Synthetic strategy for Fe-MCM-41 catalyst: a key factor for homogeneous or heterogeneous phenol oxidation
Sergey V.
Sirotin
*,
Irina F.
Moskovskaya
and
Boris V.
Romanovsky
Lomonosov Moscow State University, Moscow 119991, Russia. E-mail: ssirotin@rambler.ru; Tel: +7 495 939 35 70
First published on 25th July 2011
Abstract
Iron(III) was introduced into the mesoporous molecular sieve MCM-41 using two protocols: (i) iron nitrate was added in situ in the course of MCM-41 synthesis; (ii) a nitrate or triiron oxoacetate complex was loaded into the as-synthesized MCM-41 sieve by using incipient wetness impregnation method. In both cases the iron content was in the range of 1–11 wt%. The prepared materials were characterized by N2-BET, XRD, H2-TPR, NH3-TPD, ESR and Mössbauer spectroscopy techniques. All the catalysts were found to exhibit high enough activity in the liquid phase oxidation of phenol by hydrogen peroxide. At 25 °C the oxidation of phenol occurs in homogeneous phase and is mediated only by the Fe3+ ions leached from the solid, while at 80 °C the framework iron atoms mainly contribute into the total activity so that the mechanism of true heterogeneous catalysis is operative. For iron content of 0.25 wt% and less, the framework iron is fairly stable and highly resistant to the acidic conditions of liquid-phase reactions.
1. Introduction
The growing academic and industrial interest in Fe-containing zeolites and analogous materials arises from their high effectiveness as catalysts for the oxidation of organic substrates.1 Their use as catalysts for the liquid phase oxidation of phenol includes two main directions: (i) obtaining of catechol and hydroquinone and (ii) the full mineralization of phenol in waste waters also known as CWO process. The heterogenization of iron species can be performed using several conventional approaches. First, one can obtain nanosized oxide particles, which can be effectively stabilized by the porous system of the support.2–4 Also, iron can be introduced directly into the framework of the support5–7 and finally, the ionic form of iron can be preserved if using heterogenizing salts8,9 or performing ion-exchange treatment.10,11 In all cases it is commonly suggested in numerous papers that heterogenized iron species realize the oxidation of phenolvia a radical mechanism, which is similar to that established for the homogeneous oxidation by aqueous ions Fe3+ (Fenton-like system).12–14However, while performing the liquid phase oxidation in the presence of iron-containing heterogeneous catalysts the leaching of active iron often takes place.12,15,16 This undesirable feature makes both the separation of valuable catalytic products and the following runs of the catalyst harder. At the same time, it was strongly evidenced that iron(III) ions, which are being transferred from the catalyst surface into the reaction media possess remarkable activity in the oxidation of organic substrates.16,17 Unfortunately, there is still no unambiguous and reliable concept to circumscribe the contributions of different forms of iron to the total activity.
In the present work we define the conditions when the heterogeneous and homogeneous scenarios of oxidation are accomplished, respectively. The concrete recommendations for obtaining stable and active catalysts that provide the true heterogeneous oxidation of phenol by hydrogen peroxide are also given.
2. Experimental
2.1. Synthesis
Iron was introduced into the MCM-41 molecular sieve using two different approaches. First, iron was added in situ as a nitrate at the stage of MCM-41 synthesis (denoted as s series). The aim was to obtain the catalyst with isolated iron ions in the framework of the support, i.e. containing isomorphically substituted iron. Second, iron was added as a nitrate (n series) or an oxoacetate complex (a series) via simple wet impregnation of the freshly prepared and calcined MCM-41 support. In this case the presence of nanosized oxide particles were expected after calcination.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0,3 CTMABr:11 NH3:60 EtOH:144 H2O (TEOS = tetraorthoethoxysilane, CTMABr = cetyltrimethylammonium bromide). The mixture was stirred for 2 h at RT, then the amorphous precipitate was separated, washed with hot water (80 °C) and calcined at 550 °C for 24 h in air.
:0,3 CTMABr:11 NH3:60 EtOH:144 H2O (TEOS = tetraorthoethoxysilane, CTMABr = cetyltrimethylammonium bromide). The mixture was stirred for 2 h at RT, then the amorphous precipitate was separated, washed with hot water (80 °C) and calcined at 550 °C for 24 h in air.
Then 1 g of freshly prepared and calcined MCM-41 support was dispersed in the solution of 0.09 g Fe3O(AC6) (calculated for 2 wt% Fe) in 25 mL of water so as to be fully covered. The mixture was slightly acidified in order to prevent the fast hydrolysis of the complex. The impregnated MCM-41 powder was dried and calcined at 550 °C for 24 h in air. The yellow-colored catalysts were denoted as Fe(n)-MCM-41a, where n in brackets is equal to 2, 5 and 7 and refers to iron content (approximately).
2.2. Characterization
Iron content was measured using AA technique. A sample was treated by the mixture of concentrated HF, HNO3 and HCl acids. Then the resulting solution was dry-evaporated and the residue was dissolved in HCl. Before analysis this procedure was done twice.The N2-BET specific surface area, the pore volume, and the average pore diameter were calculated from the nitrogen adsorption isotherms recorded at 77 K on a Micromeritics ASAP 2000N apparatus.
The X-ray diffraction measurements were made on a DRON-3M diffractometer. Diffraction patterns were recorded in the angle range of 2θ = 15°–60° with filtered CoKα radiation (λ = 1.79 Å). The diffraction peaks were identified using JCPDS database.
57Fe Mössbauer spectra for oxide-containing catalysts were recorded at RT on an electrodynamic type spectrometer in a “constant acceleration” mode with 57Co(Rh) as a γ-radiation source. The recorded spectra were processed using standard software. IS values were given relative to standard α-Fe.
FTIR spectra of the samples (KBr pellets) were recorded in the range of 400–4000 cm−1 on a Nicolet Protege 460 E.S.P. Fourier-transform spectrometer. The recorded IR spectra were processed with the Nicolet OMNIC E.S.P. software package.
Temperature-programmed reduction of the samples with hydrogen (H2-TPR) was carried out according to the following procedure. 20–50 mg of a sample was loaded into a quartz tube reactor, heated at the rate of 6.4 °C min−1 up to 400 °C in the flow of dry Ar (30 mL min−1) and kept at this temperature for 1 h. After cooling to RT, the Ar flow was replaced by the reducing gas mixture (3.5 vol% H2 in Ar, 30 mL min−1), and the temperature was increased to 1000 °C at the rate of 8.0 °C min−1. The total hydrogen uptake was determined from the area under the TPR curve.
