Design of silver(I)-heteropolyacids: toward the molecular control of reactivity in organic chemistry†
Sophie
Borghèse
,
Benoît
Louis
,
Aurélien
Blanc
* and
Patrick
Pale
*
Laboratoire de Synthèse et Réactivité Organiques, associé au CNRS, Institut de Chimie, Université de Strasbourg, 4 rue Blaise Pascal, CS 90032, 67081 Strasbourg, France. E-mail: ablanc@unistra.fr; ppale@unistra.fr; Fax: +33 3 6885 1517; Tel: +33 3 6885 1517
First published on 25th July 2011
Abstract
A series of silver polyoxometalates (POMs) have been prepared, characterized and studied as catalysts. By tuning the amount of silver and POM acidity, a very effective and reusable catalyst was obtained, allowing the synthesis of various furans and pyrroles in high yields under mild conditions.
1 Introduction
The design and control of chemical systems over multiple-length scales from the molecule to the crystal represent one of the greatest challenges in science,1 especially with specific applications in mind. This could be achieved by associating chemical entities through non-covalent interactions.2 By selecting the appropriate building blocks and the nature of the interaction, one can thus tune the so-obtained supramolecular structures, leading to new chemical and physical properties.3Catalysis is now reaping benefits from this concept, especially in homogeneous catalysis, in which new catalysts with dual properties have been obtained by self-assembling different species.4 In heterogeneous catalysis, the rational design of multifunctional catalysts is still a challenge,5 although few self-assembled catalysts have begun to emerge.5,6
In this context, polyoxometalates (POMs) hold a special position, due to their preparation by self-assembly offering numerous variations of their macrostructural organization (secondary and tertiary structures) through tailoring size, structure and elemental composition of the primary unit (Keggin, Wells–Dawson, Anderson, etc…). Consequently, POMs exhibit a large diversity of properties, leading to various applications from medicine7 to magnetism,8 and more recently, as heterogeneous9 and homogeneous10catalysts in organic synthesis.
In such structures, Keggin-type POMs are readily obtained by condensing two different oxoanions, i.e. silicate or phosphate, the tetrahedron of which serves as a condensing point to assemble tungstate or molybdate in extremely stable and compact structures. Depending on the conditions used, these assemblies form heteropolyanions or their conjugated acids, the so-called heteropolyacids (HPAs) such as H3PW12O40, H3PMo12O40, and H4SiW12O40. The latter exhibits strong Brönsted acidities, due to weak interactions between the protons and the large counter-anion, as for liquid superacids.11
We thus wondered if we could tune and amplify Keggin POM reactivity by replacing step by step these protons with metals known for their catalytic properties in homogeneous catalysis. To the best of our knowledge, only substituted iron,12copper,13 and silver,14titanium, tin, bismuth or ruthenium15POMs have been reported and used as monofunctional heterogeneous catalysts. Such a conceptually novel strategy would provide catalysts with at least dual activity, offering new opportunities in organic chemistry by combining two or more chemical properties on the same POM catalyst.16 This would lead to new versatile and bi(multi)functional POMs as catalytic and recoverable tools toward sustainable organic chemistry.
Due to our interest in coinage metals in organic synthesis,17 we have chosen silver and prepared a series of Ag–H–POMs and applied them, for the first time, to a typical reaction, i.e. the rearrangement of alkynyloxiranes to furans.18 This reaction proceeds under homogeneous conditions in methanol using a mixture of silver triflate and p-toluenesulfonic acid as catalysts (Scheme 1, top). The acid co-catalyst increases the selective oxirane opening caused by the nucleophilic attack of methanol, while silver salts promote the cyclization into furan of the major opened product (Scheme 1, bottom). Ag–H–POMs combining both silver and acid in a single catalyst would thus be perfect for this transformation, and their inherent heterogeneous nature would facilitate recovery of both the product and the catalyst.
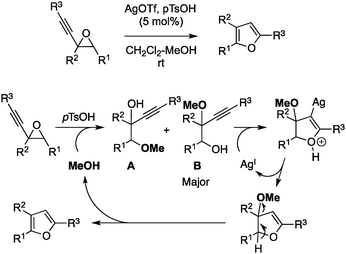 | ||
| Scheme 1 Mechanism for the Ag- and acid catalyzed rearrangement of alkynyloxiranes to furans. | ||
We report herein the synthesis, the characterization of HPA–silver catalysts and the evaluation of their performance in the synthesis of furans.
