Oxidative dehydrogenation of propane using lattice oxygen of vanadium oxides on silica
Kenta
Fukudome
,
Na-oki
Ikenaga
,
Takanori
Miyake
and
Toshimitsu
Suzuki
*
Department of Chemical, Energy, and Environmental Engineering, and High Technology Research Center, Kansai University, 3-3-35 Yamate, Suita, Osaka 564-8680, Japan. E-mail: v902360@kansai-u.ac.jp; k027802@kansai-u.ac.jp; ikenaga@kansai-u.ac.jp; tmiyake@kansai-u.ac.jp
First published on 20th June 2011
Abstract
Oxidative dehydrogenation of propane using lattice oxygen of vanadium oxide was carried out with a fix-bed flow reactor at 450 °C under atmospheric pressure. Vanadium oxide loaded on SiO2 from V(t-BuO)3O afforded a high propylene selectivity of 88.3% with a propane conversion of 26.5% based on the lattice oxygen of vanadium oxide. The catalyst prepared with V(t-BuO)3O exhibited higher propane conversion and propylene selectivity than that prepared with NH4VO3. Raman, XPS, and XRD analyses revealed that isolated VO43− species seem to be active sites in the ODHP. For the dehydrogenation of propane, the lattice oxygen of the isolated VO43− species was used to give propylene and H2O. The isolated VO43− species was reduced to the isolated V3+ species and was regenerated by oxidation with oxygen. The activity was maintained for at least 10 repeated cycles, suggesting that VOx/SiO2 would be a promising catalyst for ODHP.
1. Introduction
Polypropylene is one of the most important general purpose plastics, and currently the demand for it is markedly increasing. Propylene is co-produced with ethylene by the steam cracking of naptha, and with gasoline by the fluidized catalytic cracking of gas oil. However, these processes are carried out at high temperatures and consume a large amount of energy with low selectivities to the desired alkenes.The development of more selective, simple, and energy-saving processes to produce alkenes starting from less valuable alkanes is of current interest in petrochemical industries. One such process is an oxidative dehydrogenation (ODH) of light alkanes (ODHE, reaction (1); ODHP, reaction (2)). ODH, which is an exothermic reaction, has potential advantages because it consumes smaller amounts of energy and requires little catalyst regeneration.
| C2H6 + 1/2O2 → C2H4 + H2O, ΔH = −149 kJ mol−1 | (1) |
| C3H8 + 1/2O2 → C3H6 + H2O, ΔH = −118 kJ mol−1 | (2) |
From the perspective of space-time yield (STY) in tables summarized by Cavani et al.,1 ODHP to obtain propylene is more difficult than ODHE to obtain ethylene. In general, in the case of ODHE, the selectivity to ethylene increases with temperature due to the ethyl radical from ethane forming ethylene in the gas phase at higher temperatures (600–700 °C); in addition, the higher stability of ethylene leads to the observed high selectivity.2 On the other hand, in the ODHP, the apparent activation energy for propylene combustion is lower than that for propylene formation. This is due to the fact that the bond dissociation energy of allylic hydrogen in propylene is smaller than that for the C–H bonds of propane.1 For example, in the ODHE, the MoVTeNbO catalyst has been reported to exhibit over 80% ethylene selectivity with ethane conversion higher than 80% (340–400 °C).3 In contrast, in the ODHP, Buyevskaya et al. have reported that VOx/MCM-48 exhibits a propylene selectivity of 53.0% with 32.8% propane conversion at 500 °C.4 This catalyst afforded the highest STY, as indicated in the table summarized by Cavani et al.1
In general, vanadium-based catalysts are reported to be active and selective for ODHP. As vanadium-based catalysts, V2O5/MgO,5–10 magnesium vanadates (MgV2O6, Mg2V2O7, Mg3V2O8),11–16 V2O5/Al2O3,17–19 V2O5/SiO2,20,21 V2O5/ZrO2,22,23 and V2O5/TiO224,25 have been reported to exhibit a high activity for ODHP. In particular, the V2O5/MgO catalyst has been reported to exhibit a propylene selectivity higher than 80% with propane conversion above 10% at 450 °C.10
The redox properties of these vanadium-based catalysts are of importance for reactions which proceed via the Mars-van-Krevelen mechanism. Hydrocarbon molecules react with oxygen of the vanadium oxides to give alkene and H2O, and the reduced vanadium oxides are re-oxidized by gas phase molecular oxygen to regenerate the active vanadium oxides. The extent of reduction of the surface V5+ species also depends on the specific oxide support.26,27
The selectivity to propylene is not sufficient for industrial application due to parallel and consecutive combustion of propane and propylene to CO2 and H2O (reactions (3) and (4)), even though most cases were conducted at a low propylene partial pressure by diluting it with an inert gas. In addition to the lower selectivity to propylene described above, oxygen becomes the limiting reactant, since the higher propane/oxygen feed ratio is usually employed, resulting in oxygen-starved conditions with large oxygen consumption by combustion reactions (3) and (4). Consequently, low propane conversion is obtained.
| C3H8 + 5O2 → 3CO2 + 4H2O, ΔH = −2220 kJ mol−1 | (3) |
| C3H6 + 9/2O2 → 3CO2 + 3H2O, ΔH = −2018 kJ mol−1 | (4) |
To avoid combustion of propylene, mild oxidants such as N2O and CO2 are used in ODHP. Kondratenko et al. have reported that VOx/γ-Al2O3 and VOx/MCM-41 give high propylene selectivity in the ODHP with N2O as an oxidant.28,29 However, industrialization of this process would not be possible because of the high N2O cost. From an industrial perspective, catalytic oxidative dehydrogenation of propane using oxygen as an oxidant should be exploited without diluting feed gas with an inert gas.
We have reported that the use of CO2 as an oxidant markedly promotes the dehydrogenation of propane over the oxidized diamond-supported Cr2O3 and V2O5 catalysts.30 With mild oxidants such as N2O and CO2, however, it is difficult to re-oxidize completely reduced VOx species as compared with oxygen. As a result, propylene yields decrease with increases in the reaction time on stream.
Recently, the V2O5–SiO2 catalyst has been reported to exhibit a propane conversion of 10% with a propylene selectivity of 80% at 550 °C under anaerobic conditions.20 However, after the reaction and regeneration, the catalytic activity is slightly lower than that observed for the first reaction.
Lattice oxygen of Fe2O3 and/or NiO has effectively been used to give synthesis gas from CH4 by using ternary mixed metal oxide catalysts such as Fe2O3–Rh2O3–Y2O3 or NiO–Cr2O3–MgO.31,32 This development has led us to exploit catalysts that use lattice oxygen in the ODHP.