Temperature-programmed desorption of ammonia (NH3-TPD) was performed as follows. 50–100 mg of the sample (0.25–0.50 mm mesh size) was loaded into the quartz tube reactor between layers of grinded quartz (0.5–1.0 mm mesh size). The sample was heated up to 400 °C for 1 h in the flow of dry air with the following treatment by He for 1 h. After cooling to RT the sample was saturated with ammonia, which was fed in a mixture with nitrogen (1:1) for 30 min. The removal of physically adsorbed ammonia was carried out at 100 °C in the flow of dry He for 1 h. Then the sample was rapidly cooled to RT, reactor attached to the thermal conductivity detector (30 mL min−1He flow). The TPD curves were recorded at linearly increasing temperature (rate of 8.0 °C min−1) up to 800 °C.
ESR spectra were recorded on a Bruker EMX-6 spectrometer in X-region (9.5 GHz) at RT. The sample was placed into a glass tube with diameter of 5 mm. The spectrum of an empty tube was subtracted from the spectra of all samples. All spectra were recorded at the modulation amplitude of 5 G and microwave radiation of 6.34 mW.
X-ray fluorescence analysis for the iron content in both reaction mixtures and recovered solid catalysts was carried out on a Spectroscan spectrometer. The instrument was preliminarily calibrated with standard iron solutions.
2.3. Catalytic tests
1 g of phenol, 30 mg of catalyst and 10 mL of H2O were added in a static reactor equipped with a thermostat. Reaction media was heated to 60 °C and then 2 mL of 25% H2O2 was added. Products were analyzed using GC with a capillary SE-30 column (30 m × 0,22 mm) and a FID. After the reaction catalysts were separated, washed with hot water (∼80 °C), dried and calcined at 550 °C in air, and then tested in the repeated cycle.3. Results and discussion
3.1. Physicochemical properties
The textural parameters of Fe-MCM-41 catalysts were calculated from the nitrogen adsorption isotherms and are summarized in Table 1.| Catalyst | Fe/wt% | S BET, m2 g−1 | V BJH/cm3 g−1 | D av./Å |
|---|---|---|---|---|
| a Including pores with diameter more than 50 nm. | ||||
| Fe(1)-MCM-41s | 0.9 | 1300 | 0.74 | 23 |
| Fe(1)-MCM-41se | 1.1 | 1080 | 0.63 | 26 |
| Fe(2)-MCM-41s | 2.0 | 1060 | 0.73 | 24 |
| Fe(3)-MCM-41s | 3.3 | 830 | 0.58a | 24 |
| Fe(6)-MCM-41s | 6.3 | 470 | 0.42a | 25 |
| MCM-41 | — | 1100 | 0.71 | 25 |
| Fe(2)-MCM-41n | 2.4 | 1080 | 0.63 | 24 |
| Fe(6)-MCM-41n | 5.6 | 1010 | 0.60 | 23 |
| Fe(11)-MCM-41n | 11.2 | 940 | 0.55 | 23 |
| Fe(2)-MCM-41a | 2.0 | 1100 | 0.65 | 25 |
| Fe(5)-MCM-41a | 4.5 | 1030 | 0.61 | 24 |
| Fe(7)-MCM-41a | 7.2 | 1010 | 0.60 | 24 |
The s series stands for the group of catalysts synthesized via in situ route so as to obtain catalytic materials with iron localized directly in the framework of the support. However, this synthetic procedure requires a calcination step, which makes the siliceous framework unstable and inevitably leads to the removal of iron from the framework.6,16 Since the very calcination step is responsible for such disordering of the Fe-MCM-41 structure, the sample Fe(1)-MCM-41se was synthesized using 4-fold extraction of template with NH4Cl/(EtOH+H2O) mixture instead of a standard calcination. This synthetic strategy was chosen to preserve the highest possible amount of iron in the MCM-41 framework. It should be noted that the double extraction procedure allows 50% removal of template only, while the 4-fold one is sufficient for its complete removal.
In comparison with Fe(1)-MCM-41s with the template removed by thermal treatment, the sample Fe(1)-MCM-41se possesses higher pore size. Thus, the non-thermal removal of the template does not lead to the shrinkage of the support channels as earlier was reported in ref. 20 and 21.
When adding up to 2 wt% of Fe to the MCM-41 synthetic mixture (s series), the textural properties of the resulting catalytic materials do not significantly differ from those of an individual MCM-41 support. However, the increase of iron content results in the consequent and remarkable decrease of both surface area and pore volume. This undesirable effect does not relate to the destruction of the siliceous framework, rather it might be ascribed to the decreased yield of the Fe-MCM-41 material itself. This conclusion is based on the FTIR spectra (Fig. 1). For all samples the bands characteristic of the MCM-41 framework can be clearly seen. The shift of the 1095 cm−1 band (asym. T–O–T vibrations) to 1083 cm−1 allowed the conclusion that independently of iron content a part of it is very similarly localized in the framework of MCM-41.22–24
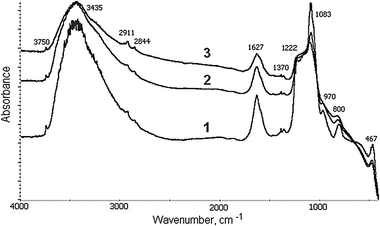 | ||
| Fig. 1 FTIR spectra of Fe-MCM-41s catalysts, containing 2.0 (1), 3.3 (2) and 6.3 (3) wt% Fe. | ||
The textural deterioration of materials on going to higher iron content is caused by the growing instability of the MCM-41 framework because of a considerable size difference between Fe3+ and Si4+ ions. As a result after calcination the Fe atoms leave the framework to form iron oxide or complex Fe–O–Si species, which can eventually block the channels of the support. Hence, the iron concentration in the framework of the MCM-41 material seems to be strongly limited. Amounts of 1 wt%6 and 2 wt%7 were reported to be successfully introduced into the MCM-41 framework. Our study confirms these observations since the Fe(1)-MCM-41s sample with 1 wt% Fe was visibly colored, which clearly indicates the presence of the oxides on the outer matrix surface.25
In contrast to the s series, the incipient wetness impregnation method used for iron loading into the MCM-41 support (n or a series) results in obtaining materials with the textural properties similar to those of an individual support regardless of both iron content and precursor type (Table 1). It is to note that the use of nitrate salt does not lead to the blockage of channels by oxide particles even at iron content up to 11.2 wt%. This synthetic procedure offers nanosized oxide particles within the pores of MCM-41 where they seem to be uniformly distributed. This suggestion is indirectly supported by the fact that increasing the iron content influences the surface area and pore volume so that they decrease slightly, though insignificantly. The yellow color of the samples indicates again that at least some part of oxides is localized on the outer surface of the catalyst.