2 Results and discussion
Four silver-substituted Keggin POMs were easily prepared by the so-called “titration method”19 from the parent H4SiW12O40 HPA. This exchange method requires very mild conditions compared to the hydrothermal synthesis, based on the condensation of all the building blocks—usually metal oxides––in water in an autoclave at high temperature, under autogeneous pressure. The former allowed for a better control of the organization of the Keggin units at the microscale level.6d,20The chemical composition and the crystallinity of the so-obtained AgxH4−xSiW12O40 materials were determined by several physico-chemical techniques. All these materials exhibited infrared bands at similar frequencies, close to those of the parent HPA (see ESI†). These bands were typical from a Keggin structure, assigned in agreement with the literature21 as follows: 1010–1020 cm−1 (νas Si–O), 963–975 cm−1 (νas W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O,), 870–880 cm−1 (νas W–O–W inter-octahedral), 728–742 cm−1 (νas W–O–W intra-octahedral). The bands located at around 1600 and 3500 cm−1 corresponded to the presence of water in the coordination sphere (secondary structure). These infrared data confirmed the retention of the Keggin structure throughout the Ag–H–POM series.
O,), 870–880 cm−1 (νas W–O–W inter-octahedral), 728–742 cm−1 (νas W–O–W intra-octahedral). The bands located at around 1600 and 3500 cm−1 corresponded to the presence of water in the coordination sphere (secondary structure). These infrared data confirmed the retention of the Keggin structure throughout the Ag–H–POM series.
To investigate more in depth the structural transformations accompanying Ag doping, the materials were also examined by powder X-ray diffraction (XRD). The XRD patterns of the Ag–H–POMs exhibited the (220), (310), (222), (400), (411), (420), (332) and (510) main reflections typical from the body-centered cubic secondary structure present in the parent H4SiW12O40 acid.22 In addition, a continuous shift toward higher 2θ diffraction angles, while ranging from H3AgSiW12O40 to HAg3SiW12O40viaH2Ag2SiW12O40, was observed, consistent with a cationic exchange.23 A refinement study of the dominant 25.5–26.5° reflection revealed this effect (Fig. 1). This shift showed that doping the SiW12O404− secondary structure with Ag+ cations viaproton exchange induced the crystallization of a Keggin structure possessing the same symmetry as the parent material but with a contracted unit cell.
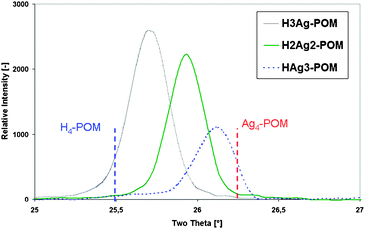 | ||
| Fig. 1 Refined XRD pattern of dominant reflection for all AgxH4−xSiW12O40 polyoxometalates. | ||
These data showed that a single crystalline phase of the Ag–H–POM was formed, whatever the amount of Ag+ introduced. These results are in line with those from Haber et al. showing that silver salts of tungstophosphoric acid were obtained with high crystallinity when the silver content was high.23
The microstructures of AgxH4−xSiW12O40 polyoxometalates were investigated by SEM to assess the powder XRD refined data. Fig. 2 shows the morphology of Ag2H2SiW12O40 crystals, which exhibit a well-defined dodecahedral shape, with ten microns in size. The dodecahedral shape was also observed for all Ag–H–POMs and is a characteristic feature for analogous materials.24 At higher magnification of the POM crystal, the elemental composition was obtained by EDX analysis. An extremely homogeneous distribution of Si, W, O, and Ag elements throughout the material was observed by statistical EDX mapping within a crystal (Fig. 3). The atomic ratios calculated from EDX analysis were equal to the expected stoichiometries for Ag2H2SiW12O40 and Ag4SiW12O40, with, respectively, W/Ag = 6 and W/Ag = 3. These observations agreed with the XRD diffraction study, thus confirming the presence of a single crystalline phase for these Ag–H–POMs.
 | ||
| Fig. 2 SEM images of Ag2H2SiW12O40 (left) and Ag4SiW12O40 (right). | ||
 | ||
| Fig. 3 EDX mapping of Ag, O, Si, and W elements within the Ag2H2SiW12O40 POM crystal. | ||
The chemical composition of AgxH4−xSiW12O40 (x = 0–4) materials was further confirmed by titration of the remaining protons, using our H/D exchange isotope technique.25
With the full characterization of these Ag–H–POMs in hand, we then evaluated their potential as bifunctional heterogeneous catalysts in the rearrangement of alkynyloxiranes to furans.18 Each one was applied to the transformation of the alkynyloxirane 1a into the corresponding furan 2a (Table 1).