We have reported that CO2 acts as an oxidant for the selective oxidation of methane and ethane to formaldehyde and acetaldehyde via the lattice oxygen of vanadium oxide loaded on oxidized diamond.33,34
The objective of the present study was to avoid deep oxidation of propane and to produce propylene by restricting an oxidant to the lattice oxygen of metal oxides having labile oxygen (reaction (5)). Regeneration of the reduced metal oxides with air (reaction (6)) was also studied.
| C3H8 + MOx → C3H6 + H2O + MOx−1 | (5) |
| MOx−1 + 1/2O2 (air) → MOx | (6) |
2. Experimental
2.1 Materials
The catalyst supports used in this study were SiO2 (Fuji Silicia Chemical Ltd, Q-3 (629 m2 g−1), Q-6 (517 m2 g−1), Q-10 (283 m2 g−1), Q-30 (118 m2 g−1), and Q-50 (63 m2 g−1)), γ-Al2O3 (Merck, 120 m2 g−1), TiO2 (P25; Japan Aerosil Co., 50 m2 g−1), CeO2 (JRC-CEO-2; Catalysis Society of Japan, 123 m2 g−1), and MgO (100A; Ube Industries Ltd, 144 m2 g−1). NH4VO3, V2O5, Co(OAc)2·4H2O, Fe(NO3)3·9H2O, Cr(NO3)3·9H2O, Ni(OAc)2·4H2O, oxalic acid, tert-butanol, benzene, toluene, and molecular sieves of 5 Å 1/8 were purchased from Wako Pure Chemical Industries, Ltd.2.2 Catalyst preparation
| V2O5 + 6(CH3)3COH ⇄ 2V(OC(CH3)3)3O + 3H2O | (7) |
In the progress of the reaction, solid V2O5 was dissolved into benzene by changing its color from orange to red to give alkoxide. Unreacted V2O5 was removed from the solution by centrifugation. Benzene and unreacted tert-butanol were removed by distillation under an Ar atmosphere, and then under vacuum. After removing excess solvent and tert-butanol, a white crystal was obtained. The product was further heated under vacuum at 313 K overnight to remove trace amounts of solvent.
An SiO2-supported vanadium oxide catalyst was prepared as follows: the entire procedure was carried out in a glovebox filled with N2. The SiO2 support was dried at 200 °C to remove the adsorbed water before impregnation. Onto dried 1.00 g of SiO2, 0.29 g of V(t-BuO)3O (1.0 mmol) dissolved in 7 mL toluene was added. The sample was kept still for 24 h and subsequently dried at 120 °C for 1 h under N2 flow. The supported catalyst precursor was calcined at 600 °C for 5 h in air (hereafter denoted as V(OR)).
2.3 Catalyst test
The ODHP was carried out using a fixed-bed flow-type quartz reactor (10 mm i.d.) at 400–600 °C at atmospheric pressure. After placing 200 mg of the catalyst in the reactor, the ODHP was carried out under 5 mL min−1 of propane and 20 mL min−1 of Ar. Prior to the reaction, the catalyst was treated with air at the reaction temperature for 30 min. Products (CH4, C2H4, C2H6, C3H6, C3H8, H2, CO, CO2, and O2) were analyzed with an on-line gas chromatograph equipped with a TCD detector (PC-Chrom, M2000 Chromato Analyzer) using a molecular sieve of 5 Å and Poraplot Q columns. After the reaction, regeneration of the reduced catalyst was carried out with 5 mL min−1 of O2 and 20 mL min−1 of Ar at a temperature range of 400–600 °C. During the regeneration stage, formed CO and CO2 were analyzed by an FID gas chromatograph (Shimadzu GC14B) equipped with a methanizer using an activated carbon column.2.4 Catalyst characterization
The surface area of the catalyst was measured by the BET method at 77 K using nitrogen as the adsorbate, with a Quantachrome model AUTOSORB-1.The powder X-ray diffraction (XRD) pattern was obtained with a Shimadzu model XRD-6000 diffractometer with monochromatized Cu Kα radiation. X-Ray photoelectron spectra (XPS) were obtained with a Jeol model JPS-9000MX using Mg Kα radiation as the energy source.
Raman spectra were obtained with a Jasco model NSR-3100 laser Raman spectrometer using 532 nm diode laser excitation with a CCD detector. ESR data were obtained using a Jeol model JES-RE1X operated at the X-band.
3. Results and discussion
3.1 Propane-temperature programmed reaction (TPR)
Selection of active metal oxides was performed using SiO2 as a support. The following metal oxides were selected: NiO, Co3O4, Fe2O3, and V2O5 from our previous studies on partial oxidation of methane (POM)31,32 and selective oxidation of methane and ethane to oxygenates with CO2.33,34Fig. 1 shows the TPR profiles of NiO, Co3O4, Fe2O3, and VOx loaded on SiO2 using propane as a reactant. On all the metal oxides, sharp rises in H2 formation were observed starting at different temperatures, indicating that non-oxidative dehydrogenation or cracking of propane (reactions (8) and (9)) proceeded, respectively. The behavior of H2O formation was much more complicated, and the temperature at which H2O or H2 was detected was defined as the formation temperature.| C3H8 → C3H6 + H2, ΔH = 124 kJ mol−1 | (8) |
| C3H8 → 3C + 4H2, ΔH = 104 kJ mol−1 | (9) |
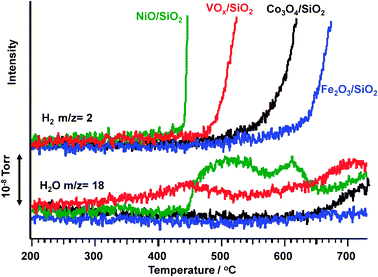 | ||
| Fig. 1 Temperature-programmed reaction of propane over various metal oxides. Ramping rate: 5 °C min−1, sample: 200 mg, metal oxide: 1.0 mmol g−1 support, flow rate: C3H8/Ar = 5/20 (mL mL−1 min−1). | ||
Over NiO/SiO2, the H2 formation temperature was 420 °C, and this temperature was lower than that for H2O at 440 °C. This result indicates that non-oxidative dehydrogenation or cracking of propane proceeded before oxygen transfer from the lattice of NiO to propane. Similarly, over Co3O4/SiO2 and Fe2O3/SiO2 catalysts, H2 formation was observed below the H2O formation temperature. On the other hand, over VOxV(NH)/SiO2, H2O formation started at approximately 360 °C, and this temperature was lower than that of H2 formation at 470 °C. This result indicates that ODHP on VOx/SiO2 proceeded at a lower temperature than non-oxidative dehydrogenation or the cracking of propane.