Fig. 2 shows H2-TPR profiles for the catalysts containing 2–11 wt% of Fe. Vertical lines denote temperatures corresponding to the reduction steps of a bulk oxide: Fe2O3 → Fe3O4 (380 °C) and Fe3O4 → Fe0 (465 °C).26 The first maximum of hydrogen uptake is shifted to 400–450 °C for all samples and can be considered as indicating the iron oxide particles to be hosted most likely inside the pores of MCM-41 and thus reduced at higher temperatures. Non-stoichiometric hydrogen uptake above 900 °C is also characteristic of all samples. Earlier, this effect has been observed for the Fe-containing molecular sieve materials27 and was suggested to be caused by the noticeable hydrogen absorption by nanosized metal iron particles.
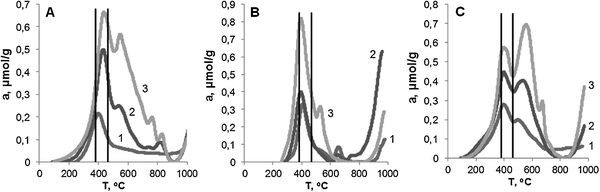 | ||
| Fig. 2 TPR curves for Fe-MCM-41 catalysts. (A) s series: 1 – 2.0; 2 – 3.3; 3 – 6.3 wt% Fe; (B) n series: 1 – 2.4; 2 – 5.6; 3 – 11.2 wt% Fe; (C) a series: 1 – 2.0; 2 – 4.5; 3 – 7.2 wt% Fe. | ||
The Fe(2)-MCM-41s sample shows a high-temperature shoulder on the peak corresponding to an Fe3O4 → Fe0 reduction step. This shoulder becomes the well-resolved maximum for the samples with higher iron content (Fig. 2A). In the first case, the only maximum is seen supposedly due to the even distribution of oxides inside the nanopores of the support, which makes them equally accessible to hydrogen and reducible at the same temperature, i.e., of the first reduction step. This behavior has been earlier observed in ref. 28.
In contrast, for the samples with increased iron content the shape of the high-temperature maxima implies the oxide particles to possess rather different accessibility so that a part of them can be reduced at higher temperatures only. Probably, these latter are the large oxide particles, which are localized inside the MCM-41 channels and were formed after iron atoms left the framework in the course of calcination and then aggregated. The hydrogen uptake below 200 °C for Fe(3)-MCM-41s and Fe(6)-MCM-41s samples can be ascribed to the easy reduction of low dispersion oxides on the outer surface of the support.
The hydrogen uptake for all s series samples (Table 2) is lower than the stoichiometric value H2/Fe of 1.5. A part of the highly dispersed and poorly accessible oxides could remain unreduced2 as well as the framework Fe(III) atoms, which are hardly reduced to zero-valent state at temperatures below 900 °C.19 Since in all cases a H2/Fe value is not much less than 1.5, we assume that in these samples iron presents mainly as low dispersion oxides, which can be fully reduced.
| Catalyst | Peak maxima, °C | H2/Fe | |||
|---|---|---|---|---|---|
| a Not including the hydrogen uptake at 800–1000 °C. | |||||
| Fe(2)-MCM-41s | 401 | — | — | — | 1.2 |
| Fe(3)-MCM-41s | 432 | 535 | 820 | — | 1.4 |
| Fe(6)-MCM-41s | 437 | 546 | 762 | 829 | 1.3 |
| Fe(2)-MCM-41n | 405 | 658 | — | — | 1.0a |
| Fe(6)-MCM-41n | 392 | 651 | — | — | 0.5a |
| Fe(11)-MCM-41n | 394 | 529 | — | — | 0.6a |
| Fe(2)-MCM-41a | 398 | 498 | 886 | 2.1 | |
| Fe(5)-MCM-41a | 399 | 538 | — | 1.7a | |
| Fe(7)-MCM-41a | 396 | 559 | 673 | 1.5a | |
Only one narrow peak is observed for samples Fe(2)-MCM-41n and Fe(6)-MCM-41n obtained by the incipient wetness impregnation with nitrate (Fig. 2B). It indicates the formation of accessible oxides, which possess uniform dispersion and are easily and fully reduced at the temperature of the first reduction step. The peak at 530 °C on the TPR curve of Fe(11)-MCM-41n sample implies an additional reduction step and might account for the relative growth of nanosized oxides with the increase of iron content.29 Additional peaks at 700 °C can be attributed to the various Fe–O–Si species, e.g. framework iron as a result of interaction between an oxide particle and a channel of the support. The appearance of these peaks with the increase of iron was earlier observed in ref. 30.
The experimental values of H2/Fe (mol.) were obtained attributing the area below the TPR curve to the total amount of iron in the catalyst. For the n series samples this value is less than the stoichiometric one of 1.5 (Table 2) required for the full reduction of Fe2O3 present in the sample. Hence, the oxides were not fully reduced. This conclusion is additionally confirmed by the yellow color of the samples after the TPR experiment. The TPR profile for Fe(2)-MCM-41n and Fe(6)-MCM-41n samples are similar except that the latter has a higher uptake in the high temperature region. If calculating the H2/Fe values per iron content in the catalysts, it becomes evident that in both catalysts the same amount of iron oxide was reduced regardless of the three-fold difference in total iron amount. The increase in iron content up to 6 wt% influences the amount of oxide particles but not their size, which remains constant. Therefore, the reduction of the Fe(6)-MCM-41n sample results in a higher formation of metal iron, which leads to the higher hydrogen uptake above 800 °C. The increase of iron content up to 11.2 wt% results in the larger size of oxide particles, which now can be easily reduced. So that, for the Fe(11)-MCM-41n sample the highest amount of reduced oxide was found. The large metal iron particles being formed from the low dispersion oxides have small hydrogen uptake at high temperatures.
If the oxoacetate complex is used as a source of iron (a series), then the oxide species of different size and accessibility are formed in the channels of MCM-41. This conclusion is supported by the fact that the uptake peaks on TPR profiles are widened in comparison with that of the n series samples (Fig. 2C). The uptake below 200 °C for the a series catalysts indicates that a part of low dispersion oxides are present on the outer surface.