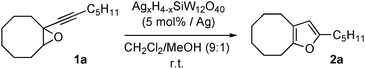
The parent and purely acidic H4SiW12O40 material furnished approximately 10% of furan (entry 1), while the main product was the alcohol resulting from epoxide opening (60%). As expected, the POMs combining Brönsted acidity and silver(I) were effective catalysts, quantitatively leading to the furan within 5 h, whatever the H+/Ag+ ratio (entries 2–4). In contrast, the reaction only proceeded slowly in the presence of the non-acidic Ag4SiW12O40 POM and required 24 h to be nearly complete (entry 5).
These results were perfectly consistent with those obtained under homogeneous conditions, since the presence of both silver and protons was required to achieve the rearrangement to furan.18
Interestingly, the rate of furan formation was much higher for the three Ag–H–POMs containing 1, 2 or 3 Ag+ cations around the Keggin unit. Among them, Ag2H2SiW12O40 POM proved to be the most efficient catalyst giving the furan in 92% yield after only 2 h of reaction, whilst 61% and 69% were reached with, respectively, Ag3HSiW12O40 and AgH3SiW12O40 (entry 3 vs. 2 and 4). Both the acidity and the silver content thus represent key parameters for performing the reaction. While too many acid sites had a detrimental effect on the reaction efficiency, the absence of these acid sites dramatically reduced the reaction rate. Ag-containing POM having an appropriate molecular composition has therefore to be tuned for the targeted reaction.
It is noteworthy that the leaching test revealed unambiguously the heterogeneous nature of the catalysts, as no conversion was observed using the filtrate after removal of the solid catalyst.26
With these conditions in hand, we briefly investigated the scope of this heterogeneous rearrangement compared to the homogeneous version (Table 2). The general trend of this screening showed that an Ag2H2SiW12O40 catalyst was at least as efficient in terms of yields and reaction rates as the combination of silver trifluoromethanesulfonate and p-toluenesulfonic acid (entries 1–3 and 5–7).18 However, the homogeneous conditions were more effective in the transformation of 1-(hex-1-ynyl)-7-oxabicyclo[4.1.0]heptane 1d to 2d (entry 4). Under Ag2H2SiW12O40 catalysis, this epoxide gave the expected furan 2d with only 40% yield, while an 80% yield was obtained under homogeneous conditions. An unexpected regioselectivity at the epoxide opening step was responsible for the difference, as shown by NMR monitoring. After 10 min, the ratio between the opening products A and B (Scheme 1) was, respectively, 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 and 11:89 for the heterogeneous and homogeneous versions, respectively. Since it has been shown that Ag-catalysts could only readily cyclize the B regioisomer,18 the overall yield could only be at best 50% starting from a 50:50 mixture of regioisomers. Similar reasons may also explain the slight difference for the transformation of 1f to 2f (entry 6).
:50 and 11:89 for the heterogeneous and homogeneous versions, respectively. Since it has been shown that Ag-catalysts could only readily cyclize the B regioisomer,18 the overall yield could only be at best 50% starting from a 50:50 mixture of regioisomers. Similar reasons may also explain the slight difference for the transformation of 1f to 2f (entry 6).