Table 1 summarizes the results of the ODHP over NiO, Co3O4, Fe2O3, and VOx/SiO2. Since the H2O formation temperature varied according to the oxide species employed, as seen in Fig. 1, the reaction temperature was set at 425 and 600 °C according to the reactivities of the oxides. NiO/SiO2 (run 1) afforded a high H2 yield together with a large amount of carbon (selectivity of 78.4%), and a small propane conversion of 2.0%, indicating that the cracking of propane occurred (reaction (9)). Co3O4/SiO2 (run 2) exhibited a high propylene selectivity of 75.9% and a H2 yield of 0.29 mmol with a higher propane conversion of 9.3%. The results are interpreted based on the progress of non-oxidative dehydrogenation of propane (reaction (8)). Similarly, over Fe2O3/SiO2 (run 3), non-oxidative dehydrogenation again proceeded. VOx/SiO2 (run 4) afforded the highest propylene selectivity of 81.5% with a low propane conversion of 2.2% and with the lowest H2 yield, indicating that ODHP proceeded. The amount of propane fed to the catalyst during the reaction (8 min) and that of lattice oxygen in the VOx/SiO2 (200 mg) were 1.63 and 0.46 mmol, respectively. The stoichiometry of the reaction is shown in reaction (10), and evidence for VO43− will be discussed later. When V5+ was reduced to V3+, the maximum conversion of propane was estimated to be 11.3%. A propane conversion of 2.2% over VOx/SiO2 corresponds to ca. 20% conversion of the lattice oxygen. The propane conversion based on propane feed and the lattice oxygen (V5+ → V3+) is, hereafter, denoted as propane conversion (feed) and propane conversion (VO), respectively. Similarly, propylene yield based on propane feed and the lattice oxygen is denoted as propylene yield (feed) and propylene yield (VO), respectively.
| C3H8 + VO43− → C3H6 + H2O + VO33− | (10) |
| Run | Catalyst | Precursor | Reaction temp./°C | Conversion/% | Selectivity/% | Yield/μmol | Yield/% | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C3H8b | C3H8c | C3H6 | CO | CO2 | C2 + CH4 | Carbon | C3H6 | H2 | C3H6d | C3H6e | ||||
| a Catalyst: 200 mg, loading as metal: 1.0 mmol g−1 support, pretreatment: O2/Ar = 5/20 (mL mL−1 min−1), 30 min at the reaction temperature. Flow rate: C3H8/Ar = 5/20 (mL mL−1 min−1), reaction time: 8 min. b C3H8 conversion based on feed. c C3H8 conversion based on lattice oxygen. d C3H6 yield based on feed. e C3H6 yield based on lattice oxygen. f Vanadium: 1.0 mmol + 1 g SiO2 sand. g Flow rate: C3H8 = 5 mL min−1 neat. | ||||||||||||||
| 1 | NiO/SiO2 | Ni(OAc)2 | 425 | 2.0 | 17.5 | 14.8 | 0.0 | 1.4 | 5.5 | 78.4 | 2.1 | 103.0 | 0.3 | 2.6 |
| 2 | Co3O4/SiO2 | Co(OAc)2 | 600 | 9.3 | 61.6 | 75.9 | 1.7 | 0.3 | 5.7 | 16.5 | 116.2 | 286.0 | 7.1 | 47.0 |
| 3 | Fe2O3/SiO2 | Fe(NO3)3 | 600 | 3.1 | 18.2 | 64.6 | 0.0 | 5.2 | 20.0 | 2.1 | 33.2 | 22.3 | 1.9 | 11.2 |
| 4 | VOx/SiO2 | NH4VO3 | 450 | 2.2 | 19.5 | 81.5 | 10.4 | 5.7 | 0.1 | 2.3 | 29.5 | 0.3 | 1.8 | 15.9 |
| 5 | V2O5 + SiO2f | V2O5 | 450 | 1.7 | 15.0 | 55.7 | 28.6 | 13.7 | 0.2 | 1.8 | 15.6 | 0.2 | 1.0 | 8.8 |
| 6 | VOx/SiO2 | V(t-BuO)3O | 450 | 3.0 | 26.5 | 88.3 | 4.0 | 3.0 | 0.3 | 4.4 | 44.2 | 0.3 | 2.7 | 23.9 |
| 7g | VOx/SiO2 | V(t-BuO)3O | 450 | 4.0 | 35.4 | 83.6 | 2.6 | 6.0 | 0.1 | 7.7 | 55.1 | 0.3 | 3.4 | 30.1 |
At this point, vanadium oxide was selected as the metal oxide in the ODHP with lattice oxygen and used in the following detailed studies.
3.2 Effect of supports and vanadium precursors on the ODHP
Fig. 2 shows the effect of support materials on the ODHP over VOx-loaded catalysts prepared with V(NH) at 450 °C. VOx-loaded SiO2, Al2O3, and TiO2 afforded the same propane conversion (VO) of ca. 20%, but propylene yield (VO) decreased in the following order: SiO2 > Al2O3 > TiO2. Over VOx-loaded Al2O3 and TiO2, lower propylene yields (VO) were obtained due to the cracking of propane and oxidation of propane and propylene to CO and CO2. In contrast, VOx-loaded CeO2 and MgO afforded low propane conversions and propylene selectivities. Basic supports seem to be not appropriate for the oxygen transfer from vanadium oxide on these supports. Thus, VOx/SiO2 showed the highest performance in the ODHP using lattice oxygen of VOx.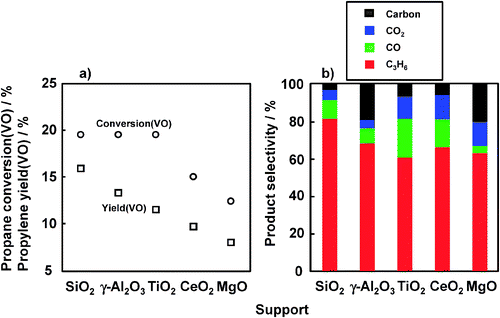 | ||
| Fig. 2 Effect of supports on the ODHP. Catalyst: V loading 1 mmol g−1 support, 200 mg; pretreatment: O2/Ar = 5/20 (mL mL−1 min−1), 30 min, 450 °C; flow rate: C3H8/Ar = 5/20 (mL mL−1 min−1); reaction temperature: 450 °C; reaction time: 8 min. | ||
Fig. 3 shows the effect of the surface area of SiO2 on the ODHP at 450 °C. Five different SiO2 supports having specific surface areas from 63 to 629 m2 g−1 with average pore diameters from 3 to 50 nm and narrow pore size distribution profiles were compared. These catalysts were prepared with V(NH) and V(OR) as vanadium precursors. SiO2 having lower surface areas of 63, 118, and 283 m2 g−1 exhibited higher catalytic activities and slightly increased propane conversion (VO) with an increase in the surface area of SiO2. Propylene yield (VO) reached a maximum value with SiO2, having a surface area of 283 m2 g−1, and a further increase in the surface area of SiO2 markedly decreased propane conversion (VO). These tendencies were pronounced when bulkier V(OR) was loaded onto high surface area SiO2 as seen in Fig. 3b. In contrast, the selectivity to propylene slightly increased, and the selectivities to CO and CO2 decreased with increasing surface areas of SiO2. Table 2 summarizes the surface areas of SiO2 and VOx/SiO2 catalysts. When VOx was loaded onto SiO2, the surface area decreased for the catalysts except for the lowest surface area of SiO2 (SA = 63 m2 g−1), indicating that VOx was supported on the inner surface of the mesopores. It should be noted that SiO2 having higher surface areas of 517 and 629 m2 g−1 had very small mesopores and that surface areas significantly decreased to 378 and 462 m2 g−1, respectively, by the loading of VOx. This seems to indicate that certain amounts of vanadium species were loaded on the mouth of narrow pores, and as a result, access of propane into active sites inside pores would be hindered. Therefore, together with the results shown in Fig. 3b, the amount of effective VOx on the catalyst surface became smaller than that on the lower surface area catalysts. As a result, the higher surface area catalysts exhibited low propane conversions (VO).
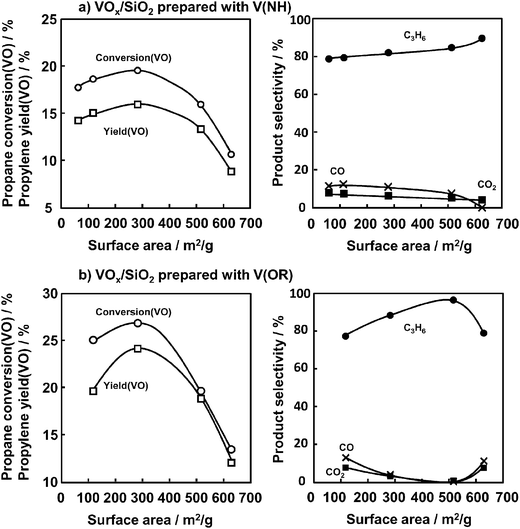 | ||
| Fig. 3 Effect of SiO2 surface area on the ODHP over VOx/SiO2. (a) Propane conversion, propylene yield and selectivity, catalyst prepared with V(NH), (b) propane conversion, propylene yield and selectivity, catalyst prepared with V(OR). Conditions are the same as indicated in the caption of Fig. 2. | ||
In Table 1, runs 4, 5 and 6 provide a comparison of the effects of vanadium precursors on the ODHP. When V(NH) was used as a precursor (run 4), VOx/SiO2 exhibited a propylene selectivity of 81.5% with a propane conversion (VO) of 19.5%. A mixture of bulk V2O5 and SiO2 sand (run 5) exhibited the lowest propane conversion (VO) and propylene selectivity of 15.0 and 55.7%, respectively. As seen in run 6, the use of V(t-BuO)3O (hereafter V(OR)) as a precursor resulted in the highest propylene selectivity of 88.3% with a propane conversion (VO) of 26.5%. Propane conversion based on lattice oxygen and propylene selectivity decreased in the following order: V(OR) > V(NH) > bulk V2O5. In addition, over VOx/SiO2 prepared with V(NH) and bulk V2O5, deep oxidations of propane and propylene to CO and CO2 proceeded. Pak et al. have reported that the V2O5/MgO catalyst prepared with V(OR) shows higher activity than that prepared with V(NH) in the ODHP with molecular oxygen.7 Thus, further studies were carried out with SiO2 having a surface area of 283 m2 g−1 using V(OR) as a vanadium precursor for VOx.
In Table 1, runs 6 and 7 compare the effect of dilution of propane on the ODHP. When neat propane was fed at the same W/F, conversion (VO) increased from 26.5 to 35.4% due to longer contact time, with a slight decrease in propylene selectivities (88.3 to 83.6%). Due to experimental limitation, such as a large dead volume of experimental setup, including an on-line analysis system, accuracy of analyses decreases with a small feed rate, and then further studies were carried out under Ar dilution conditions.
3.3 Effect of VOx loading level on the ODHP
Fig. 4 shows the effects of VOx loading level on the ODHP at 450 °C. With an increase in the VOx loading from 0.5 to 2.0 mmol as V metal on 1 g of SiO2, propane conversion (VO) reached a maximum of 26.5%, and a further increase in the VOx loading markedly decreased the propane conversion (VO), together with a decrease in propylene selectivity. In contrast, CO and CO2 selectivities increased with increasing VOx loading. The reason for this increase will be discussed later.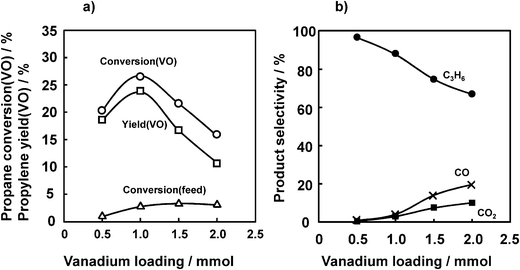 | ||
| Fig. 4 Effect of VOx loading on the ODHP over VOx/SiO2 prepared with V(t-BuO)3O. (a) Propane conversion and propylene yield, (b) selectivity. Conditions are the same as indicated in the caption of Fig. 2 except for the V loading. | ||
3.4 Effect of reaction temperature on the ODHP
Fig. 5 shows the effects of reaction temperature on the ODHP. With an increase in the reaction temperature from 400 to 475 °C, propane conversion (VO) increased from 7.1 to 42.5%. In contrast, propylene selectivity decreased to some extent (95.1 to 85.6%). At 475 °C, a large amount of H2 was produced, indicating that non-oxidative dehydrogenation of propane proceeded without consuming lattice oxygen of vanadium oxide. Therefore, 450 °C was considered to be the optimal temperature.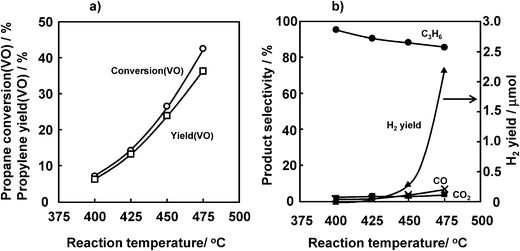 | ||
| Fig. 5 Effect of the reaction temperature on the ODHP over VOx/SiO2 prepared with V(t-BuO)3O. (a) Propane conversion and propylene yield, (b) selectivity. Conditions are the same as indicated in the caption of Fig. 2 except for the reaction temperature. | ||
3.5 Effect of the propane supply on the ODHP
Fig. 6 shows the effects of the amount of propane supplied to the catalyst on the propane conversion (feed) and propylene selectivity. With an increase in the propane feed from 0.82 to 3.28 mmol against 0.18 mmol of lattice oxygen of VOx (V5+ → V3+), namely a propane/lattice oxygen ratio between 4.5 and 17.8, the propane conversion (feed) and propylene yield (feed) reached maximum values at a propane/lattice oxygen ratio of 8.9, and further increases in the propane feed decreased the propane conversion (feed) and propylene yield (feed). On the other hand, the selectivity to propylene slightly increased and the selectivities to CO and CO2 decreased with increases in the propane/lattice oxygen ratio.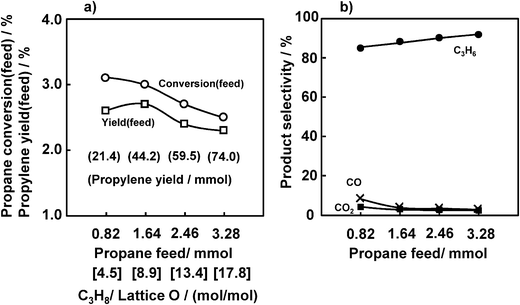 | ||