At the same time, the iron oxides in the a series samples can be readily reduced to metal below 800 °C (Table 2). This feature seems to occur due to the relatively large size of the oxide particles even though they are still nanosized. The metal particles thus produced are able to absorb hydrogen in excess above 800 °C so that the total amount of H2 consumed per Fe atom at 100–1000 °C is higher than 1.5.
The NH3-TPD curves for the catalysts with different iron content are shown in Fig. 3. In the case of any Fe-containing solid, this technique cannot attribute unambiguously the ammonia adsorption to Brønsted or Lewis type acid sites. Tentatively, the largest amount of ammonia desorbed from the Fe(2)-MCM-41s sample could be due to the presence of iron mainly in the framework of MCM-41 since these tetracoordinated Fe atoms give rise to the true Brønsted centers. However, at higher iron content (sample Fe(6)-MCM-41s) the iron tends to form oxide particles, which expose rather Lewis acidity. These aprotic centers are usually characterized by the extended shoulder at higher temperatures.31 At the same time, Lewis acid sites are poorly presented in our high-iron content samples because Fe atoms inside the large oxide particles are inaccessible for ammonia molecules.
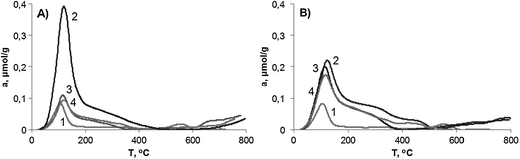 | ||
| Fig. 3 TPD-NH3 curves for the Fe-MCM-41 catalysts. (A) 1 – MCM-41; 2 – Fe(2)-MCM-41s; 3 – Fe(2)-MCM-41n; 4 – Fe(2)-MCM-41a; (B) 1 – MCM-41; 2 – Fe(6)-MCM-41s; 3 – Fe(11)-MCM-41n; 4 – Fe(7)-MCM-41a. | ||
For samples of both n and a series with low iron content, the ammonia desorption profiles are nearly the same indicating the equal accessibility of oxides and their uniform size. With the increase of iron content, the number of acid sites also increases. The ammonia desorption at 350–500 °C from the Fe(7)-MCM-41n sample is noticeably higher than that from Fe(11)-MCM-41n and can be attributed to the presence of a lower dispersion of oxides in comparison to the a series catalysts. Oxide particles with a larger size possess a surface of lower curvature, so that the complex [Fe⋯NH3] becomes more stable and decomposes at higher temperatures. The same reason stands for the increased ammonia desorption from the Fe(6)-MCM-41s sample at 350–500 °C.
The ESR spectrum for the Fe(1)-MCM-41se catalyst, which was synthesized using no calcination procedure but template extraction instead is given in Fig. 4. It shows the signal at g ≈ 4.2, which is drastically widened in the case of the Fe(2)-MCM-41s sample. But this signal cannot be detected for Fe(2)-MCM-41n while it is hardly seen for Fe(2)-MCM-41a even at 1:10 scale.
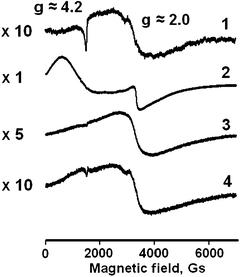 | ||
| Fig. 4 ESR spectra for Fe-MCM-41 catalysts: 1 – Fe(1)-MCM-41se; 2 – Fe(2)-MCM-41s; 3 – Fe(2)-MCM-41n; 4 – Fe(2)-MCM-41a. | ||
At the same time, the signal at g ≈ 2.0 was detected for all catalyst samples. It can be attributed to the iron in oxides whereas the signal 4.2 refers to the isolated Fe-containing species.32 These latter might presumably be the iron atoms in the framework of MCM-41, which in the case of Fe(2)-MCM-41s sample are formed at the hydrolysis stage during the synthesis of the MCM-41 support. For the Fe(2)-MCM-41a sample, they may be explained as resulting from the interaction between oxide particles and the walls of support channels. It is evident that such an assignment cannot be unambiguous without additional data provided by other independent techniques. For instance, the signal at g ≈ 2.0 for the Fe(1)-MCM-41se sample with no oxide species might be attributed either to non-distorted octahedral coordination atoms in the oxide phase or to tetrahedrally coordinated atoms in the framework of MCM-41.33
The identity of the oxide phases as well as their size distribution for the samples with 6–11 wt% was determined by Mössbauer spectroscopy (see Fig. 5). The resonance parameters are summarized in Table 3. The sextets on the recorded spectra at RT are characteristic of low dispersion oxides29 though their phases can be hardly identified. Sample Fe(6)-MCM-41s contains 80% of low dispersion oxides, the share of γ-Fe2O3 being 30% only. Since the nanosized phase of α-Fe2O3 (ca. 21%) can be also seen, not all iron is incorporated in the framework of MCM-41. Moreover, it is located mainly out of the framework where it forms oxide particles of both low and high dispersion. The doublet with IS = 0.31 mm s−1 and QS = 1.76 mm s−1 can be supposedly attributed to the iron with tetrahedral coordination. However, the QS value allows us to attribute it to neither framework iron nor iron in magnetite since all iron atoms are in Fe(III) oxidation state.