| Entry | Furans or Pyrroles | Heterogeneous conditionsa | Homogeneous conditionsb | |||
|---|---|---|---|---|---|---|
| Yieldc | Time/h | Yieldd | Time/h | |||
| a Yields of 2a were calculated from 1H NMR integration relative to naphthalene as an internal standard. Ag2H2SiW12O40 (5 mol%/Ag). b AgOTf/pTsOH (5 mol%). c Yields of 2a were calculated from 1H NMR integration relative to naphthalene as an internal standard. d Isolated yields. e Degradation occurs leading to unidentified by-products. f 10 mol% catalyst loading. g Reactions run at reflux. | ||||||
| 1 |
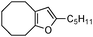
|
2a | 92 | 2 | 92 | 3 |
| 2 |
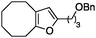
|
2b | 89 | 1.75 | 90 | 1.75 |
| 3 |
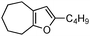
|
2c | 84 | 5 | 72 | 3 |
| 4 |
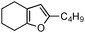
|
2d | 40 | 24 | 80 | 24 |
| 5 |
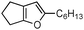
|
2e | 7e | 2.5 | Trace | 24 |
| 6 |
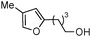
|
2f | 41 | 24 | 51 | 2 |
| 7 |
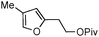
|
2g | 70 | 24 | 81 | 16 |
| 8 |
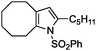
|
2h | 51f | 48 | 61f | 16g |
Interestingly, analogous aziridines could also be rearranged, for the first time, with such catalysts. The aziridine 1h was converted to the pyrrole 2h, although longer contact time and higher catalyst loading (10 mol%) were necessary (entry 8). It is worth noting that the homogeneous version required heating with this starting material.
An important and useful feature of any heterogeneous catalyst is its recovery and recyclability. Thus, we engaged the optimized bifunctional catalyst, i.e.Ag2H2SiW12O40, into a recycling process in the rearrangement of 1a into furan 2a (Fig. 4). For each recycling run, the catalyst was recovered from the reaction mixture by filtration, washed with n-pentane and dried in an oven before the next run. Two to three successive runs could be performed without loss in activity if the reaction time was increased to 5 or 24 h. The furan yields were mostly quantitative in each run (100%, 100% in runs 1–2 for 5 h, and 100%, 100%, 96% in runs 1–3, Fig. 4). At the fourth run, an activity decrease was observed. Nonetheless, the furan was still obtained with high yield after 24 h.27 This could so far not be explained since control experiments did not show significant modification of the catalyst structure28 nor a real mass loss of the catalyst after each run due to filtration.
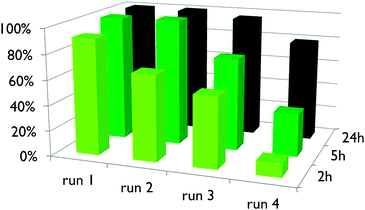 | ||
| Fig. 4 Ag2H2SiW12O40 (5 mol%/Ag) recycling and reuse for the transformation of alkynyloxirane 1a in furan 2a. | ||
Conclusions
In conclusion, we designed bifunctional POMs exhibiting both Brönsted acidity and metallic properties, and applied them in an organic transformation as catalytic and recoverable tools toward a more sustainable chemistry. Following a rational approach, a catalyst, having an optimal molecular composition i.e. proper acidity versus silver content (Ag2H2SiW12O40), could easily be prepared and used for the transformation of alkynyloxiranes to furans.Further synthesis and applications of bifunctional HPAs containing transition-metal cations are ongoing in our laboratory.
Experimental
General experimental details
All starting materials were commercial and were used as received; alkynyloxiranes 1a–g and alkynylaziridine 1h were synthesized according to the procedure reported elsewhere.18a,29Furans 2a, 2b–g were already fully characterized in the literature.18a,b The reactions were monitored by thin-layer chromatography carried out on silica plates (silica gel 60 F254) using UV-light and molybdophosphoric acid/Ce(SO4)2 · 4H2O for visualization. Column chromatographies were performed on silica gel 60 (0.040-0.063 mm) using mixtures of diethyl ether and n-pentane or ethyl acetate and cyclohexane as eluents. Evaporations of solvents were conducted under reduced pressure at temperatures less than 30 °C. FT-IR measurements were performed neat on an ALPHA FT-IR apparatus. 1H and 13C NMR spectra were recorded on an Avance 300 spectrometer at 300 and 75 MHz, respectively. Chemical shifts δ and coupling constants J are given in ppm and Hz, respectively. Chemical shifts δ are reported relative to residual solvent as an internal standard (chloroform-d1: 7.24 ppm for 1H and 77.23 ppm for 13C). Electrospray (ES-MS) low/high-resolution mass spectra were obtained from the mass spectrometry department of the Service Commun d'Analyses, Institut de Chimie, Strasbourg.Notes and references
- (a) A. Ramanan and M. S. Whittingham, Cryst. Growth Des., 2006, 6, 2419 CrossRef CAS; (b) A. Stein, S. W. Keller and T. E. Mallouk, Science, 1993, 259, 1558 CAS; (c) M. J. Zaworotko, Cryst. Growth Des., 2007, 7, 4 CrossRef CAS.
- (a) J. M. Lehn, Science, 1985, 227, 849 CAS; (b) J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 29, 1304 CrossRef.
- J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89 CrossRef.
- (a) R. Noyori, I. Tomido, Y. Tanimoto and M. Nishizawa, J. Am. Chem. Soc., 1984, 106, 6709 CrossRef CAS; (b) R. Noyori, I. Tomido, M. Yamada and M. Nishizawa, J. Am. Chem. Soc., 1984, 106, 6717 CrossRef CAS; (c) Y. M. A. Yamada, N. Yoshika, H. Sasai and M. Shibasaki, Angew. Chem., Int. Ed. Engl., 1997, 36, 1871 CrossRef CAS; (d) E. M. Vogl, H. Gröger and M. Shibasaki, Angew. Chem., Int. Ed., 1999, 38, 1570 CrossRef CAS.
- (a) M. P. Pileni, Acc. Chem. Res., 2007, 40, 685 CrossRef CAS; (b) D. Farrusseng, S. Aguado and S. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502 CrossRef CAS.
- (a) B. Louis and P. Mothé-Esteves, J. Phys. Chem. B, 2006, 110, 16793 CrossRef; (b) S. Ivanova, B. Louis, M. J. Ledoux and C. Pham-Huu, J. Am. Chem. Soc., 2007, 129, 3383 CrossRef CAS; (c) Y. Hu, T. Mei, J. Guo and T. White, Inorg. Chem., 2007, 46, 11031 CrossRef CAS; (d) P. Mothe-Esteves, M. M. Pereira, J. Arichi and B. Louis, Cryst. Growth Des., 2010, 10, 371 CrossRef CAS.
- D. L. Long, R. Tsunasshima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736 CrossRef CAS.
- U. Kortz, S. Nellutla, A. C. Stowe, N. S. Dalal, J. Vantol and B. S. Bassil, Inorg. Chem., 2004, 43, 144 CrossRef CAS.
- For selected examples see: (a) Y. Kamiya, T. Okuhara, M. Misono, A. Miyaji, K. Tsuji and T. Nakajo, Catal. Surv. Asia, 2008, 12, 101 CrossRef CAS; (b) J. S. Yadav, B. V. Subba Reddy, D. N. Chaya, G. G. K. S. Narayana Kumar, P. Naresh and B. Jagadeesh, Tetrahedron Lett., 2009, 50, 1799 CrossRef CAS.
- (a) C. Boglio, G. Lemière, B. Hasenknopf, S. Thorimbert, E. Lacôte and M. Malacria, Angew. Chem., Int. Ed., 2006, 45, 3324 CrossRef CAS; (b) Y. Kikukawa, S. Yamaguchi, Y. Nakagawa, K. Uehara, S. Uchida, K. Yamaguchi and N. Mizuno, J. Am. Chem. Soc., 2008, 130, 15872 CrossRef CAS; (c) N. Dupré, P. Rémy, K. Micoine, C. Boglio, S. Thorimbert, E. Lacôte, B. Hasenknopf and M. Malacria, Chem.–Eur. J., 2010, 16, 7256 Search PubMed.
- (a) K. Y. Lee and M. Misono, in Handbook of heterogeneous Catalysis, Wiley-VCH, Weinheim, 1997, vol. 1, p. 118 Search PubMed; (b) M. Misono, Chem. Commun., 2001, 1141 RSC; (c) A. K. Ghosh and J. B. Moffat, J. Catal., 1986, 101, 238 CrossRef CAS.
- K. T. Venkateswara Rao, P. S. Sai Prasad and N. Lingaiah, J. Mol. Catal. A: Chem., 2009, 312, 65 CrossRef CAS.
- (a) J. S. Yadav, B. V. Subba Reddy, K. V. Purnima, K. Nagaiah and N. Lingaiah, J. Mol. Catal. A: Chem., 2008, 285, 36 CrossRef CAS; (b) N. Pasha, N. Sheshu Babu, K. T. Venkateswara Rao, P. S. Sai Prasad and N. Lingaiah, Tetrahedron Lett., 2009, 50, 239 CrossRef CAS.
- (a) J. S. Yadav, B. V. Subba Reddy, S. Praveenkumar, K. Nagaiah, N. Lingaiah and P. S. Sai Prasad, Synthesis, 2004, 901 CrossRef CAS; (b) J. S. Yadav, B. V. Subba Reddy, P. Sreedhar, R. Srinivasa Rao and K. Nagaiah, Synthesis, 2004, 2381 CrossRef CAS; (c) J. S. Yadav, B. V. Subba. Reddy, P. Sridhar, J. S. S. Reddy, K. Nagaiah, N. Lingaiah and P. S. Sai Prasad, Eur. J. Org. Chem., 2004, 552 CrossRef CAS; (d) K. Mohan Reddy, N. Seshu Babu, I. Suryanarayana, P. S. Sai Prasad and N. Lingaiah, Tetrahedron Lett., 2006, 47, 7563 CrossRef; (e) J. S. Yadav, B. V. Subba. Reddy, K. V. Purnima, S. Jhansi, K. Nagaiah and N. Lingaiah, Catal. Commun., 2008, 9, 2361 CrossRef CAS; (f) N. Lingaiah, N. Seshu Babu, K. Mohan Reddy, P. S. Sai Prasad and I. Suryanarayana, Chem. Commun., 2007, 278 RSC.
- K.-i. Shimizu, K. Niimi and A. Satsuma, Catal. Commun., 2008, 9, 980 CrossRef CAS.
- For a recent and pioneering example of bifunctional POM hybrids as organocatalysts, see: J. Li, X. Li, P. Zhou, L. Zhang, S. Luo and J.-P. Cheng, Eur. J. Org. Chem., 2009, 4486 CrossRef CAS.
- (a) J.-M. Weibel, A. Blanc and P. Pale, Chem. Rev., 2008, 108, 3149 CrossRef CAS; (b) J.-M. Weibel, A. Blanc and P. Pale, in Silver in Organic Chemistry, ed. M. Harmata, Wiley-VCH, Hoboken, 2010, ch. 3 and 10, p. 83 and p. 285 Search PubMed; (c) U. Halbes-Letinois, J.-M. Weibel and P. Pale, Chem. Soc. Rev., 2007, 36, 759 RSC.
- (a) A. Blanc, K. Tenbrink, J.-M. Weibel and P. Pale, J. Org. Chem., 2009, 74, 4360 CrossRef CAS; (b) A. Blanc, K. Tenbrink, J.-M. Weibel and P. Pale, J. Org. Chem., 2009, 74, 5342 CrossRef CAS.
- S. Tatematsu, T. Hibi, T. Okuhara and M. Misono, Chem. Lett., 1984, 865 CrossRef CAS.
- H. Yang, S. Guo, J. Tao, J. Lin and R. Cao, Cryst. Growth Des., 2009, 9, 4735 CAS.
- (a) R. P. F. De Bonfim, L. C. de Moura, H. Pizzala, S. Caldarelli, S. Paul, J. G. Eon, O. Mentré, M. Capron, L. Delevoye and E. Payen, Inorg. Chem., 2007, 46, 7371 CrossRef CAS; (b) T. Okuhara, N. Mizuno and M. Misono, Adv. Catal., 1996, 41, 113 CrossRef CAS.
- L. Pesaresi, D. R. Brown, A. F. Lee, J. M. Montero, H. Williams and K. Wilson, Appl. Catal., A, 2009, 360, 50 CrossRef CAS.
- J. Haber, K. Pamin, L. Matachowski, B. Napruszewska and J. Połtowicz, J. Catal., 2002, 207, 296 CrossRef CAS.
- K. Okamoto, S. Uchida, T. Ito and N. Mizuno, J. Am. Chem. Soc., 2007, 129, 7378 CrossRef CAS.
- S. Borghèse, A. Blanc, P. Pale and B. Louis, Dalton Trans., 2011, 40, 1220 RSC.
- Ag2H2SiW12O40 was stirred for 24 h at room temperature in a mixture of CH2Cl2/MeOH (9/1) before filtration.
- It is worth noting that for run 4, a yield of 100% was reached after 36 h of reaction.
- XRD pattern of the catalyst after recycling was identical, revealing the preservation of the POM structure, while SEM imaging showed slight external modifications. However, EDX mapping surprisingly revealed a less homogeneous Ag distribution.
- M. J. Södergren, D. A. Alonso, A. V. Bedekar and P. G. Andersson, Tetrahedron Lett., 1997, 38, 6897 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cy00154j |
| This journal is © The Royal Society of Chemistry 2011 |