysts and the catalyst can be adsorbed onto the surface or chemically bonded.67 The nature of the support can also affect the morphology of the polymer produced. Kaminsky has reported examples of metallocene catalysts being heterogenized onto a silica surface and then used for the production of polypropylene.68
Methylaluminoxane (MAO) co-catalyst can be immobilized onto silica rather than the metallocene.69 Alt et al. also produced self-immobilized catalysts where the metallocene contained an olefin or alkyne moiety that participated as a co-monomer in the polymerization reaction.70 Thus, the catalyst becomes heterogenized as the polymerization progresses.
Metallocenes can also be immobilized onto an organic support. This is generally achieved by covalently attaching a ligand precursor to a polymere.g.polystyrene then adding the metal. Grubbs et al. immobilized a titanocene onto a PS-DVB for olefin reduction reactions.71 Fréchet et al. immobilized a metallocene inside a polystyrene matrix, excapsulating it while still enabling co-polymerization of ethylene and 1-hexene.72
Polymers can be used to encapsulate catalyst complexes by entangling them in the polymer structure, which is equivalent to a restriction of motion inside a zeolite. A degree of flexibility aids retention of the metal complex conformation, but the flexibility should not be too great since this can lead to leaching of the complex if the solvent causes the structure to swell. This means that polymer encapsulated complexes often have limited solvent tolerance.39 Similarly, sufficiently high catalyst loading is only achievable if the metal complex is soluble in the synthesis of the polymer structure. The accessibility of the catalyst within the polymer structure can offer advantages over inorganic structures such as zeolites. The pore size of a zeolite can be too small for some metal complexes and the ionic interactions can also significantly affect the complex conformation. The larger “pore” size of the polymer matrix results in a more accessible active site.
The Pd(II) species of the encapsulated Pd(OAc)2, known as [PdEnCat]™ could be reduced to the polyurea encapsulated Pd(0) species. The [Pd0EnCat]™ successfully reduced various aryl ketones and nitro groups without any by-products resulting from over reduction of the aromatic ring or further hydrogenolysis to generate methylene compounds.65 No specific leaching data was reported but there was no observed drop in reaction rate with catalyst recycling.
The “polymer incarceration” is a method to microencapsulate the catalyst into an epoxide containing copolymer of styrene, 4-vinylbenzene and methacrylic acid or alcohol.73 These materials were then isolated and further cross-linked. The most active catalyst was prepared by encapsulation of Ph(PPh3)4 in a copolymer of styrene, 4-vinylbenzyl glycidyl ether and tetraethyleneglycol mono-2-phenyl-2-propenyl ether. These catalysts were used in both hydrogenation and allylation reactions. No leaching was observed in the hydrogenation, but some was observed in the allylation reactions. In comparison to the more commonly used homogeneous versions, these immobilized Pd(0) catalysts can be stored at room temperature and have exhibited higher activity in some cases.
To date, there are several examples of heterogenization of hydrogenation catalysts but those have focused on classic hydrogenation reactions using H2 gas. Although the immobilized Shvo catalyst (Fig. 17)74a was used for alcohol dehydrogenation,74b its application for transfer hydrogenation has yet to be achieved. Heterogenization of a transfer hydrogenation catalyst could facilitate far more efficient, milder reaction conditions for hydrogenation.
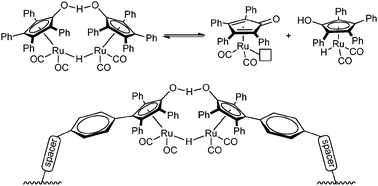 | ||
| Fig. 17 The Shvo catalyst and its heterogenization. | ||
Finally, molecular cross-linking of solid particles of soluble homogeneous catalysts could be another approach to heterogenization, though only enzymes have been used so far to prepare catalytically active heterogenized materials.75
Notes and references
- B. Cornils, W. A. Herrmann, I. T. Horváth, W. Leitner, S. Mecking, H. Olivier-Bourbigou and D. Vogt, Multiphase Homogeneous Catalysis, Wiley-VCH, Weinheim, 2005 Search PubMed.
- B. C. Gates, Catalytic Chemistry, John Wiley & Sons, New York, 1992 Search PubMed.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, , VCH, Weinheim, 1990 Search PubMed.
- (a) A. F. M. Barton, Chem. Rev., 1975, 75, 731–753 CrossRef CAS; (b) A. F. M. Barton, CRC Handbook of Solubility Parameters and Other Cohesion Parameters, CRC Press, Inc, Boca Raton, Florida, 1983 Search PubMed.
- M. Makosza, Pure Appl. Chem., 2000, 72, 1399 CrossRef CAS.
- W. Keim, F. H. Kowaldt, R. Goddard and C. Krüger, Angew. Chem., Int. Ed. Engl., 1978, 17, 466 CrossRef.
- (a) E. G. Kuntz, CHEMTECH, 1987, 17, 570 CAS; (b) B. Cornils and E. G. Kuntz, Development of Commercial Biphasic Catalysis in Aqueous Phase Organometallic Catalysis, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, 1998, p. 271m Search PubMed.
- (a) I. T. Horváth, R. V. Kastrup, A. A. Oswald and E. J. Mozeleski, Catal. Lett., 1989, 2, 85 CrossRef; (b) R. L. Pruett, Adv. Organomet. Chem., 1979, 17, 1 CrossRef CAS.
- Aqueous phase organometallic catalysis, ed. B. Cornils and W. A. Herrmann, Wiley VCH, Weinheim, 1998 Search PubMed.
- I. T. Horváth, Catal. Lett., 1990, 6, 43 CrossRef.
- J. P. Arhancet, M. E. Davis, J. S. Merola and B. E. Hanson, Nature, 1989, 339, 454 CrossRef CAS.
- E. Fache, C. Mercier, N. Pagnier, B. Despeyroux and P. Panster, J. Mol. Catal., 1993, 79, 117 CrossRef CAS.
- T. Jongsma, G. Challa and P. W. N. M. Van Leeuwen, J. Mol. Catal., 1993, 83, 37 CrossRef CAS.
- I. T. Horváth, Angew. Chem., Int. Ed. Engl., 1991, 30, 1009 CrossRef.
- (a) C. P. Mehnert, J. Am. Chem. Soc., 2002, 124, 12932 CrossRef CAS; (b) C. P. Mehnert, Chem.–Eur. J., 2005, 11, 50 CrossRef.
- (a) M. Haumann, K. Dentler, J. Joni, A. Riisager and P. Wasserscheid, Adv. Synth. Catal., 2007, 349, 425 CrossRef CAS; (b) A. Riisager, R. Fehrmann, S. Flicker, R. van Hal, M. Haumann and P. Wasserscheid, Angew. Chem., Int. Ed., 2005, 44, 815 CrossRef CAS.
- A. Riisager, B. Jørgensen, P. Wasserscheid and R. Fehrmann, Chem. Commun., 2006, 994 RSC.
- J. H. Hildebrand, J. M. Prausnitz and R. L. Scott, Regular and Related Solutions, Van Nostrand Reinhold Co., New York, 1970, ch. 10 Search PubMed.
- (a) I. T. Horváth and J. Rábai, Science, 1994, 266, 72–75 Search PubMed; (b) I. T. Horváth and J. Rábai, US 5,463,082, 1995.
- J. A. Gladysz, D. P. Curran and I. T. Horváth, Handbook of Fluorous Chemistry, Wiley/VCH, Weinheim, 2004 Search PubMed.
- V. Herrera, P. J. F. de Rege, I. T. Horváth, T. L. Husebo and R. P. Hughes, Inorg. Chem. Commun., 1998, 1, 197 CrossRef CAS.
- I. T. Horváth, G. Kiss, R. A. Cook, J. E. Bond, P. A. Stevens, J. RabáI and E. J. Mozeleski, J. Am. Chem. Soc., 1998, 120, 3133 CrossRef.
- D. F. Foster, D. Gudmensen, D. J. Adams, A. M. Stuart, E. G. Hope, D. J. Cole-Hamilton, G. Schwarz and P. Pogorzelec, Tetrahedron, 2002, 58, 3901 CrossRef CAS.
- K. S. A. Vallin, Q. Zhang, M. Larhed, D. P. Curran and A. Hallberg, J. Org. Chem., 2003, 68, 6639 CrossRef CAS.
- (a) M. Wende and J. A. Gladysz, J. Am. Chem. Soc., 2001, 123, 11490 CrossRef CAS; (b) K. Ishihara, S. Kondo and H. Yamamoto, Synlett, 2001, 1371 CrossRef CAS; (c) M. Wende and J. A. Gladysz, J. Am. Chem. Soc., 2003, 125, 5861 CrossRef CAS.
- L. V. Dinh and J. A. Gladysz, Angew. Chem., Int. Ed., 2005, 44, 4095 CrossRef CAS.
- (a) D. E. Bergbreiter, L. Zhang and V. M. Mariagnanam, J. Am. Chem. Soc., 1993, 115, 9295 CrossRef CAS; (b) D. E. Bergbreiter, Chem. Rev., 2002, 102, 3345 CrossRef CAS.
- A. Berger, R. J. M. Klein Gebbink and G. Van Koten, Top. Organomet. Chem., 2006, 20, 1 CrossRef CAS.
- D. Rechavi and M. Lemaire, J. Mol. Catal. A: Chem., 2002, 182–183, 239 CrossRef CAS.
- US Pat., 6 288 253, 2001.
- G. Barre, D. Taton, D. Lastécouéres and J.-M. Vincent, J. Am. Chem. Soc., 2006, 71, 8680 Search PubMed.
- N. Candelon, D. Lastécouéres, A. K. Diallo, J. R. Aranzes, D. Astruc and J.-M. Vincent, Chem. Commun., 2008, 741 RSC.
- Y. Shen, S. Zhu and R. Pelton, Macromolecules, 2001, 34, 3182 CrossRef CAS.
- W. J. Shaw, J. C. Linehan, A. Gutowska, D. Newell, T. Bitterwolf, J. L. Fulton, Y. S. Chen and C. F. Windisch, Inorg. Chem. Commun., 2005, 8, 894 CrossRef CAS.
- W. J. Shaw, Y. S. Chen, J. Fulton, J. Linehan, A. Gutowska and T. Bitterwolf, J. Organomet. Chem., 2008, 693, 2111 CrossRef CAS.
- Y. Y. Huang, Y. M. He, H. F. Zhou, L. Wu, B. L and Q. H. Fan, J. Org. Chem., 2006, 71, 2874 CrossRef CAS.
- R. J. Sowden, M. F. Sellin, N. De Blasio and D. J. Cole-Hamilton, Chem. Commun., 1999, 2511 RSC.
- J. A. Moulijn, P. W. N. M. van Leeuwen and R. A. van Santen, Catalysis. An Integrated Approach to Homogeneous Heterogeneous and Industrial Catalysis, in Studies in Surface Science and Catalysis, ed. B. Delmon and T. Yates, Elsevier, Amsterdam, 1993, vol. 79 Search PubMed.
- I. Tóth and P. van Geem, Encyclopedia of Catalysis, John Wiley & Sons, Hoboken, 2002 Search PubMed.
- R. A. Houghten, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 5131 CrossRef CAS.
- (a) P. J. Comina, A. K. Beck and D. Seebach, Org. Process Res. Dev., 1998, 2, 18 CrossRef CAS; (b) H. Sellner and D. Seebach, Angew. Chem., Int. Ed., 1999, 38, 1918 CrossRef CAS.
- R. H. Grubbs and L. R. Kroll, J. Am. Chem. Soc., 1971, 93, 3062 CrossRef CAS.
- J. P. Collman, L. S. Hegedus, M. P. Cooke, J. R. Norton, G. Dolcetti and D. N. Marquardt, J. Am. Chem. Soc., 1972, 94, 1789 CrossRef CAS.
- S. B. T. Nguygen and R. H. Grubbs, J. Organomet. Chem., 1995, 497, 195 CrossRef.
- (a) E. Le Roux, M. Taoufik, C. Copéret, A. de Mallmann, J. Thivolle-Cazat, J. M. Basset, B. M. Maunders and G. J. Sunley, Angew. Chem., Int. Ed., 2005, 44, 6755 CrossRef CAS; (b) R. Blanc, C. Copéret, J. Thivolle-Cazat, J. M. Basset, A. Lesage, L. Emsley, A. Sinha and R. R. Schrock, Angew. Chem., Int. Ed., 2006, 45, 1216 CrossRef; (c) M. Chabanas, A. Baudouin, C. Copéret and J. M. Basset, J. Am. Chem. Soc., 2001, 123, 2062 CrossRef CAS.
- P. McMorn and G. J. Hutchings, Chem. Soc. Rev., 2004, 33, 108 RSC.
- J. M. Fraile, J. I. García and J. A. Mayoral, Chem. Rev., 2009, 109, 360 CrossRef CAS.
- G. A. E. Oxford, D. Dubbeldamb, L. J. Broadbelt and R. Q. Snurr, J. Mol. Catal. A: Chem., 2011, 334, 89 CrossRef CAS.
- C. S. Gill, K. Venkatasubbaiah and C. W. Jones, Adv. Synth. Catal., 2009, 351, 1344 CrossRef CAS.
- X. Tu, X. Fu, X. Hu and Y. Li, Inorg. Chem. Commun., 2010, 13, 404 CrossRef CAS.
- J. M. Fraile, J. I. García, J. Massam and J. A. Maoral, J. Mol. Catal. A: Chem., 1998, 136, 47 CrossRef CAS.
- J. M. Fraile, J. I. García, J. A. Mayoral, T. Tarnai and M. A. Harmer, J. Catal., 1999, 186, 214 CrossRef CAS.
- R. J. Clarke and I. J. Shannon, Chem. Commun., 2000, 1936 Search PubMed.
- M. I. Burguete, J. -M. Fraile, J. I. García, E. García-Verdugo, C. I. Herrerias, S. V. Luis and J. A. Mayoral, J. Org. Chem., 2001, 66, 8893 CrossRef CAS.
- D. Rechavi and M. Lemaire, J. Mol. Catal. A: Chem., 2002, 182–183, 239 CrossRef CAS.
- D. Rechavi and M. Lemaire, Org. Lett., 2001, 3, 2493 CrossRef CAS.
- D. Hoegaerts, B. F. Sels, D. E. De Vos, F. Verpoort and P. A. Jacobs, Catal. Today, 2000, 60, 209 CrossRef CAS.
- D. P. Curran, Synlett, 2001, 1488 CrossRef CAS.
- E. G. Hope, A. M. Stuart and A. J. West, Green Chem., 2004, 6, 345 RSC.
- M. T. Reetz, Adv. Mater., 1997, 9, 943 CrossRef CAS.
- A. Corma and H. García, Chem. Rev., 2002, 102, 3837 CrossRef CAS.
- M. Dams, L. Drijkoningen, B. Pauwels, G. Van Tendeloo, D. E. De Vos and P. A. Jacobs, J. Catal., 2002, 209, 225 CrossRef CAS.
- S. V. Ley, C. Ramarao, R. S. Gordon, A. B. Holmes, A. J. Morrison, I. F. McConvey, I. M. Shirley, S. C. Smith and M. D. Smith, Chem. Commun., 2002, 1134 RSC.
- C. Ramarao, S. V. Ley, S. C. Smith, I. M. Shirley and N. DeAlmeida, Chem. Commun., 2002, 1132 RSC.
- J.-Q. Yu, H.-C. Wu, C. Ramarao, J. B. Spencer and S. V. Ley, Chem. Commun., 2003, 678 RSC.
- Q.-H. Fan, Y.-M. Li and A. S. C. Chan, Chem. Rev., 2002, 102, 3385 CrossRef CAS.
- G. H. Fink, B. Steinmetz, J. Zechlin, C. Przybyla and B. Tesche, Chem. Rev., 2000, 100, 1377 CrossRef CAS.
- (a) W. Kaminsky, Macromol. Chem. Phys., 1996, 197, 3907 CrossRef CAS; (b) W. Kaminsky and F. Renner, Makromol. Chem. Rapid Commun., 1993, 14, 239 CrossRef CAS.
- (a) US Pat., 4 912 075; (b) US Pat., 5 529 965.
- B. Pfeifer, W. Milius and H. G. Alt, J. Organomet. Chem., 1998, 553, 205 CrossRef.
- R. H. Grubbs, C. Gibbons, L. R. C. Kroll, W. D. Bonds Jr., and C. H. Brubaker Jr.,, J. Am. Chem. Soc., 1973, 95, 2373 CrossRef CAS.
- S. B. Roscoe, J. M. J. Frechet, J. F. Walzer and A. J. Dias, Science, 1998, 280, 270 CrossRef CAS.
- R. Akiyama and S. Kobayashi, J. Am. Chem. Soc., 2003, 125, 3412 CrossRef CAS.
- (a) Y. Shvo, E. Salanu and N. Menashe, J. Organomet. Chem., 1996, 514, 97–102 CrossRef; (b) J. H. Choi, N. Kim, Y. J. Shin, J. H. Park and J. Park, Tetrahedron Lett., 2004, 45, 4607 CrossRef CAS.
- (a) F. A. Quiocho and F. M. Richards, Biochemistry, 1966, 5, 4062 CrossRef CAS; (b) N. L. St. Clair and M. A. Navia, J. Am. Chem. Soc., 1992, 114, 7314 CrossRef CAS; (c) R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289 CrossRef CAS.
Footnotes |
| † This paper is based on the plenary lecture of ITH at EuropaCat X, Glasgow, Scotland, 28 August–2 Sept 2011. |
| ‡ While the letter S in the abbreviation of SLP catalyst can stand either for supported or stationary liquid phase, the latter is perhaps a more accurate and precise description for a catalyst of constant performance. |
| This journal is © The Royal Society of Chemistry 2011 |