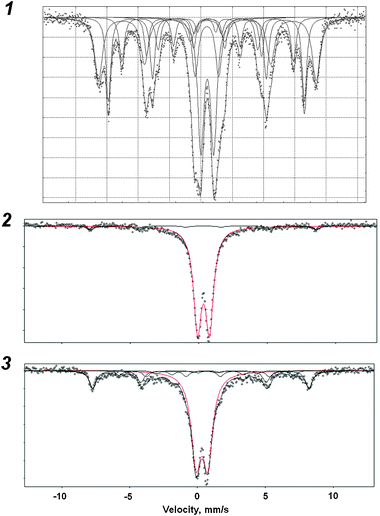 | ||
| Fig. 5 Mössbauer spectra for the Fe-MCM-41 catalysts with 6–11 wt% Fe. 1 – Fe(6)-MCM-41s; 2 – Fe(11)-MCM-41n; 3 – Fe(7)-MCM-41a. | ||
| Catalyst | Signal | Coord. | Attribution | IS a/mm s−1 | QSa/mm s−1 | Sa/% |
|---|---|---|---|---|---|---|
| a Signal parameters: IS – isomer shift, QS – quadrupole splitting, S – relative area. | ||||||
| Fe(6)-MCM-41s | Sextet | Oct. | Fe3+, γ-Fe2O3, >20 nm | 0.34 | −0.02 | 27 |
| Sextet | Tetr. | Fe3+ (1), >20 nm | 0.37 | −0.23 | 22 | |
| Sextet | Oct. | Fe3+ (2), >20 nm | 0.36 | −0.02 | 12 | |
| Sextet | Oct. | Fe3+ (3), >20 nm | 0.19 | −0.20 | 9 | |
| Doublet | Tetr. | Fe3+, >20 nm | 0.31 | 1.76 | 9 | |
| Doublet | Oct. | Fe3+, α-Fe2O3, <20 nm | 0.34 | 0.96 | 21 | |
| Fe(11)-MCM-41n | Doublet | Oct. | Fe3+, α-Fe2O3, <10 nm | 0.34 | 0.88 | 95 |
| Sextet | Oct. | Fe3+, >20 nm | 0.33 | — | 5 | |
| Fe(7)-MCM-41a | Doublet | Oct. | Fe3+, α-Fe2O3, <10 nm | 0.31 | 0.86 | 68 |
| Sextet | Oct. | Fe3+, γ-Fe2O3, >20 nm | 0.32 | −0.19 | 20 | |
| Sextet | Oct. | Fe3+ (1), >20 nm | 0.63 | −0.02 | 6 | |
| Sextet | Oct. | Fe3+ (2), >20 nm | 0.74 | — | 7 |
The impregnation of the MCM-41 support material with nitrate (n series) results in the specific formation of nanosized α-Fe2O3 only. These high dispersion particles are less than 10 nm in size, which was also reported in ref. 34.
In the case when an oxoacetate complex was used as an iron source, low dispersion oxides (30%) are detected. We expected the large organic ligands in the Fe3O(AC)6 molecule to prevent the aggregation of the precursor when drying. However, this did not occur and seemingly the [Fe3O] fragment in Fe3O(AC)6 played the role of a crystallization centre, which increased significantly the probability of aggregation. In fact, though the high dispersion α-Fe2O3 is predominant (70%) in the Fe(7)-MCM-41a sample, the rest of the iron is in low dispersion particles.
Some part of the iron oxide in the high dispersion state can interact with the channels of MCM-41 forming an [Fe–O–Si] species.30 This is the case for the Fe(11)-MCM-41n and Fe(7)-MCM-41a catalysts. Nevertheless, no signals from framework iron or analogous [Fe–O–Si] structures were detected in the Mössbauer spectra, maybe because of their low concentration.
The Mössbauer data are in close agreement with the H2-TPR results, which showed the presence of low dispersion oxides for both a and s series. These oxides were also revealed directly by XRD technique (Fig. 6). In the case of the Fe(11)-MCM-41n sample, all iron is contained in oxide particles sized less than 10 nm, i.e., below the coherent-scattering region.
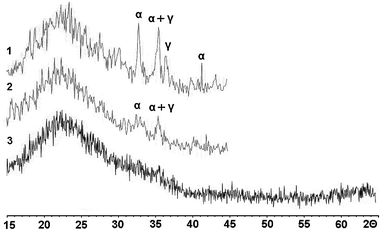 | ||
| Fig. 6 XRD patterns for the Fe-MCM-41 catalysts with 6–11 wt% Fe: 1 – Fe(6)-MCM-41s; 2 – Fe(7)-MCM-41a; 3 – Fe(11)-MCM-41n. Reflections of bulk oxides α-Fe2O3 and γ-Fe2O3 are marked with α and γ, respectively. | ||
3.2. Liquid phase oxidation of phenol at 25 °C
All the catalysts prepared exhibited a remarkable activity in the liquid phase oxidation of phenol by hydrogen peroxide. The experimental results are presented in Table 4. The turnover frequency values (TOF, min−1) were calculated for the phenol conversion level not exceeding 5%.| Catalyst | Conversion/% | CTa/% | HQ a/% | Conversion (%) | CT (%) | HQ (%) | TOF |
|---|---|---|---|---|---|---|---|
| a CT – catechol, HQ – hydroquinone. | |||||||
| Reaction time 15 min | Reaction time 2 h | ||||||
| Fe(2)MCM-41s | 10 | 0 | 0 | 40 | 10 | 2 | 7.9 |
| Fe(3)MCM-41s | 8 | 0 | 0 | 31 | 7 | 0 | 3.4 |
| Fe(6)MCM-41s | 7 | 0 | 0 | 30 | 6 | 0 | 1.7 |
| Reaction time 1.5 h | Reaction time 4 h | ||||||
| Fe(2)MCM-41n | 11 | 0 | 0 | 29 | 3 | 1 | 2.3 |
| Fe(6)-MCM-41n | 9 | 0 | 0 | 35 | 10 | 8 | 2.1 |
| Fe(11)-MCM-41n | 11 | 3 | 0 | 38 | 23 | 5 | 0.8 |
| Reaction time 1 h | Reaction time 4 h | ||||||
| Fe(2)MCM-41a | 12 | 5 | 0 | 48 | 21 | 6 | 4.3 |
| Fe(5)MCM-41a | 16 | 5 | 0 | 53 | 23 | 9 | 3.4 |
| Fe(7)MCM-41a | 16 | 1 | 0 | 48 | 27 | 11 | 1.7 |
As seen, the sample Fe(2)–MCM-41s, which contains both framework Fe and iron oxides has the highest TOF = 7.9 min−1. However, this catalyst showed fast deactivation, so that in the second run its activity was 5 times lower than for the fresh sample (Table 5). This loss in activity might be unambiguously explained by the partial leaching of iron during the first experiment.
| Catalyst | ΔTOF a/% | ΔMe b/% |
|---|---|---|
| a Activity in the second cycle as compared with that in the first one. b Iron leached after the first cycle. c Catalysts of a series did not show appreciable activity in the second cycle for 30 h. | ||
| Fe(2)-MCM-41s | 14 | 57 |
| Fe(3)-MCM-41s | 35 | — |
| Fe(6)-MCM-41s | 24 | 38 |
| Fe(2)-MCM-41n | 52 | 26 |
| Fe(6)-MCM-41n | 72 | 17 |
| Fe(11)-MCM-41n | >90 | 6 |
| Fe(2)-MCM-41a | 0c | 44 |
| Fe(5)-MCM-41a | 0 | 29 |
| Fe(7)-MCM-41a | 0 | 27 |
We suggest that oligomerization products being formed in the course of the reaction and blocking the pores of the MCM-41 support could account for this partial leaching of iron. On the other hand, adsorption measurements showed that the increase of iron content in the s series samples resulted in the partial amorphization of the support mesoporous structure. This process can lead to the formation of low active iron silicates or complex oxides, which are resistant to iron leaching even in strong acid media.