| Fig. 6 Effect of the amount of C3H8 feed on the ODHP over VOx/SiO2 prepared with V(t-BuO)3O. (a) Propane conversion and propylene yield, (b) selectivity. Conditions are the same as indicated in the caption for Fig. 2 except for the propane feed. | ||
Fig. 7 shows the effects of the propane/lattice oxygen ratio, by changing the amount of catalyst, on the conversion and selectivity. With an increase in the propane/lattice oxygen ratio, propane conversion (feed) decreased with a small increase in propylene selectivity. However, even at the lowest propane/lattice oxygen ratio of 4.5, a high propylene selectivity of approximately 85% was maintained. If ODHP was carried out at a lower propane/lattice oxygen ratio, higher propane conversion (feed) was expected. Our present reactor, however, did not allow us to test ODHP below a propane/lattice oxygen ratio of 4.5 due to a limit of the reactor volume.
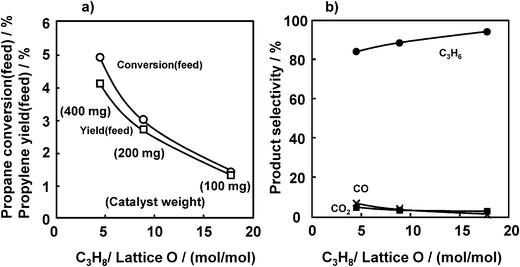 | ||
| Fig. 7 Effect of propane/lattice oxygen ratio on the ODHP over VOx/SiO2 prepared with V(t-BuO)3O. (a) Propane conversion and propylene yield, (b) selectivity. Conditions are the same as indicated in the caption of Fig. 2 except for the catalyst weight. | ||
3.6 Repetition of ODHP and catalyst regeneration cycle
In the studies above, a freshly prepared catalyst was used in a once-through reaction mode. In these runs, the catalyst itself was a reactant that supplied oxygen to abstract hydrogen from propane. The amount of lattice oxygen in the vanadium oxide decreased with increases in the amount of propane fed, and finally the catalyst lost its activity. Therefore, regeneration of the catalyst after the ODHP was essential. Fig. 8 shows the results of 10 repeated ODHP and regeneration cycles at 450 °C with propane–Ar (ODHP) and O2–Ar (regeneration). The reaction times for both were fixed at 8 min. VOx/SiO2 prepared with V(OR) exhibited a constant propane conversion (VO) up to the 10th ODHP and a regeneration cycle with a slight decrease in the propylene selectivity.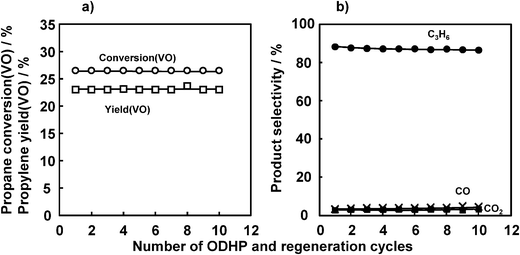 | ||
| Fig. 8 Repetition of ODHP and catalyst regeneration cycle. (a) Propane conversion and propylene yield, (b) selectivity. Catalyst: V loading 1 mmol g−1 SiO2, 200 mg; pretreatment: O2/Ar = 5/20 (mL mL−1 min−1), 30 min, 450 °C; flow rate: C3H8/Ar = 5/20 (mL mL−1 min−1); reaction temperature: 450 °C; reaction time: 8 min; regeneration: O2/Ar = 5/20 (mL mL−1 min−1), 8 min, 450 °C. | ||
In order to understand the conversion of lattice oxygen in the ODHP, Fig. 9 shows the pulsed oxidation of the used VOx/SiO2 catalyst with O2 at 450 °C. When a series of O2 pulses (98.2 μmol) were supplied to VOx/SiO2 after the ODHP, no response was observed against the first O2 pulse, with very small responses of CO (m/z = 28) and CO2 (m/z = 44), presumably due to the combustion of deposited carbon. With the introduction of a second O2 pulse, a large O2 response was observed, and nearly the same responses were observed for the successive O2 pulses. A conversion of lattice oxygen in vanadium oxide was then calculated from the amount of O2 consumed by correcting for the amounts of CO and CO2 formed. The amount of O2 used to re-oxidize vanadium oxide was evaluated as 102 μmol. This value corresponds to 55.2% of lattice oxygen of VOx (V5+ to V3+) on the fresh catalyst, and 6.3% of propane feed.
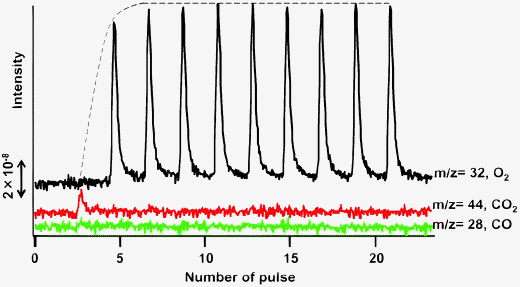 | ||
| Fig. 9 Response to pulsed oxidation of the VOx/SiO2 prepared with V(OR) after ODHP. Catalyst: V loading 1 mmol g−1 SiO2, 200 mg; pretreatment: O2/Ar = 5/20 (mL mL−1 min−1), 30 min, 450 °C; flow rate: C3H8/Ar = 5/20 (mL mL−1 min−1); reaction temperature: 450 °C; reaction time: 8 min; regeneration: Ar = 20 mL min−1, O2 pulse 98.2 mmol, 450 °C. | ||
These results seem to indicate that the regeneration cycle by oxygen could be shortened from 8 min to less than 30 s. Fig. 10 shows the results of 5 repeated ODHP and regeneration cycles at 450 °C with an 8 min propane–Ar reaction period and then 30 s of O2–Ar regeneration. Even when the amount of oxygen decreased from 3.28 to 0.20 mmol, VOx(V(OR))/SiO2 exhibited a constant propane conversion (VO) with a slight decrease in the propylene selectivity. This result indicates that lattice oxygen consumed by dehydrogenation can be regenerated by only a small excess of molecular oxygen, and that deposited carbon was eliminated from the catalyst.
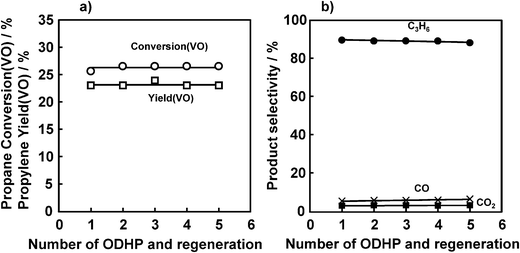 | ||
| Fig. 10 Repetition of ODHP and catalyst regeneration cycle. (a) Propane conversion and propylene yield, (b) selectivity. Catalyst: V loading 1 mmol g−1 SiO2, 200 mg; pretreatment: O2/Ar = 5/20 (mL mL−1 min−1), 30 min, 450 °C; flow rate: C3H8/Ar = 5/20 (mL mL−1 min−1); reaction temperature: 450 °C; reaction time: 8 min; regeneration: O2/Ar = 5/20 (mL mL−1 min−1), 30 s, 450 °C. | ||
For the application of this system to industrial processes, use of a moving bed or a circulating fluidized bed reactor will be appropriate.
3.7 Catalyst characterization
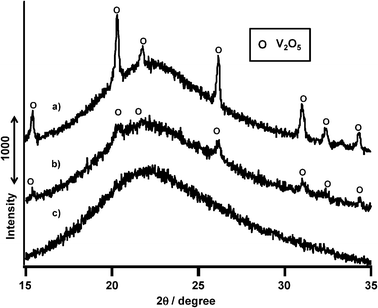 | ||
| Fig. 11 XRD patterns of VOx-based catalysts. (a) V2O5 + SiO2 sand, (b) VOx/SiO2 prepared with NH4VO3, (c) VOx/SiO2 prepared with V(t-BuO)3O. | ||
Fig. 12 shows XRD patterns of VOx/SiO2 having different vanadium loading levels from 0.5 to 2.0 mmol g−1 SiO2 prepared with V(OR). V(0.5)/SiO2 and V(1.0)/SiO2 showed no diffraction peaks (Fig. 12a and b). In contrast, V(1.5)/SiO2 and V(2.0)/SiO2 showed diffraction peaks assignable to V2O5 (Fig. 12c and d). Table 3 summarizes the surface areas of catalysts and the surface densities of vanadium species on these catalysts. Assuming homogeneous dispersion of V species on the SiO2 support, at a vanadium loading of 1.5 mmol g−1 SiO2, the surface density of vanadium is calculated to be 3.2 V atom nm−2 of SiO2 surface. Gao et al. have estimated the maximum monolayer dispersion to be 2.6 V atom nm−2.35 Therefore, higher surface densities of 3.2 and 4.3 V atom nm−2 on SiO2 led to the appearance of polymeric V2O5 crystallites. On the other hand, loading from V(NH) exhibited diffraction peaks of V2O5 at a lower loading level of 1 mmol g−1 SiO2. Therefore, higher propane conversion and propylene selectivity could be achieved with isolated VOx species on SiO2.
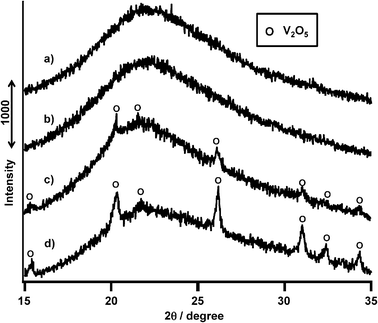 | ||
| Fig. 12 XRD patterns of VOx(0.5–2.0 mmol)/SiO2 catalysts prepared with V(t-BuO)3O. (a) V(0.5 mmol), (b) V(1.0 mmol), (c) V(1.5 mmol), (d) V(2.0 mmol). | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of isolated monovanadate VO43−,35 and the bands at 996, 704, 526, 487, 405, 289, 205, and 147 cm−1 correspond to V2O5 crystallites.36 After the ODHP, the peaks at 704, 526, 487, 405, 289, 205, and 147 cm−1 disappeared, and the intensities of the peaks at 1042 and 996 cm−1 decreased (Fig. 13b).
O stretching vibration of isolated monovanadate VO43−,35 and the bands at 996, 704, 526, 487, 405, 289, 205, and 147 cm−1 correspond to V2O5 crystallites.36 After the ODHP, the peaks at 704, 526, 487, 405, 289, 205, and 147 cm−1 disappeared, and the intensities of the peaks at 1042 and 996 cm−1 decreased (Fig. 13b).
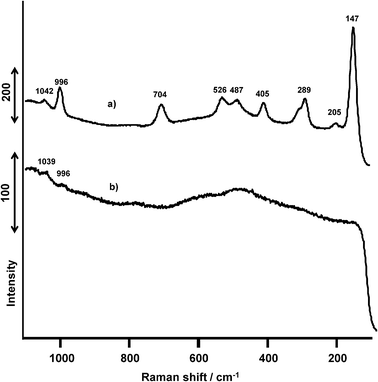 | ||
| Fig. 13 Raman spectra of VOx(1.0 mmol)/SiO2 catalysts prepared with NH4VO3. (a) Before the ODHP, (b) after the ODHP. | ||
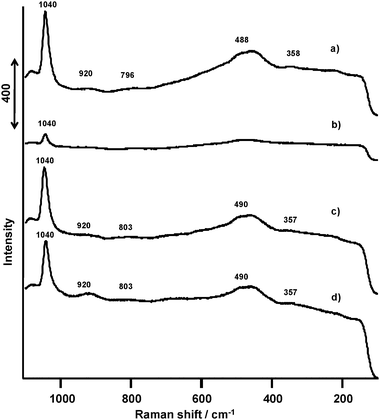 | ||
| Fig. 14 Raman spectra of VOx(1.0 mmol)/SiO2 catalysts prepared with V(t-BuO)3O. (a) Before the ODHP, (b) after the ODHP, (c) after the regeneration, (d) after the 10th regeneration. | ||
On the other hand, VOx/SiO2 catalysts prepared with V(OR) (Fig. 14a) exhibited a sharp peak at 1040 cm−1 and weak broad peaks at 920, 796, 488, and 358 cm−1. The Raman band at 920 cm−1 is assigned to the perturbed silica vibration indicative of formation of the V–O–Si bond,35 and the other bands at 796, 488, and 358 cm−1 are ascribed to those of the SiO2 support.36 After the ODHP, the band at 1040 cm−1 decreased in its intensity (Fig. 14b) and no new peaks were observed. The VO bond in the isolated VO43− seems to have abstracted hydrogen from propane. After regeneration of the once-reacted catalyst and the 10th regeneration cycle (Fig. 14c and d), the intensity of the peak at 1040 cm−1 recovered its original state. The catalyst therefore maintained a constant catalytic performance up to ten ODHP and regeneration cycles.
These results indicate that the reactive lattice oxygen in the ODHP corresponds to the VO double bond of both isolated VO43− and V2O5 crystallites. Based on the ODHP results, the reactivity of the VO double bond in the isolated VO43− is higher than that in V2O5 crystallites.
Fig. 15 compares Raman spectra of VOx/SiO2 catalysts prepared with V(OR) having different vanadium loadings (0.5–2.0 mmol). The band at 1040 cm−1 was observed for all the loadings, and the intensity of this band increased from 0.5 to 1.0 mmol. Above 1.5 mmol, its intensity decreased, and a new band appeared at 997 cm−1. Changes in the intensities of the band at 1040 cm−1 correspond to the growth of crystallite size observed in the XRD shown in Fig. 12d.
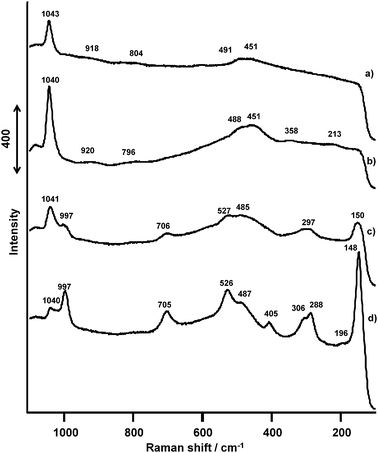 | ||
| Fig. 15 Raman spectra of VOx(0.5–2.0 mmol)/SiO2 catalysts prepared with V(t-BuO)3O. (a) V(0.5 mmol), (b) V(1.0 mmol), (c) V(1.5 mmol), (d) V(2.0 mmol). | ||
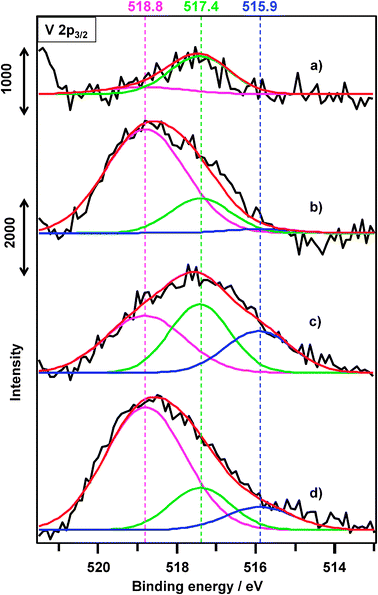 | ||
| Fig. 16 XPS patterns of VOx/SiO2 catalysts prepared with V(t-BuO)3O. (a) After H2 reduction of VOx(0.5)/SiO2, (b) before the ODHP of VOx(1.0)/SiO2, (c) after the ODHP of (b), (d) after the 10th regeneration of (b). | ||
To confirm the presence of V4+ in VOx/SiO2 catalysts prepared with V(OR), the same samples shown in Fig. 16 were characterized with ESR spectroscopy (spectra are not shown). The fresh and H2-treated catalysts showed no ESR signal from V4+ paramagnetic species (note that V5+ and V3+ are diamagnetic). After the ODHP, the catalyst showed a weak signal with a hyperfine structure (hfs) typical of isolated V4+ ions. A bulk or V4+ crystallite exhibits a broad singlet without hfs which is typical to magnetically interacting V4+ ions.38 From these results, we can safely say the existence of only a small amount of V4+, which cannot be detected by XPS.
After the ODHP, the fraction of isolated VO43− decreased to 39.8%, whereas the fraction of VO33−, VOn(OH)m, or crystalline V2O5 and V2O3 increased to 35.8 and 24.4% (Fig. 16c), respectively. Even after the 10th ODHP and regeneration cycle, the relative abundance of the three vanadium species was 70.7, 18.2, and 11.1%, which was not a major change (Fig. 16d). This result indicates that the catalyst maintained more than 70% of isolated VO43− species, suggesting that isolated VO43− species on SiO2 did not aggregate during repeated redox cycles. Based on the XPS analyses, changes in the relative abundance of VO43−, VO33−, V2O5, and V2O3 were significant before and after ODHP, indicating that ODHP proceeded by the redox cycle between V5+ (VO43−, V2O5) and V3+ (VO33−, V2O3), respectively.
From these findings, we propose the following reaction mechanisms shown in Scheme 1. The first step is H-abstraction from a secondary carbon atom of propane by oxygen of a VO double bond of isolated VO43−. The second H-abstraction may occur from a primary carbon atom by the bridging V–O–Si oxygen bond, and propylene is then released from the vanadium active site, together with the production of H2O. Blasco and Lopez Nieto have reported similar catalytic behavior of supported vanadium oxide catalysts in the dehydrogenation of butenes and n-butane.39 In the regeneration cycle, reduced VO33− species is oxidized with molecular oxygen to give the active VO43−.
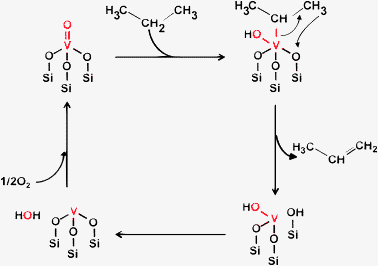 | ||
| Scheme 1 Proposed reaction scheme for the ODHP over VOx/SiO2 catalysts prepared with V(OR). | ||
4. Conclusions
ODHP was carried out using the lattice oxygen of metal oxide in the absence of gas-phase oxygen. Vanadium oxide supported on SiO2 prepared with V(t-BuO)3O as the vanadium source afforded a high C3H6 selectivity of 88.3% with a moderate C3H8 conversion of 26.5% based on the lattice oxygen of vanadium oxide. The results of Raman, XPS and XRD analyses suggested that the isolated VO43− on SiO2 was the active site in the ODHP.The active VO43− was reduced to VO33−, and this VO33− on SiO2 was regenerated with O2 at 450 °C without deactivation in repeated uses.
Acknowledgements
A part of this work was financially supported by the “High-Tech Research Center (HRC) Project” (2007–2011) by MEXT. KF expresses his thanks for the research assistantship from the HRC project.References
- F. Cavani, N. Ballrini and A. Cericola, Catal. Today, 2007, 127, 113–131 CrossRef CAS.
- F. Cavani and F. Trifiro, Catal. Today, 1995, 24, 307–313 CrossRef CAS.
- P. Botella, E. Garcia-Gonzalez, A. Dejoz, J. M. Lopez Nieto, M. I. Vazquez and J. Gonzalez-Calbet, J. Catal., 2004, 225, 428–438 CrossRef CAS.
- O. V. Buyevskaya, A. Bruckner, E. V. Kondratenko, D. Wolf and M. Baerns, Catal. Today, 2001, 67, 369–378 CrossRef.
- M. A. Chaar, D. Patel and H. H. Kung, J. Catal., 1988, 109, 463–467 CrossRef CAS.
- A. Corma, J. M. López Nieto and N. Paredes, J. Catal., 1993, 144, 425–438 Search PubMed.
- C. Pak, A. T. Bell and T. D. Tilley, J. Catal., 2002, 206, 49–59 CrossRef CAS.
- A. Klishinska, K. Samson, I. Gressel and B. Grzybowska, Appl. Catal., A, 2006, 309, 10–16 CrossRef CAS.
- A. Klishinska, S. Loridant, B. Grzybowska, J. Stoch and I. Gressel, Appl. Catal., A, 2006, 309, 17–27 Search PubMed.
- Z. Chao and E. Ruckenstein, Catal. Lett., 2004, 94, 217–221 Search PubMed.
- D. Siew Hew Sam, V. Soenen and J. C. Volta, J. Catal., 1990, 123, 417–435 CrossRef CAS.
- V. Soenen, J. M. Herrmann and J. C. Volta, J. Catal., 1996, 159, 410–417 Search PubMed.
- A. Burrows, C. J. Kiely, P. E. Højlund-Nielsen, G. Vorbeck, J. J. Calvino and C. López-Cartes, Catal. Lett., 1999, 57, 121–128 Search PubMed.
- S. Sugiyama, Y. Iizuka, N. Fukuda and H. Hayashi, Catal. Lett., 2001, 73, 137–140 Search PubMed.
- S. Sugiyama, T. Hashimoto, N. Shigemoto and H. Hayashi, Catal. Lett., 2003, 89, 229–233 Search PubMed.
- S. Sugiyama, T. Hashimoto, Y. Morishita, N. Shigemoto and H. Hayashi, Appl. Catal., A, 2004, 270, 253–260 Search PubMed.
- K. Routray, K. R. S. K. Reddy and G. Deo, Appl. Catal., A, 2004, 265, 103–113 Search PubMed.
- B. Mitra, I. E. Wacha and G. Deo, J. Catal., 2006, 240, 151–159 CrossRef CAS.
- I. Rossetti, L. Fabbrini, N. Ballarini, C. Oliva, F. Cavani, A. Cericola, B. Bonelli, M. Piumetti, E. Garrone, H. Dyrbeck, E. A. Blekkan and L. Forni, Catal. Today, 2009, 141, 271–281 Search PubMed.
- I. Rossetti, L. Fabbrini, N. Ballarini, C. Oliva, F. Cavani, A. Cericola, B. Bonelli, M. Piumetti, E. Garrone, H. Dyrbeck, E. A. Blekkan and L. Forni, J. Catal., 2008, 256, 45–61 Search PubMed.
- S. A. Karakoulia, K. S. Triantafyllidis, G. Tsilomelekis, S. Boghosian and A. A. Lemonidou, Catal. Today, 2009, 141, 245–253 Search PubMed.
- C. L. Pieck, M. A. Banares and J. L. G. Fierro, J. Catal., 2004, 224, 1–7 Search PubMed.
- R. Sasikala, V. Sudarsan, T. Sakuntala, Jagannath, C. Sudakar, R. Naik and S. R. Bharadwaj, Appl. Catal., A, 2008, 350, 252–258 Search PubMed.
- J. B. Stelzer, J. Caro and M. Fait, Catal. Commun., 2005, 6, 1–5 Search PubMed.
- R. P. Singh, M. A. Banares and G. Deo, J. Catal., 2005, 233, 388–398 Search PubMed.
- X. Gao, M. A. Banares and I. E. Wachs, J. Catal., 1999, 188, 325–331 CrossRef CAS.
- M. A. Banares, M. V. Martinez-Huerta, X. Gao, J. L. G. Fierro and I. E. Wachs, Catal. Today, 2000, 61, 295–301 CrossRef CAS.
- E. V. Kondratenko and M. Baerns, Appl. Catal., A, 2001, 222, 133–143 CrossRef CAS.
- E. V. Kondratenko, M. Cherian, M. Baerns, D. Su, R. Schlogl, X. Wang and I. E. Wachs, J. Catal., 2005, 234, 131–142 CrossRef CAS.
- K. Nakagawa, C. Kajita, N. Ikenaga, M. Nishitani-Gamo, T. Ando and T. Suzuki, Catal. Today, 1998, 84, 149–157 Search PubMed.
- O. Nakayama, N. Ikenaga, T. Miyake, E. Yagasaki and T. Suzuki, Catal. Today, 2008, 138, 141–146 Search PubMed.
- O. Nakayama, N. Ikenaga, T. Miyake, E. Yagasaki and T. Suzuki, Ind. Eng. Chem. Res., 2010, 49, 526–534 Search PubMed.
- K. Okumura, K. Nakagawa, T. Shimamura, N. Ikenaga, M. Nishitani-Gamo, T. Ando, T. Kobayashi and T. Suzuki, J. Phys. Chem. B, 2003, 107, 13419–13424 Search PubMed.
- T. Shimamura, K. Okumura, K. Nakagawa, T. Ando, N. Ikenaga and T. Suzuki, J. Mol. Catal. A: Chem., 2004, 211, 97–102 Search PubMed.
- X. Gao, S. R. Bare, B. M. Weckhuysen and I. E. Wachs, J. Phys. Chem. B, 1998, 102, 10842–10852 CrossRef CAS.
- S. Xie, E. Iglesia and A. T. Bell, Langmuir, 2000, 16, 7162–7167 CrossRef CAS.
- C. Hess, G. Tzolova-Müller and R. Herbert, J. Phys. Chem. C, 2007, 111, 9471–9479 Search PubMed.
- A. V. Kucherov, A. V. Ivanov, T. N. Kucherova, V. D. Nissenbaum and L. M. Kustov, Catal. Today, 2003, 81, 297–305 Search PubMed.
- T. Blasco and J. M. Lopez Nieto, Appl. Catal., A, 1997, 157, 117–142 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |