Recent advances in alcohol dehydrogenase-catalyzed asymmetric production of hydrophobic alcohols
Musa M.
Musa
a and
Robert S.
Phillips
*b
aDepartment of Chemistry, King Fahd University of Petroleum and Minerals, Dhahran 31261, KSA
bDepartment of Chemistry and Department of Biochemistry and Molecular Biology, University of Georgia, 1001 Cedar Street, Athens, GA 30602, USA. Fax: +1-706-542-9454; Tel: +1-706-542-1996
First published on 19th July 2011
Abstract
The efficiency of biocatalytic redox reactions catalyzed by alcohol dehydrogenases (ADHs) have been the subject of considerable research recently. Two major challenges have restricted their application in asymmetric synthesis until now. First of all, most of the interesting substrates are either insoluble or sparingly soluble in aqueous media, the natural medium for enzymes. This drawback has been overcome by using non-aqueous media like organic solvents, ionic liquids, and supercritical carbon dioxide in mono- and biphasic reactions, and several ADHs show high activity at high concentrations of such reaction media. The second challenge is the strict substrate specificity for most ADHs. The continuous search for new ADHs, together with random and rational mutagenesis to widen substrate specificity, will help in attracting organic chemists to consider utilizing them in organic synthesis more often. The aim of this perspective is to highlight recent efforts to overcome the above-mentioned limitations.
 Musa M. Musa | Musa M. Musa (born in 1976 in KSA) received his BSc in Chemistry in 1997 from Yarmouk University, Jordan. In 2000, he received his MSc in Applied Chemistry from Jordan University of Science and Technology (JUST), Jordan. In 2003, he moved to USA and started his PhD at the University of Georgia under the supervision of Prof. Robert S. Phillips working in performing biotransformations in non-aqueous media. He then carried out research with Prof. Mark D. Distefano as a postdoctoral associate at University of Minnesota, where he focused in synthesis and evaluation of protein farnesyltransferase inhibitors and protein labeling. In 2009, he joined King Fahd University of Petroleum and Minerals (KFUPM) as Assistant Professor. His research interests include employing enzymes in organic synthesis. |
 Robert S. Phillips | Robert S. Phillips (born in 1952 in USA) received his BS in Chemistry in 1974 and PhD in 1979 from Georgia Institute of Technology, Atlanta Georgia, USA. From 1980–1985, he was a staff fellow at the National Institutes of Health in Bethesda, Maryland, USA. In 1985, he moved to the University of Georgia as an Assistant Professor of Chemistry and Biochemistry, and was promoted to Associate Professor in 1990 and Professor in 1996. His research interests are in structure and mechanisms of tryptophan-metabolizing enzymes and biocatalysis with alcohol dehydrogenases. |
1. Introduction
The development of new methods for asymmetric synthesis of alcohols is of great interest to satisfy the increasing demand for enantiomerically pure compounds. This can be done either using chiral chemical catalysts or biocatalysts. Interest in biocatalysts has been increasing recently for several reasons.1 First of all, biocatalysts have inherently high chemo-, regio- and enantioselectivities. Second, biocatalytic reactions may be safer because they do not require toxic reagents or solvents. Third, they are environmentally benign (“Green”) because they are natural catalysts. Fourth, the reaction conditions are mild, which minimize side products by preventing isomerization, racemization, epimerization and rearrangement reactions. Even with all these advantages, organic chemists are still not confident to consider a biocatalytic method for a synthetic problem for several reasons. Organic chemists are not trained in handling biological systems, and the sensitivity of enzymes as well as their high cost can be daunting. However, recent advances in biocatalysis have increased the stability of a large number of enzymes and therefore increased their availability and simplified their handling. Thus, a wide number of enzymes, including alcohol dehydrogenases, are now just as commercially available as any other catalyst. One of the major remaining disadvantages of using biocatalysts is the difficulty encountered in large scale synthesis because the natural environment of enzymes is water. However, it has been shown recently that some biotransformations can be done in non-aqueous media. Although the activity of most enzymes is usually lowered in such environments, the many advantages of using non-aqueous media and the overall increase in efficiency for many processes lessen the effect of this disadvantage.A recent comprehensive review covered the latest progress in asymmetric biocatalytic redox reactions.2 Several other reviews also were devoted to this topic.3 In the present perspective, we focus on recent developments in biocatalytic production of asymmetric hydrophobic alcohols. We will focus on the use of non-aqueous media not only to enhance the solubility of hydrophobic substrates, but also to alter the activity and selectivity of such reactions. A few examples of site-directed mutagenesis to widen substrate specificity, and in some cases tune the selectivity, of ADHs will be discussed as a potential solution to enhance their substrate specificity. The high thermal stability of some ADHs, together with their high tolerance to non-aqueous media, allow for ADH-catalyzed redox reactions to be conducted in an efficient manner, and sometimes in situ with organic reactions in media that are harsh for enzymes.
2. Alcohol dehydrogenases
Alcohol dehydrogenases (EC 1.1.1.X, X = 1 or 2, ADHs) are enzymes that catalyze the reversible reduction of ketones and aldehydes to their corresponding alcohols. They require a coenzyme such as nicotinamide-adenine dinucleotide (NAD+) or its phosphate (NADP+).4 Because these coenzymes are costly due to their high hydride equivalent weight, a successful regeneration of the coenzyme is crucial to make ADH-catalyzed transformations practical.5 Several methods have been employed to regenerate the cofactor in ADH-catalyzed reactions. However, the most applicable ones in synthesis are (1) using a coupled enzyme approach, which requires two different enzymes, or (2) a simple coupled substrate approach.6 In the second method, a cheap and readily available cosubstrate, such as 2-propanol or ethanol in the reduction pathway, or acetone or acetaldehyde in the oxidation pathway, is required. Cofactor recycling can also be done using electrochemical, photoelectrochemical, or chemical methods.In the asymmetric reduction of a prochiral ketone, there are four possible pathways to deliver the hydride from NAD(P)H, as shown in Fig. 1. The pro-(R) or pro-(S)-hydride can attack from the re face of a prochiral ketone, to produce the (S)-alcohol, according to Prelog's Rule, or it can also attack from the si face of a ketone to produce the (R)-alcohol. (It should be noted that sometimes the products have opposite assignments because the small substituent, if alkenyl or alkynyl, has a higher Cahn–Ingold–Prelog priority than the larger alkyl one). The majority of commercially available ADHs, like yeast ADH (YADH), horse liver ADH (HLADH), and Thermoanaerobium brockiiADH (TbADH), fall in the first category (i.e. they deliver the pro-(R)-hydride from the re face of a prochiral ketone).7 A few ADHs are known to have anti-Prelog stereopreference; however, very few of them are commercially available, one of which is Lactobacillus kefirADH (LkADH).
 | ||
| Fig. 1 Stereochemistry of the hydride transfer from NAD(P)H to the carbonyl carbon on a ketone substrate. ADPR = adenosine diphosphoribose, R1 is more sterically hindered and has higher Cahn–Ingold–Prelog priority than R2. | ||
In an effort to overcome the limited substrate specificity for most ADHs, which has restricted their use in asymmetric synthesis, new ADHs with broad substrate specificities as well as new mutants of known ADHs have been reported. Increasing substrate specificity for ADHs to include sterically hindered substrates, most of the time hydrophobic in nature, is accompanied by switching the reaction media of ADH-catalyzed transformations from predominantly aqueous to non-aqueous ones.
3. Synthetic applications
A practical biocatalytic asymmetric reduction using “designer cells” without adding an external cofactor was reported.8Cells containing either Rhodococcus erythropolisADH, an (S)-selective ADH, or LkADH, an (R)-selective ADH, as well as Bacillus subtilisglucose dehydrogenase, which is used for in situregeneration of the cofactor NAD(P)H, were employed in the asymmetric reduction of hydrophobic substrates in high concentrations (>100 g L−1). At such highly elevated substrate concentrations, emulsions were formed. This method was applied to produce optically active alcohols with high conversions and enantioselectivities in pure aqueous media using D-glucose as the reducing agent (Scheme 1). | ||
| Scheme 1 Enantioselective reduction of ketones using “designer cells”. | ||
The reduction of ketones with two bulky substituents catalyzed by Ralstonia sp. DSM 6428 was accomplished at substrate concentrations of 10 g L−1.9,10 This enzyme was not only active on these bulky ketones, but also on ketones containing bulky and small substituents with high enantioselectivity (Scheme 2). The enzyme-coupled approach to recycle NADPH was found to be more efficient than the cosubstrate approach using 2-propanol. This ADH should be of great interest in asymmetric synthesis because of its broad substrate specificity, and there are very few reports on biocatalytic reduction of such aryl alkyl ketones with two bulky substituents.
 | ||
| Scheme 2 E. coli/Ralstonia sp.ADH-catalyzed reduction of ketones. | ||
A series of diaryl ketones was reduced to their corresponding optically active diarylmethanols with high yields and good to high enantioselectivities using commercially available ADHs (Scheme 3).11 It was noticed that there is no need for a substituted benzene ring or highly electronically dissymmetric substrates to achieve high enantioselectivity, an advantage that is not easily achieved by conventional chemical methods. All ADHs used in this study are NADPH-dependent and an enzyme-coupled approach was used to regenerate NADPH. The availability of ADHs with such high steric and electronic discrimination is valuable in making pharmaceutically important compounds like the alcohols produced from 2-(4-chlorobenzoyl)pyridine, a precursor of histamine H1antagonist (S)-carbinoxamine.

3.1. Application of site-directed mutagenesis (SDM) to tune substrate specificity and selectivity of ADHs
The activity and stereoselectivity of wild-type enzymes on unnatural substrates are not always sufficiently high enough to be practical. One approach to alter the catalytic properties of enzymes is to use structure-based site-directed mutagenesis. This can be done most efficiently if the detailed structure of the active site of an enzyme is known. However, there have been relatively few reports that employ this approach to broaden the substrate specificity of biocatalytic enzymes.Thermoanaerobacter ethanolicus secondary ADH (TeSADH, EC 1.1.1.2), is an NADP+-dependent, thermostable (up to 85 °C) oxidoreductase, which has been isolated and characterized.12 It also tolerates the presence of organic solvents and exhibits high activity towards a wide range of cyclic and acyclic secondary alcohols and ketoesters.13 This enzyme is very similar to the commercially available Thermoanerobium brockiiADH (TbADH).14 Keinan et al. proposed a model for the active site of TbADH suggesting both large and small hydrophobic binding pockets with different affinities toward the alkyl groups of ketone substrates, the small site having higher binding affinity.15 An interesting size-dependent reversal of stereoselectivity was observed. (R)-Alcohols were produced from the asymmetric reductions of ketones with two small alkyl groups attached to the carbonyl (e.g.methyl ethyl, methyl isopropyl, or methyl cyclopropyl). However, (S)-alcohols were produced with methyl ketones containing alkyl substituents larger than propyl (i.e., for these substrates the enzyme follows Prelog's rule). It was noticed that the enantioselectivity increased significantly with larger ketones like 2-hexanone and 2-heptanone, presumably because they can only fit in one mode within the active site. We observed similar results in the asymmetric reduction of ethynyl ketones and ethynyl ketoesters using wild-type TeSADH.16
The crystal structure of TbADH has been determined to be a tetramer of identical 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 652 Da subunits.17 Each subunit is composed of 352 amino acids, and contains a catalytic Zn2+ ion. Its binary complex of the apoenzyme with 2-butanol as a substrate has also been determined.18 The crystal structure shows a crevice between the surface and the active site, which allows the substrates and products to move in and out. This crevice is formed by the side chains of the hydrophobic residues, Ile49, Leu107, Trp110, Tyr267 and Cys283, as well as Met285 from another subunit. The two residues His59 and Asp150 have been shown to be essential for catalysis, because they are ligands to the catalytic Zn2+. The structure of the active site of TbADH, which is the same as TeSADH,19 is shown in Fig. 2.
652 Da subunits.17 Each subunit is composed of 352 amino acids, and contains a catalytic Zn2+ ion. Its binary complex of the apoenzyme with 2-butanol as a substrate has also been determined.18 The crystal structure shows a crevice between the surface and the active site, which allows the substrates and products to move in and out. This crevice is formed by the side chains of the hydrophobic residues, Ile49, Leu107, Trp110, Tyr267 and Cys283, as well as Met285 from another subunit. The two residues His59 and Asp150 have been shown to be essential for catalysis, because they are ligands to the catalytic Zn2+. The structure of the active site of TbADH, which is the same as TeSADH,19 is shown in Fig. 2.
 | ||
| Fig. 2 Active site of TbSADH with NADP+ and bound Zn. From Prot. Eng. Des. Sel., 2007, 20, 47–55. | ||
In an effort to broaden substrate specificity of TeSADH, W110A TeSADH was designed as a mutation that enlarges the large pocket in the active site of TeSADH, and the mutant enzyme was generated by SDM.20 This mutant protein is able to accommodate substrates with aromatic substituents, which are not substrates for wild-type TeSADH. W110A TeSADH was then employed in the asymmetric production of phenyl-ring containing alcohols with high activity, comparable to that of TeSADH on its substrates like 2-butanol. SDM was also employed to generate I86A TeSADH, which was designed to enlarge the small pocket of the active site of TeSADH and to reverse the stereoselectivity of TeSADH.21 As predicted, this single point mutation was able not only to switch the stereopreference of TeSADH, but also to accommodate the aromatic ring of sterically hindered substrates like phenyl and heteroaryl ketones within the small pocket of TeSADH. I86A TeSADH was employed in the asymmetric reduction of these hydrophobic ketones to non-racemic alcohols in anti-Prelog fashion as will be discussed later. It was also shown that the inverse stereoselectivity of I86A TeSADH is a result of delivering the pro-(R)-hydride of NADPH to the si-face. The search for ADHs with anti-Prelog stereopreference is very important because there are very few known ADHs with this preference. The use of W110A and I86A TeSADHs in the asymmetric production of alcohols will be described in later sections of the current perspective.
Zhu and Hua reported an NADPH-dependent carbonyl reductase from Sporobolomyces salmonicolor (SSCR) that is able to reduce ketones with two bulky substituents.22 Thus, a series of aryl alkyl ketones were reduced using SSCR to produce their non-racemic alcohols in low to good enantioselectivities. SSCR has relatively broad substrate specificity, as it accepts ketones with diverse structures as substrates. As was seen above with TbADH, a clear reversal in the enantiopreference was noticed when increasing the chain length of the alkyl group in phenyl alkyl ketones from ethyl to propyl (Scheme 4). A dramatic enhancement in the enantioselectivity of reduction was noticed when the alkyl group is branched.
 | ||
| Scheme 4 Enantioselective reduction of aryl alkyl ketones catalyzed by Sporobolomyces salmonicolorcarbonyl reductase (SSCR). | ||
Substrate-enzyme docking studies for SSCR showed that M242 and Q245 in the catalytic site are mutation sites with potential to enhance the enantioselectivity of SSCR-catalyzed reduction of para-substituted acetophenones. Three Q245 mutants of SSCR were introduced and shown to invert the stereopreference of the produced alcohols in the asymmetric reduction of para-substituted acetophenones from (R)- to (S)-configured alcohols.23 Several double mutants of SSCR were then created by structure-based site-saturation mutagenesis.24 Of these, M242L/Q245P SSCR showed the highest enantioselectivity in the asymmetric reduction of para-substituted acetophenones to their corresponding (S)-alcohols (>99% ee, Scheme 5). In contrast, these substrates were reduced with wild-type SSCR to the corresponding (R)-alcohols with low to modest enantioselectivities.
 | ||
| Scheme 5 Enantioselective reduction of para-substituted acetophenones with WT SSCR and M242L/Q245P SSCR. | ||
3.2. The use of organic solvents in ADH-catalyzed transformations
In 1986, Klibanov and co-workers demonstrated for the first time that an ADH-catalyzed asymmetric redox reaction can proceed in an organic solvent.25 They deposited HLADH and its cofactor NAD+ onto the surface of glass beads, which were then used for asymmetric oxidoreductions in isopropyl ether presaturated with Tris-HCl buffer solution, using ethanol in the reduction pathway, and isobutyraldehyde in the oxidation pathway, to regenerate NADH and NAD+, respectively. The drawback of this method was the low percent conversion obtained (only about 20% after 6 days).Subsequent studies have shown that there is no doubt that ADH-catalyzed processes can perform well in organic solvents.26 These reactions have been employed in high concentrations of organic solvents (up to >99%). The use of organic solvents varies from the use of substrates like 2-propanol and acetone as co-solvents to the use of other water-miscible solvents in high concentrations, or to the use of water-immiscible organic solvents. The use of organic solvents has not only enhanced the efficiency of ADH-catalyzed reactions by allowing these biotransformations to be conducted at high substrate concentrations, but also altered the activity and enantioselectivity of ADHs.
541, which is exceptionally stable toward organic solvents.27 They were able to achieve asymmetric reductions of prochiral ketones by using 2-propanol as a cosubstrate and cosolvent at concentrations up to 50% (v/v) 2-propanol and 20% (v/v) acetone, producing (S)-alcohols (Scheme 6).28 They were also able to produce the (R)-alcohols through enantiospecific kinetic resolution by stereospecific oxidation of the corresponding racemic alcohols (Scheme 6). In a later report, it has been shown that ADH-A can tolerate up to 80% of 2-propanol and 50% v/v of acetone, a property that no other ADH has shown.29 Very recently, the structure of ADH-A from Rhodococcus ruber has been solved as a homotetramer, and it revealed that the two dimer interfaces are connected by ten salt bridges, which could explain the high tolerance to organic solvents based on electrostatics.30 Knowing the details of the structure, especially the active site of such a highly chemotolerant ADH, will help in increasing the substrate specificity of ADH-A to include interesting substrates like ketones with bulky substituents.
 | ||
| Scheme 6 Asymmetric production of alcohols catalyzed by lyophilized cells of Rhodococcus ruberDSM 44541. | ||
The high tolerance of TeSADH to organic solvents allowed its use in asymmetric production of alcohols using 2-propanol or acetone as both cosubstrates and cosolvents at the same time to enhance the solubility of hydrophobic substrates. This was very valuable in the asymmetric redox reactions catalyzed by W110A TeSADH. Phenyl-ring-containing ketones were reduced to the corresponding Prelog products, (S)-alcohols, with good to high yields and high enantioselectivities.31 The W110A TeSADH-catalyzed asymmetric reductions were conducted in Tris-HCl buffer solutions containing 2-propanol (30%, v/v) as cosubstrate and cosolvent at substrate concentrations of 35 mM (Scheme 7). The anti-Prelog products, (R)-alcohols, were produced by enantiospecific oxidation of (S)-alcohols in the racemate to the corresponding ketones, leaving the desired (R)-alcohol as the slowly reacting enantiomer, i.e., by kinetic resolution. Starting from a racemic mixture of the phenyl-ring-containing alcohol and using acetone (10%, v/v) as a cosubstrate as well as cosolvent at substrate concentration of 70 mM, it was possible to employ W110A TeSADH-catalyzed oxidation to produce (R)-alcohols with maximum percent conversion of 50% with high ee (Scheme 7).

The high tolerance of TeSADH to organic solvents allowed TeSADH-catalyzed redox reactions for hydrophobic substrates to be carried out at concentrations that are acceptable to synthetic organic chemists by using either water-miscible or -immiscible organic solvents. Stereoselective reductions and stereospecific oxidations catalyzed by W110A TeSADH were conducted in reaction media containing high concentrations of hydrophilic organic solvents (low log P).32 Both yield and enantioselectivity were shown to be dependent on the reaction medium. For example, a clear enhancement in the enantioselectivity of W110A TeSADH-catalyzed asymmetric reduction of phenylacetone was noticed when water-miscible organic solvents, DMF or acetonitrile, Tris-HCl buffer solution, and 2-propanol (40:40:20, v/v/v) were used as a ternary mono-phasic solvent system, in comparison with using the Tris-HCl buffer solution and 2-propanol (70:30, v/v) binary solvent system (Scheme 8). On the other hand, stereospecific kinetic resolutions of racemic alcohols were conducted in different reaction media to show that toluene is the best solvent in terms of stereoselectivity, but the poorest in terms of activity (Scheme 8). This study for TeSADH, together with other studies,33 demonstrated that it is possible to tune both the yield and enantioselectivity for a given ADH-catalyzed redox reaction; however, there is no clear correlation between the physicochemical properties of the non-aqueous solvent and the yield or enantioselectivity of the reaction.
 | ||
| Scheme 8 W110A TeSADH-catalyzed asymmetric production of phenyl-ring-containing alcohols in media containing water-miscible and -immiscible organic solvents. | ||
A variety of sterically bulky diaryl ketones was reduced to the corresponding chiral alcohols using SSCR and its mutant Q245P (Scheme 9).34 These reactions were conducted in phosphate buffer solution containing an organic cosolvent (10% v/v). Both yield and enantioselectivity were dependent on the organic cosolvent used. A switch in the enantiopreference for the asymmetric reduction of 4-chlorobenzophenone and 4-methylbenzophenone was noticed when Q245P SSCR was used.

Gröger et al. reported a practical method for asymmetric reductions of poorly water-soluble ketones using Rhodococcus erythropolisADH, an NAD+-dependent ADH, in water/n-heptane (4:1, v/v) biphasic systems with satisfactory conversions,35,36 using formate dehydrogenase (FDH) from Candida boidinii to regenerate NADH. This method was used to reduce hydrophobic ketones at concentrations up to 200 mM to produce their corresponding (S)-alcohols with moderate to good conversions and high ee (Scheme 10).
 | ||
| Scheme 10 Rhodococcus erythropolis ADH-catalyzed asymmetric reductions in biphasic media. | ||
Anti-Prelog benzylic and heteroaryl alcohols were produced by I86A TeSADH in good to excellent yields and very high enantioselectivities (Scheme 11).21 These reactions were conducted in biphasic systems containing hexane as an organic hydrophobic solvent, using 2-propanol as a cosubstrate. Addition of ZnCl2 (10 μM) to the reaction mixtures showed an increase in the activity of I86A TeSADH, as the mutation affects Zn binding.
 | ||
| Scheme 11 I86A TeSADH-catalyzed asymmetric production of benzylic and heteroaryl alcohols in biphasic system. | ||
As a result of its exceptional tolerance to organic solvents, ADH-A from Rhodococcus ruber (both as E. coli/ADH-A cells and as purified recombinant enzyme) was successfully applied in organic solvents at 50% (v/v) and even in micro-aqueous organic systems (99%, v/v) (Scheme 12).37 The robustness of this ADH not only allowed bioreductive transformations to be conducted at concentrations as high as 2.0 M, but also allowed the recovery of E. coli/ADH-A by filtration, which showed activity without a change in selectivity until the fifth cycle. In contrast to other studies, a correlation was noticed in this study between log P and the biocompatibility of the organic solvent, with the higher the hydrophobicity of the solvent the better.

An exceptionally organic solvent-tolerant ADH from Paracoccus pantotrophus DSM 11072 (PpADH) was recently reported.38 It was employed in the asymmetric reduction of hydrophobic ketones in non-conventional organic solvents, including mono- and biphasic organic/aqueous solvent systems, as well as micro-aqueous media (Scheme 13). Unexpectedly, DMSO, a highly hydrophilic solvent (i.e. low log P value), was found to be the best cosolvent for PpADH-catalyzed bioreduction reactions. This high tolerance of PpADH to DMSO was explained by the ability of the Paracoccus pantotrophus DSM 11072 to grow in high sulfur content media.
 | ||
| Scheme 13 Asymmetric reduction of ketones employing lyophilized cells of E. coli/PpADH. | ||
Asymmetric reduction of rac-3-methylcyclohexanone to (1S,3S)-3-methylcyclohexanol by thermophilic ADH from Thermus sp. (TADH) was reported employing an electrochemical regeneration method for NADHvia[Cp*Rh(bpy)(H2O)]+2 as a selective mediator (Scheme 14).39Octane was used as a second organic phase (octane: aqueous, 1:1.5) to provide continuous extraction of the produced alcohol, and therefore avoid enzyme inhibition caused by the product. Using this method, 1.32 g L−1 of the product was obtained in 10 h.
 | ||
| Scheme 14 Electroenzymatic TADH-catalyzed asymmetric reduction of rac-3-methylcyclohexanone. (bpy: bipyridyl). | ||
3.3. ADH-catalyzed asymmetric redox reactions in ionic liquids (ILs)
Ionic liquids (ILs) are liquid salts with low melting points. Those that are liquid at room temperature are called room-temperature ILs. The unique characteristics of ILs, including their reusability, low vapor pressure, and non-flammability, make them good alternatives to environmentally non-benign organic solvents. Thus, performing enzyme-catalyzed reactions, which are also environmentally friendly, in ILs is of great interest from an environmental standpoint.40 A recent perspective presented an overview of activation of enzymes in ILs.41The most popular ILs used in biocatalysis are imidazolium-based, like 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIM][BF4]), 1-butyl-3-methylimidazolium bis[(trifluoromethyl)sulfonyl]imide ([BMIM][NTf2]), and 1-butyl-3-methylimidazolium hexafluorophosphate ([BMIM][PF6]). Among these, [BMIM][BF4] is water-miscible, and [BMIM][NTf2] as well as [BMIM][PF6] are water-immiscible. In 2004, Eckstein et al. conducted the first example of an ADH-catalyzed asymmetric reduction in a biphasic system containing [BMIM][NTf2].42 They reported that, by taking advantage of the partition coefficients of 2-propanol and acetone, 2-propanol preferably remained in the aqueous phase, and improved ADH-catalyzed reduction yields were obtained. Since then, a number of ADH-catalyzed transformations have been conducted in both water-miscible and -immiscible ILs.43
Mono- and biphasic systems containing ILs were employed in the asymmetric reduction of hydrophobic ketones catalyzed by ADH-A from Rhodococcus ruber.44Hydroxy functionalized water-miscible tris-(2-hydroxyethyl)-methylammonium methylsulfate ([MTEOA]MeSO4) was employed in concentrations up to 90% (v/v). Employing such high concentrations of ILs allowed ADH-catalyzed reduction to be conducted at substrate concentrations of 1.5 M.
A series of water-immiscible ILs were tested as potential solvents in asymmetric whole cell bioreduction of ketones.45,46 This was done by using a recombinant E. coli containing overexpressed Lactobacillus brevisADH and Candida boidiniiformate dehydrogenase for cofactor regeneration. Using 1-hexyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide ([HMPL][NTF]), (R)-2-octanol was produced from the corresponding prochiral ketone in both high yield and enantiomeric excess in space-time-yield up to 180 g L−1 day−1.
The influence of [BMIM][PF6] on the enantioselectivity of Saccharomyces cervisiaeADH-catalyzed reduction of ethyl 2-oxo-4-phenylbutyrate was investigated (Scheme 15).47 It was noticed that (R)-ethyl 2-hydroxy-4-phenylbutyrate was obtained in 70.4% ee when ethyl ether or benzene were used as solvents. However, when [BMIM][PF6] was used, a shift to the (S)-configured product was noticed (27.7% ee (S)).
![Saccharomyces cervisiae
ADH-catalyzed asymmetric reduction of 2-oxo-4-phenylbutyrate in organic solvents and [BMIM][PF]6.](/image/article/2011/CY/c1cy00160d/c1cy00160d-s15.gif) | ||
| Scheme 15 Saccharomyces cervisiae ADH-catalyzed asymmetric reduction of 2-oxo-4-phenylbutyrate in organic solvents and [BMIM][PF]6. | ||
3.4. ADH-catalyzed asymmetric redox reactions in supercritical carbon dioxide (scCO2)
Enzymatic asymmetric reduction of ketones by ADH from Geotrichum candidum in an scCO2/aqueous biphasic system in the presence of sodium bicarbonate was reported.48 It was noted that the addition of sodium bicarbonate enhanced the yield from 4% to 25% in the asymmetric reduction of acetophenone. Several ketones were reduced to the corresponding (S)-alcohols in high yields and excellent enantioselectivities using this environmentally benign method (Scheme 16). The same biphasic system was utilized in the asymmetric reduction of o-fluoroacetophenone using alginate-immobilized form of the enzyme to produce the corresponding optically active alcohol in 75% yield and >99% ee. | ||
| Scheme 16 Geotrichum candidum ADH-catalyzed asymmetric reduction of ketones in scCO2/aqueous buffer biphasic system. | ||
4. ADH-mediated dynamic reductive kinetic resolution (DRKR)
Giacomini et al. reported a highly efficient reduction of arylpropionic aldehydes by using HLADH through DRKR.49 They were able to produce (2S)-2-phenyl-1-propanol and (2S)-2-(4-iso-butylphenyl)propanol [(S)-ibuprofenol] in good yields and enantiomeric ratios from 2-phenylpropanal and Ibuprofenal, respectively, using ethanol as a cosubstrate to regenerate NADH (Scheme 17). This DRKR process involves HLADH-catalyzed stereoselective reduction of (S)-aldehyde combined with in situketo-enol racemization of the slow reacting (R)-aldehyde. Water-miscible organic co-solvents like THF and CH3CN were used to improve the solubility of these hydrophobic substrates. A slight improvement in the enantiomeric ratios of the products was noticed when THF (10%, v/v) was used in comparison with CH3CN (10%, v/v) in the DRKR of 2-phenylpropanal. Recently, the same method was used to produce six (2S)-2-arylpropanols.50 It was shown that keto-enol racemization step in DRKR can be controlled by modulating the pH of the reaction mixture. | ||
| Scheme 17 HLADH-mediated DRKR of rac-arylpropionic aldehydes in phosphate buffer/organic cosolvent. | ||
DRKR of 2-arylpropanal, Profen-type substrates, catalyzed by Sulfolobus solfataricusADH-10 (SsADH-10) was accomplished in phosphate buffer solutions containing only 5% EtOH and catalytic amounts of NADH at 80 °C.51 This method uses a higher reaction temperature and a smaller amount of organic cosolvent than other known ADH-catalyzed processes, the “thermal switching” approach. It was used to produce (S)-profenols, which are precursors for nonsteroidal anti-inflammatory drugs like naproxene, ibuprofen, flurbiprofen, fenoprofen, and ketoprofen, from their rac-aldehydes in good to high yields and high enantioselectivities (Scheme 18). The hyperthermophilicity of SsADH-10 allowed a thermal recycling approach that enabled it to be recycled five times with high enantioselectivities, taking advantage of the low solubility of products at room temperatures, which will allow them to precipitate, keeping SsADH-10 in the solution to be recycled.
 | ||
| Scheme 18 SsADH10-mediated DRKR of 2-arylpropanals. | ||
Facile enolization of α-chloro ketones allowed their ADH-mediated DRKR to produce their corresponding halohydrins with high enantiomeric and diastereomeric ratios. Stewart reported the biocatalytic reduction of an α-chloro-β-keto ester to yield the corresponding halohydrin in high yield, and enantio- as well as diastereoselectivity (Scheme 19).52 The resulting α-chloro-β-hydroxy ester was then converted to N-benzoylphenylisoserine, the Taxol side chain. (+)-(2S,3R)-3-Chloro-4-(4′-chlorophenyl)-2-butanol was produced from 3-chloro-4-(4′-chlorophenyl)-2-butanone when subjected to W110A TeSADH-catalyzed reduction in more than 50% conversion, with high enantio- and diastereoselectivity (Scheme 20). We explained this result as a DRKR that involves a KR coupled with a keto-enol racemization for the slow reacting enantiomer of the α-chloro ketone. The α-chloro alcohol product was then converted to (−)-(2S,3S)-4-(4′-chlorophenyl)-2,3-epoxybutane using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) without racemization.
 | ||
| Scheme 19 Asymmetric production an α-chloro-β-hydroxy esterviaADH-mediated DRKR. | ||
 | ||
| Scheme 20 W110A TeSADH-mediated DRKR. | ||
4.1. Immobilized ADH in asymmetric synthesis
Immobilization of an enzyme is its attachment to an inert and insoluble support. There are several ways in which an enzyme can be immobilized, such as absorption on glass or in alginate beads, covalent binding to polymer beads, or entrapment in silica gel. Immobilization of enzymes has several advantages including convenient handling, enhanced stability, ease of recovery and reuse.Geotrichum candidum cells containing ADH were immobilized on a wet water-absorbing polymer and employed in the asymmetric reduction of ketones in [emim][BF4] using 2-propanol as cosubstrate.53Acetophenone derivatives, benzylacetone, keto esters, 2-hexanone, and a fluorinated epoxy ketone were reduced to their (S)-configured alcohols with good yields and excellent enantioselectivities.
The low stability of LkADH in organic solvents was overcome by entrapment of the enzyme and its cofactor NADPH in polyvinyl alcohol gel beads.54 The entrapped enzyme was employed in transforming hydrophobic ketones to the corresponding (R)-alcohols in hexane using 2-propanol as a cosubstrate.
W110A TeSADH was encapsulated together with its cofactor using the sol–gel method then dried at room temperature to produce a xerogel.55 Xerogel-immobilized W110A TeSADH was then employed in the asymmetric reduction of hydrophobic ketones in water-immiscible organic solvents at 140 mM, with yields comparable to those obtained using the free enzyme in aqueous media containing 2-propanol as a cosolvent. Interestingly, a significant enhancement in the enantioselectivity of reduction of phenylacetone was noticed when the reaction medium was switched from Tris-HCl buffer solution using 2-propanol as a co-solvent to hydrophobic solvents like hexane, toluene, or diisopropyl ether (Scheme 21). We explained this improvement in enantioselectivity by the differences in solvation of the enzyme active site.
 | ||
| Scheme 21 Asymmetric reduction of phenylacetone by xerogel encapsulated W110A TeSADH in different reaction media. | ||
Lactobacillus brevis and Thermoanaerobium sp. ADHs were absorbed with NADP+ on a commercially superabsorbent polymer.56 Asymmetric reductions of acetophenone, 4-acetylpyridine, and ethyl acetoacetate with superabsorbed ADH were conducted using 2-propanol as a cosubstrate and cosolvent.
TbADH and its cofactor NADP+ were immobilized on mesoporous silica SBA-15, and its activity was compared with the free enzyme in different reaction media.57 This study found that the activity of the TbADH is higher in the immobilized form than the free form.
4.2. Compatibility of ADH-catalyzed redox reactions with organic reactions
The high thermal stability of some ADHs, in addition to their high tolerance to non-aqueous media, stimulated researchers to incorporate them together with organic transformations in the same pot under conditions that are generally considered harsh for enzymes. Gauchot et al. reported an ADH-catalyzed asymmetric reduction together with a Pd-catalyzed cross-coupling reaction, Suzuki coupling, in a biphasic system containing a buffer aqueous phase and an imidazolium-based, [BMIM][NTf2], IL phase.58 This cascade reaction was used to produce enantiopure biaryl alcohols in high yields and excellent enantioselectivities (Scheme 22). Both the IL phase containing the Pd catalyst and the aqueous buffer phase containing the biocatalyst can be recycled. This study showed that it is possible to incorporate biocatalytic reduction reactions in chemical cascade reactions using ILs.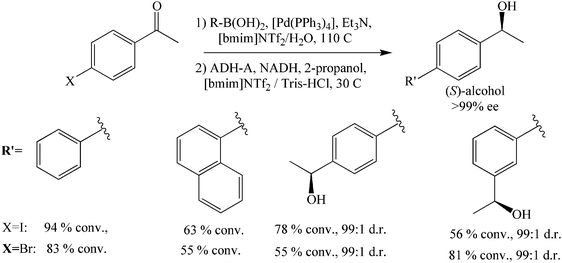 | ||
| Scheme 22 One-pot chemo-biocatalyzed synthesis of biaryl alcohols by Suzuki coupling followed by ADH-A-catalyzed reduction. | ||
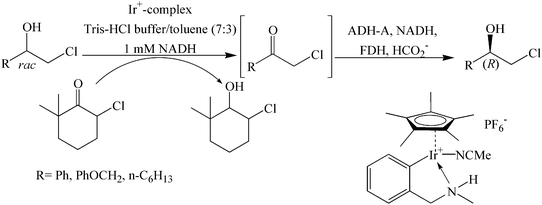
Optically active (R)-chlorohydrins were produced from their racemates by combining an iridium-catalyzed oxidation to produce their ketones, which were reduced enantioselectively employing ADH-A (Scheme 23).59 This cascade reaction was accomplish by using a 6-chloro-2,2-dimethyl-cyclohexanone as an orthogonal hydrogen acceptor that is accepted by the Ir catalyst but not by ADH-A, and using formate/FDH as the hydrogen donor. This is one of the unique examples where an enzyme-catalyzed process is combined with a metal-catalyzed process in one pot.
Summary and outlook
The availability of thermostable ADHs that have high activity in non-aqueous media has allowed their application in redox reactions in organic synthesis. Recent reports have also shown that it is possible to use ADHs in hydrophobic organic solvents that have very low portions of aqueous media (less than 1%). This achievement, together with reasonable substrate specificity that allowed sterically bulky substrates to be accepted by some ADHs, allowed redox biotransformations to be employed in high concentrations, and therefore make them efficient biocatalysts in producing optically active alcohols that can be used as building blocks in pharmaceutical, agrochemical, and food industries. More ADHs that are able to accept sterically bulky substrates and have broad substrate specificities are acquired to take ADHs to another level and make organic chemists consider ADHs as biocatalysts for stereoselective reductions. The tolerance of some ADHs to very high concentrations of non-aqueous solvents will make them a very powerful synthetic tool, and will ease their application with other reactions in one-pot transformations.References
- (a) Biocatalysis in the Pharmaceutical and Biotechnology Industry, ed. R. N. Patel, CRC Press, Boca Raton, FL, USA, 2007 Search PubMed; (b) M. Woodley, Trends Biotechnol., 2008, 26, 321–327 CrossRef CAS; (c) K. Faber, Biotransformations in organic chemistry, 5th edn, Springer, Heidelberg, 2004 Search PubMed.
- T. Matsuda, R. Yamanaka and K. Nakamura, Tetrahedron: Asymmetry, 2009, 20, 513–557 CrossRef CAS.
- E. García-Urdiales, I. Alfonso and V. Gotor, Chem. Rev., 2005, 105, 313–354 CrossRef.
- R. S. Phillips, Wiley Encyclopedia of Chemical Biology, 2009, 3, 200–209 Search PubMed.
- A. Weckbecker, H. Gröger and W. Hummel, Adv. Biochem. Eng./Biotechnol., 2010, 120, 195–242 Search PubMed.
- W. Kroutil, H. Mang, K. Edegger and K. Faber, Curr. Opin. Chem. Biol., 2004, 8, 120–126 CrossRef CAS.
- V. Prelog, Pure Appl. Chem., 1964, 9, 119–130 CrossRef CAS.
- H. Gröger, F. Chamouleau, N. Orologas, C. Rollmann, K. Draus, W. Hummel, A. Weckbecker and O. May, Angew. Chem., Int. Ed., 2006, 45, 5677–5688 CrossRef.
- I. Lavandera, A. Kern, B. Ferreira-Silva, A. Glieder, S. de Wildeman and W. Kroutil, J. Org. Chem., 2008, 73, 6003–6005 CrossRef CAS.
- I. Lavandera, G. Oberdorfer, J. Gross, S. de Wildeman and W. Kroutil, Eur. J. Org. Chem., 2008, 2539–2543 CrossRef CAS.
- M. D. Truppo, D. Pollard and P. Devine, Org. Lett., 2007, 9, 335–338 CrossRef CAS.
- (a) D. Burdette and J. G. Zeikus, Biochem. J., 1994, 302, 163–170 CAS; (b) D. S. Burdette, C. Vieille and J. G. Zeikus, Biochem. J., 1996, 316, 115–122 CAS; (c) F. O. Bryant, J. Weigel and L. G. Ljungdahl, Appl. Environ. Microbiol., 1988, 460–465 CAS.
- (a) C. Zheng, V. T. Pham and R. S. Phillips, Bioorg. Med. Chem. Lett., 1992, 2, 619–622 CrossRef CAS; (b) C. Zheng, V. T. Pham and R. S. Phillips, Catal. Today, 1994, 22, 607–620 CrossRef CAS.
- In unpublished results, M. Laivenieks, C. Vieille and J. G. Zeikus, showed that TeSADH is identical to TbADH.
- E. Keinan, E. K. Hafeli, K. K. Seth and R. Lamed, J. Am. Chem. Soc., 1986, 108, 162–169 CrossRef CAS.
- C. Heiss and R. S. Phillips, J. Chem. Soc., Perkin Trans. 1, 2000, 2821–2825 RSC.
- Y. Korkhin, A. J. Kalb, M. Peretz, O. Bogin, Y. Burstein and F. Frolow, J. Mol. Biol., 1998, 278, 967–981 CrossRef CAS.
- C. Li, J. Heatwole, S. Soelaiman and M. Shoham, Proteins, 1999, 37, 619–627 CrossRef CAS.
- C. Heiss, M. Laivenieks, J. G. Zeikus and R. S. Phillips, Bioorg. Med. Chem., 2001, 9, 1659–1666 CrossRef CAS.
- K. J. Ziegelmann, M. M. Musa, R. S. Phillips, J. G. Zeikus and C. Vieille, Protein Eng., Des. Sel., 2007, 20, 47–55 CrossRef.
- M. M. Musa, N. Lott, M. Laivenieks, L. Watanabe, C. Vieille and R. S. Phillips, ChemCatChem, 2009, 1, 89–93 Search PubMed.
- D. Zhu and L. J. Hua, J. Org. Chem., 2006, 71, 9484–9486 CrossRef CAS.
- D. Zhu, Y. Yang, S. Majkowicz, T. H.-Y. Pan, K. Kantardjieff and L. Hua, Org. Lett., 2008, 10, 525–528 CrossRef CAS.
- H. Li, Y. Yang, D. Zhu, L. Hua and K. Kantardjieff, J. Org. Chem., 2010, 75, 7559–7564 CrossRef CAS.
- J. Grunwlad, B. Wirz, M. P. Scollar and A. M. Klibanov, J. Am. Chem. Soc., 1986, 108, 6732–6734 CrossRef CAS.
- A. M. Klibanov, Curr. Opin. Biotechnol., 2003, 14, 427–431 CrossRef CAS.
- W. Stampher, B. Kosjek, C. Moitzi, W. Kroutil and K. Faber, Angew. Chem., Int. Ed., 2002, 41, 1014–1017 CrossRef CAS.
- W. Stampfer, B. Kosjek, K. Faber and W. Kroutil, J. Org. Chem., 2003, 68, 402–406 CrossRef CAS.
- B. Kosjek, W. Stampfer, M. Pogorevc, W. Goessler, K. Faber and W. Kroutil, Biotechnol. Bioeng., 2004, 86, 55–62 CrossRef CAS.
- M. Karabec, A. Lyskowski, K. C. Tauber, G. Steinkellner, W. Kroutil, G. Grogan and K. Gruber, Chem. Commun., 2010, 46, 6314–6316 RSC.
- M. M. Musa, K. I. Ziegelmann-Fjeld, C. Vieille, J. G. Zeikus and R. S. Phillips, J. Org. Chem., 2007, 72, 30–34 CrossRef CAS.
- M. M. Musa, K. I. Ziegelmann-Fjeld, C. Vieille and R. S. Phillips, Org. Biomol. Chem., 2008, 6, 887–892 RSC.
- M. V. Filho, T. Stillger, M. Muller, A. Liese and C. Wandrey, Angew. Chem., Int. Ed., 2003, 42, 2993–2996 CrossRef CAS.
- H. Li, D. Zhu, L. Hua and E. R. Biehl, Adv. Synth. Catal., 2009, 351, 583–588 CrossRef CAS.
- H. Gröger, W. Hummel, S. Buchholz, K. Drauz, T. V. Nguyen, C. Rollmann, H. Hüsken and K. Abokitse, Org. Lett., 2003, 5, 173–176 CrossRef.
- H. Gröger, W. Hummel, C. Rollmann, F. Chamouleau, H. Hüsken, H. Werner, C. Wunderlich, K. Abokitse, K. Drauz and S. Buchholz, Tetrahedron, 2004, 60, 633–640 CrossRef CAS.
- G. De Gonzalo, I. Lavandera, K. Faber and W. Kroutil, Org. Lett., 2007, 9, 2163–2166 CrossRef.
- I. Lavanderaq, A. Kern, M. Schaffenberger, J. Gross, A. Glieder, S. de Wildman and W. Kroutil, ChemSusChem, 2008, 1, 431–436 CrossRef CAS.
- V. Hollrigl, K. Otto and A. Schmid, Adv. Synth. Catal., 2007, 349, 1337–1340 CrossRef.
- (a) S. Park and R. J. Kazlauskas, Curr. Opin. Biotechnol., 2003, 14, 432–437 CrossRef CAS; (b) Z. Yang and W. Pan, Enzyme Microb. Technol., 2005, 37, 19–28 CrossRef CAS; (c) F. van Rantwijk, F. Secundo and R. A. Sheldon, Green Chem., 2006, 8, 282–286 RSC.
- M. Moniruzzaman, N. Kamiya and M. Goto, Org. Biomol. Chem., 2010, 8, 2887–2899 RSC.
- M. Eckstein, M. Villela, A. Liese and U. Kragl, Chem. Commun., 2004, 1084–1085 RSC.
- (a) W. Hussain, D. J. Pollard, M. Truppo and G. J. Lye, J. Mol. Catal. B: Enzym., 2008, 55, 19–29 CrossRef CAS.
- G. De Gonzalo, I. Lavandera, K. Durchschein, D. Wurn, K. Faber and W. Kroutil, Tetrahedron: Asymmetry, 2007, 18, 2541–2546 CrossRef CAS.
- S. Bräutigam, D. Dennewald, M. Schürmann, J. Lutje-Spelberg, W.-R. Pitner and D. Weuster-Botz, Enzyme Microb. Technol., 2009, 45, 310–316 CrossRef.
- S. Bräutigam, S. Bringer-Meter and D. Weuster-Botz, Tetrahedron: Asymmetry, 2007, 18, 1883–1887 CrossRef.
- Y.-G. Shi, Y. Fang, Y.-P. Ren, H.-P. Wu and H. L. Guan, J. Ind. Microbiol. Biotechnol., 2008, 35, 1419–1424 CrossRef CAS.
- T. Harada, Y. Kubota, T. Kamitanaka, K. Nakamura and T. Matsuda, Tetrahedron Lett., 2009, 50, 4936–4936.
- D. Giacomini, P. Galletti, A. Quintavalla, G. Gucciardo and F. Paradishi, Chem. Commun., 2007, 4038–4040 RSC.
- P. Galletti, E. Emer, G. Gucciardo, A. Quintavalla, M. Pori and D. Giacomini, Org. Biomol. Chem., 2010, 8, 4117–4123 RSC.
- J. A. Friest, Y. Maezato, S. Broussy, P. Blum and D. B. Berkowitz, J. Am. Chem. Soc., 2010, 132, 5930–5931 CrossRef CAS.
- B. D. Feske, I. A. Kaluzna and J. D. Stewart, J. Org. Chem., 2005, 70, 9654–9657 CrossRef CAS.
- T. Matsuda, Y. Yamagishi, S. Koguchi, N. Iwai and T. Kitazume, Tetrahedron Lett., 2006, 47, 4619–4622 CrossRef CAS.
- D. M.-R. De Temino, W. Hartmeier and M. B. Ansorge-Schumacher, Enzyme Microb. Technol., 2005, 36, 3–9 CrossRef.
- M. M. Musa, K. I. Ziegelmann-Fjeld, C. Vieille, J. G. Zeikus and R. S. Phillips, Angew. Chem., Int. Ed., 2007, 46, 3091–3094 CrossRef CAS.
- G. E. Jeromin, Biotechnol. Lett., 2009, 31, 1717–1721 CrossRef CAS.
- M. Vittorini, E. Dumitriu, G. Barletta and F. Secundo, Bioprocess Biosyst. Eng., 2011, 34, 247–251 Search PubMed.
- V. Gauchot, W. Kroutil and A. R. Schmitzer, Chem.–Eur. J., 2010, 16, 6748–6751 CrossRef CAS.
- F. G. Mutti, A. Orthaber, J. H. Schrittwieser, J. G. de Vries and R. Pietschnig, Chem. Commun., 2010, 46, 8046–8048 RSC.
| This journal is © The Royal Society of Chemistry 2011 |