The n series catalysts are less active than those of the s series (Table 4). In this case the active phase is made of nanosized oxide particles, which are strongly stabilized by the porous network of the support and hence slowly dissolve to produce Fe3+ ions into reaction media. On increasing the iron content, the particles grow as was evidenced by adsorption measurements and TPR experiments. The decrease of activity is accompanied with the decrease of iron leaching (Table 5). We suggest that the oxides inside the channels of MCM-41 dissolve more slowly with increased particle size.
The results presented in Tables 4 and 5 reveal that the higher the amount of iron leached in the first cycle, the higher is the rate of oxidation in this cycle. This makes it possible to conclude that the catalytic process is performed by the Fe3+ ions transferred into the reaction media from the heterogeneous catalyst. This explanation is in good agreement with the fact that when more iron was leached in the first cycle the catalyst was less active in the second one.
The supported oxides in the a series catalysts contain particles of both low and high dispersion and are more active than those of the n series. With the increase of iron content they tend to decrease in both activity and iron leaching. These effects are undoubtedly connected with the increasing size of the particles inside the channels of MCM-41. In the second cycle all catalysts of the a series show nearly zero activity even at a run time of more than 30 h. Since the iron atoms are still present in the catalytic material (Table 5), this zero activity means that these remaining iron species do not dissolve in acidic media and, more so, are inactive at 25 °C. For instance, for the sample with 7.2 wt% of Fe these iron species might be clearly seen in Mössbauer spectra.
The kinetic curves of the phenol oxidation for samples of all series with 2–11 wt% of Fe are presented in Fig. 7. The pronounced induction period is a common feature for all kinetic experiments. If a process over a heterogeneous catalyst exhibits an induction period, it can be tentatively explained by active component leaching, i.e.Fe3+ ions in our case. However, this question provides controversy in the literature. Authors12,13 also observed an induction period but did not attribute it to the iron transfer from the catalyst to solution. On the contrary, in our study the appearance of Fe3+ ions in the homogeneous solution was strongly evidenced by the XFA technique. So far, the phenol oxidation kinetics confirm the suggestion that both framework Fe and supported oxides are inactive in this reaction at 25 °C.
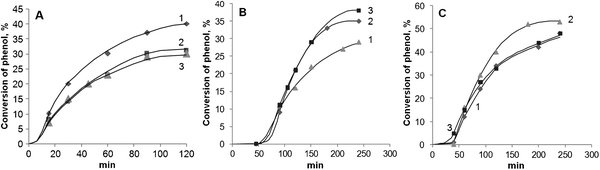 | ||
| Fig. 7 Kinetic curves of phenol oxidation at 25 °C over Fe-MCM-41 catalysts. (A) S series (1 – 2.0; 2 – 3.3; 3 – 6.3 wt% Fe); (B) n series (1 – 2.4; 2 – 5.6; 3 – 11.2 wt% of Fe); (C) a series (1 – 2.0; 2 – 4.5; 3 – 7.2 wt% of Fe). | ||
Since the phenol conversion does not exceed the accuracy of GC determination in the course of the induction period, one could suppose that the free Fe3+ ions in solution might be responsible for the catalytic process. However, their concentration is strongly limited because iron comes into the solution only from low dispersion oxides on the outer surface of the catalyst. Therefore, the drastic rise of the reaction rate is caused by the dissolution of nanosized oxides in the channels of MCM-41. For the s series catalysts the induction period is less pronounced since they predominantly contain low dispersion oxides on the outer surface, which easily dissolve at the very beginning of the reaction. On the contrary, the phenol oxidation over the n series and a samples containing nanosized oxides only (n series) or particles of both low and high dispersion (a series) are characterized by relatively long induction periods.
Correlating the oxide particle morphologies with the length of the induction period, we propose that the latter depends on the interaction between the oxide particle and the surface of the MCM-41 channel. High dispersion oxides in the n series materials are stabilized in the porous matrix of the MCM-41 support, so that they slowly dissolve with the longest induction period as a result. In contrast to the n series, a part of the oxides in the a series and s series samples is of low dispersion, so that they have weak bonds with the support. This results in their relatively fast dissolution, which leads to the shortened induction period.
The duration of the induction period can be considered as correlating with H2-TPR data (Fig. 2). The hydrogen uptake above 465 °C can be attributed to the reduction of low dispersion oxides, which cannot be completely reduced at low temperatures. This suggestion implies this uptake to increase with the increase of low dispersion oxides contribution to the total particle distribution. From the TPR data one can conclude that the share of low dispersion oxides increases as n < a < s, inverse to the duration of the induction period. This result is also confirmed by the Mössbauer data, which showed 80, 30 and <5% iron to be present as low dispersion oxides for s, a and n series with 6–11 wt% Fe, respectively.
Since the oxide particles undergo dissolution in an acid solution, this effect was investigated in a separate series of catalytic tests. The starting reaction mixture had a pH of 5 before adding the catalyst. With the Fe(2)-MCM-41s catalyst added, the solution became more acidic up to pH ∼2 in 2 h. To prove the influence of acidity on the iron leaching, we used an Fe(2)-MCM-41n sample containing small uniform oxide particles. In a buffer with pH 4 the oxidation of phenol started immediately without an induction period, though the leaching of iron was 84% in 4 h, which is 2 times higher than that under the standard conditions. Therefore, the media acidity drastically affects the iron leaching. It should be noted, however, that different explanations concerning this effect have been proposed in the literature. Authors of ref. 13 similarly related it to the acidity whereas the interaction between the organic substrates and the “heterogeneous” iron was suggested in ref. 17 to be the true reason.
To clarify whether the catalytic oxidation is performed by Fe3+ ions in homogeneous solution only, we carried out the catalytic test in the buffer at pH 7 over the Fe(2)-MCM-41s sample, which contains both oxides and framework iron. In this way, no oxides could be dissolved producing Fe3+ ions to solution. No phenol conversion was observed. This result directly evidences that at 25 °C the heterogeneous iron is not active in the oxidation of phenol whether it is incorporated in the wall of MCM-41 material as a framework Fe or it presents in an oxide form. Thus, at 25 °C the catalytic process is not heterogeneous and is realized via true homogeneous route since only Fe3+ ions are active.
3.3. Liquid phase oxidation of phenol at 80 °C
The common feature of all catalysts containing more than 2 wt% Fe is that the concentration of framework Fe is limited and also iron in the framework can be blocked by the oxide particles. To reveal specifically the activity of framework Fe we used two synthetic routes to prepare the samples containing the minimal possible amount of oxides so that to obtain predominantly iron in the framework of MCM-41.A simple increase of iron concentration in the synthetic gel does not enhance its introduction into the framework of MCM-41. The excess of iron salt remains in the channels of the support, then aggregates at drying and inevitably forms an oxide after calcination. At the same time, iron ions just introduced to the framework of freshly and uncalcined MCM-41material are stable enough. The calcination step usually applied to remove the template makes them leave the framework to form an oxide.6,16 The upper limit for the framework iron is usually estimated to be 1–2 wt%.7,35 Taking this into account, we synthesized two samples with 1 wt% Fe: (i) an Fe(1)-MCM-41se sample using an extraction procedure for template removal and (ii) an Fe(1)-MCM-41 sample using conventional thermal treatment.
The kinetic curves of the phenol oxidation over these two catalysts at 80 °C are shown in Fig. 8. The TOF values in the first cycle for both samples were estimated roughly because of the extremely high reaction rate. Such high values in both cases are undoubtedly caused by the considerable leaching of iron (about 70%) into the solution. Even though the Fe(1)-MCM-41s sample contains only 1 wt% Fe, this does not ensure the complete introduction of iron into the framework of MCM-41. Therefore, the iron atoms again leave the framework after calcination to form the oxides then dissolve and thereby release Fe3+ ions into the solution. Since the induction period is not distinctly pronounced, the accessible oxides on the outer surface seem to dissolve in this case. Their presence can be visually determined by the slight yellow coloring.
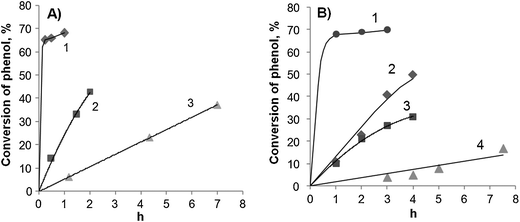 | ||
| Fig. 8 Kinetic curves of phenol oxidation at 80 °C over Fe(1)-MCM-41s (A) and Fe(1)-MCM-41se (B) catalysts. Curve number denotes the reaction cycle. Catalysts added: (A) 1 - 50; 2 - 33 and 3 - 23 mg; (B) 1 - 50; 2 - 36; 3 - 23 and 4 - 16 mg. | ||
In the case of the Fe(1)-MCM-41se sample, the loss of iron means that at 80 °C the framework Fe can also leach into the solution. Iron is well known to form stable complexes with phenol and catechol so that at higher temperatures this can also induce iron leaching, which takes place immediately at the beginning of reaction. It can be seen from the kinetic curves that they have no pronounced induction period. This observation allows us to conclude that not only the calcination step is responsible for further leaching of iron. The acidity of the reaction media seems to be an important co-factor of iron leaching regardless if is in oxides or in the framework of support.
Even though both catalysts suffer from iron leaching during the first oxidation run, they demonstrate rather low but substantially constant activity in the subsequent runs (Fig. 9). This finding enables us to conclude that it is a case of true heterogeneous catalysis as performed by the framework-hosted Fe only. This suggestion is strongly supported by the fact that the iron content in these catalysts remains also constant after each repeated run at the level of 0.25 wt%. The lower activity of the framework Fe's in comparison with that of free Fe3+ ions can be explained by their noticeably lower redox potential.36
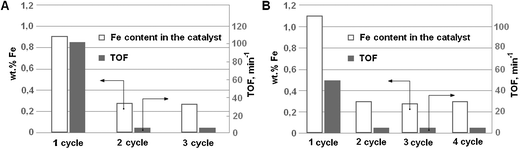 | ||
| Fig. 9 Activity and stability of (A) Fe(1)-MCM-41s and (B) Fe(1)-MCM-41se catalysts in phenol oxidation at 80 °C. | ||
Thus, the framework Fe in MCM-41 matrices is fairly stable if the iron content does not exceed 0.25 wt%. These iron species are stable not only under acidic conditions but also after the thermal treatment at 550 °C required to regenerate a used catalyst. The extraction procedure for template removal is by no means an alternative to the standard calcination step, providing low content of framework Fe in a synthesized material. This behavior of the framework Fe under liquid phase conditions has been also reported in ref. 13, except that 2.5 wt% of Fe was found to be stable and to remain in the framework.
The prepared materials are potential catalysts for the complete mineralization of phenol. To show this we used the Fe(1)-MCM-41s sample as a catalyst under the following conditions: 1 g of phenol, 6 mL of 25% H2O2, 100 mg of cat. (H2O2:phenol = 4.5:1.0 mol/mol, Fe:phenol = 1.7 × 10−3 mol/mol), 90 °C. The yield of CO2 determined by total carbon analysis was found to be 30% in 8 h, so that this material can be an effective catalyst for the phenol abatement from waste waters.
4. Conclusions
The interplay between the formation of nanosized Fe oxide particles and the inclusion of Fe as framework atoms was shown to be strongly influenced by the synthetic route. The in situ adding of an iron source at the stage of MCM-41 synthesis and the calcination of the as-synthesized material leads mainly to low dispersion oxides if the iron content exceeds 3 wt%. On loading no more than 0.25 wt% of Fe, the resulting material contains mostly framework iron, which is resistant to the leaching in both thermal treatment and acidic media. The incipient wetness impregnation of the MCM-41 support with nitrate affords nanosized particles of α-Fe2O3 even at an iron content as high as 11 wt%.The nature of the active iron species responsible for the liquid phase phenol oxidation was shown to be dependent on the reaction conditions. At 25 °C, free Fe3+ ions in solution are the only active species resulting from the partial dissolution of iron oxides. At 80 °C, the mechanism of true heterogeneous catalysis is operative, the framework Fe's being the active centers for oxidation.
Acknowledgements
This work was supported by the Russian Foundation for Basic Research, Project no. 08-03-00544, and the Federal Special Program, grant 2008-10-1.3-07-47. Authors thank Prof. P. B. Fabrichny for Mössbauer studies, Dr V. V. Yuschenko for TPR measurements, Dr A. V. Fionov for ESR investigations and Dr S. N. Nesterenko for XF analysis. S. V. Sirotin acknowledges also Haldor Topsøe A/S for financial support.References
- P. Ratnasamy and R. Kumar, Catal. Today, 1991, 9, 329–416 CrossRef.
- M. Stockenhuber, R. W. Joyner, J. M. Dixon, M. J. Hudson and G. Grubert, Microporous Mesoporous Mater., 2001, 44–45, 367–375 CrossRef CAS.
- C. Minchev, H. Huwe, T. Tsoncheva, M. Dimitrov, D. Paneva, I. Mitov and M. Fröba, Stud. Surf. Sci. Catal., 2004, 154, 841–847 CrossRef.
- A. A. Akhlebinina, J. F. Lamonier, I. F. Moskovskaya, L. M. Kustov, P. A. Chernavskii, A. Aboukaïs and B. V. Romanovsky, Russ. J. Phys. Chem. A, 2005, 79, 1946–1949 CAS.
- U. Ciesla, D. Demuth, R. Leon, P. Petroff, G. Stucky, K. Unger and F. Schüth, J. Chem. Soc., Chem. Commun., 1994, 1387–1388 RSC.
- W. A. Carvalho, M. Wallau and U. Schuchardt, J. Mol. Catal. A: Chem., 1999, 144, 91–99 CrossRef CAS.
- M. N. Timofeeva, M. S. Mel'gunov, O. A. Kholdeeva, M. E. Malyshev, A. N. Shmakov and V. B. Fenelonov, Appl. Catal., B, 2007, 75, 290–297 CrossRef CAS.
- R. Kumar, A. Raj, S. B. Kumar and P. Ratnasamy, Stud. Surf. Sci. Catal., 1994, 84, 109–116 CrossRef CAS.
- C. W. Lee, D. H. Ahn, B. Wang, J. S. Hwang and S.-E. Park, Microporous Mesoporous Mater., 2001, 44–45, 587–594 CrossRef CAS.
- J. Wang, J.-N. Park, X.-Y. Wei and C. W. Lee, Chem. Commun., 2003, 628–629 RSC.
- J.-N. Park, J. Wang, K. Y. Choi, W.-Y. Dong, S.-I. Hong and Ch. W. Lee, J. Mol. Catal. A: Chem., 2006, 247, 73–79 CrossRef CAS.
- Ch. Xiong, Q. Chen, W. Lu, H. Gao, W. Lu and Z. Gao, Catal. Lett., 2000, 69, 231–236 CrossRef CAS.
- J.-S. Choi, S.-S. Yoon, S.-H. Jang and W.-S. Ahn, Catal. Today, 2006, 111, 280–287 CrossRef CAS.
- D. Q. Khieu, D. T. Quang, T. D. Lam, N. H. Phu, J. H. Lee and J. S. Kim, J. Inclusion Phenom. Macrocyclic Chem., 2009, 65, 73–81 CrossRef CAS.
- T. Shishido, Q. Zhang, Y. Wang, T. Tanaka and K. Takehira, Phys. Scr., 2005, 115, 762–764 CrossRef.
- N. Gokulakrishnan, A. Pandurangan and P. K. Sinha, J. Chem. Technol. Biotechnol., 2007, 82, 25–32 CrossRef CAS.
- J. A. Melero, G. Calleja, F. Martínez, R. Molina and M. I. Pariente, Chem. Eng. J., 2007, 131, 245–256 CrossRef CAS.
- M. Grün, K. K. Unger, A. Matsumoto and K. Tsutsumi, Microporous Mesoporous Mater., 1999, 27, 207–216 CrossRef.
- Á. Szegedi, Z. Kónya, D. Méhn, E. Solymár, G. Pál-Borbély, Z. E. Horváth, L. P. Biró and I. Kiricsi, Appl. Catal., A, 2004, 272, 257–266 CrossRef.
- C.-Y. Chen, H.-X. L. and M. E. Davis, Microporous Mater., 1993, 2, 17–26 CrossRef CAS.
- S. Hitz and R. Prins, J. Catal., 1997, 168, 194–206 CrossRef CAS.
- H. Kosslick, G. Lischke, G. Walther, W. Storek, A. Martin and R. Fricke, Microporous Mater., 1997, 9, 13–33 CrossRef CAS.
- Ch. Wu, Y. Kong, F. Gao, Y. Wu, Y. Lu, J. Wang and L. Dong, Microporous Mesoporous Mater., 2008, 113, 163–170 CrossRef CAS.
- N. S. Nesterenko, O. A. Ponomoreva, V. V. Yuschenko, I. I. Ivanova, F. Testa, F. Di Renzo and F. Fajula, Appl. Catal., A, 2003, 254, 261–272 CrossRef CAS.
- S. K. Badamali, A. Sakthivel and P. Selvam, Catal. Lett., 2000, 65, 153–157 CrossRef CAS.
- Y. Okamoto, H. Kikuta, Y. Ohto, S. Nasu and O. Terasaki, Stud. Surf. Sci. Catal., 1997, 105, 2051–2058 CrossRef.
- A. A. Maerle, I. F. Moskovskaya, V. V. Yushchenko and B. V. Romanovskii, Russ. J. Phys. Chem., 2006, 80, 1004–1005 CrossRef CAS.
- P. Decyk, M. Trejda and M. Ziolek, C. R. Chim., 2005, 8, 635–654 CrossRef CAS.
- W. Kündig and H. Bömmel, Phys. Rev., 1966, 142, 327–333 CrossRef.
- P. Decyk, M. Trejda, M. Ziolek, J. Kujawa, K. Głaszczka, M. Bettahar, S. Monteverdi and M. Mercy, J. Catal., 2003, 219, 146–155 CrossRef CAS.
- H. Kosslick, G. Lischke, H. Landmesser, B. Parlitz, W. Storek and R. Fricke, J. Catal., 1998, 176, 102–114 CrossRef CAS.
- E. A. Zhilinskaya, G. Delahay, M. Mauvezin, B. Coq and A. Aboukaïs, Langmuir, 2003, 19, 3596–3602 CrossRef CAS.
- D. Coldfarb, M. Bemardo, K. C. Strohmaier, D. E. W. Vaughan and H. Tbomann, J. Am. Chem. Soc., 1994, 166, 6344–6453 CrossRef.
- A. Wingen, N. Anastasievič, A. Hollnagel, D. Werner and F. Schüth, J. Catal., 2000, 193, 248–254 CrossRef CAS.
- Y. Wang, Q. Zhang, T. Shishido and K. Takehira, J. Catal., 2002, 209, 186–196 CrossRef CAS.
- E. V. Kuznetsova, E. N. Savinov, L. A. Vostrikova and V. N. Parmon, Appl. Catal., B, 2004, 51, 165–170 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |