Catalytic performance of La1−xErxCoO3 perovskite for the deoxidization of coal bed methane and role of erbium in a catalyst
Received
20th April 2011
, Accepted 11th May 2011
First published on 23rd June 2011
Abstract
The effective utilization of coal bed methane (CBM) is very significant for energy utilization and environmental protection, reducing significantly greenhouse gas methane emission. In this paper, La1−xErxCoO3 perovskite (0 ≤ x ≤ 0.4) catalysts for the catalytic deoxidization of CBM were prepared by a co-precipitation method and were characterized by X-ray diffraction (XRD), laser Raman spectroscopy (LRS), H2 and CH4 temperature-programmed reduction (H2, CH4-TPR), CO pulse and N2adsorption/desorption techniques. The results show that the amount of Er affects obviously the physicochemical and catalytic properties of La1−xErxCoO3, and when x = 0.2, La0.8Er0.2CoO3 exhibits the best activity for deoxidization of CBM, because Er doping promotes the activity and migration of the lattice oxygen of La0.8Er0.2CoO3. The influences of the operation parameters (methane concentration, oxygen concentration and space velocity) on the catalytic deoxygenation of CBM and the kinetics behaviours were investigated. The reaction order of removing oxygen is 0.9 for methane partial pressure and −0.6 for oxygen partial pressure.
1. Introduction
Coal bed methane (CBM), also known as coal mine gas and coal seam gas, is a kind of flammable gas whose major component is methane.1 If CBM was not utilized and vented into the atmosphere, it not only would bring about air pollution, but also an energy resource would be wasted, because methane in CBM is 21 times more effective than carbon dioxide as a greenhouse gas.2 At a typical gassy mine, CBM is emitted in three streams: (1) gas drained from the seam before mining (60–95% methane), (2) gas drained from the worked areas of the mine, e.g. goafs3 (30–90% methane), usually mixed some amount of oxygen, and (3) methane ventilation air (0.1–1% methane).2,4 High concentration drained gas is easy to use; medium concentration CBM (30–80% methane), which mixed with some amount of oxygen, needs to be converted into methane-rich gas, containing at least 96 vol.% methane, and then it can be delivered to local natural gas supply systems. The pressure swing adsorption (PSA) method can be used in practice to recover methane from coal bed gas,5 in which both methane and oxygen in emissions are concentrated. However it is inevitable that the emissions still contain 5–15% of oxygen, within the limit of methane explosion. So it is necessary to remove oxygen of the medium concentration drained gas before using it. There are two main methods used in the CBM deoxygenation: non-catalytic and catalytic deoxygenation. There are several disadvantages of non-catalytic deoxygenation, such as not removing oxygen thoroughly, operation at high temperature, and producing some by-products. While there are several advantages of catalytic deoxygenation, such as, removing oxygen thoroughly, and operating at low temperature, the choice of catalyst is critical and some methane will be consumed. Comparing the characteristics of the two methods, we choose the latter method to catalytically remove oxygen from CBM. Catalytic deoxygenation is a method which uses methane catalytic combustion to remove oxygen.
Although methane catalytic combustion is a simple reaction, it is a strong exothermic reaction, which would induce temperature runaway of the catalyst bed, resulting in catalyst deactivation due to a catalyst sintering at high temperature. As an option to overcome the catalyst deactivation, lanthanum-transition metal perovskites (LaMO3) have attracted a lot of attention, because of their outstanding hydrothermal stability and high temperature stability.6
As a novel catalytic material, perovskite composite oxides (general formula ABO3) are being extensively researched, especially for the development of combustion catalysts and environmental purification catalysts.7Lanthanum cobaltite perovskite as an important one of the perovskite structures oxides attracts wide interest because of its excellent catalytic activity for the oxidation reactions,8–12 such as for the total oxidation of methane.11,12 Rare earth elements have some special chemical properties and are often used as a promoter in a variety of catalysts, to improve the performance of a catalyst. Doping Ce or Nd as an additive in LaCoO3 perovskite can vary its physicochemical properties and improve the catalytic activity.9,13 Although the catalytic oxidation of methane has been studied extensively, in almost all studies the methane concentration is very low, such as <0.1%, and the oxygen concentration is very high (>15%), in fact, this is a de-methane process. Being different with the catalytic de-methane process above, this paper is focused on the deoxidization of coal bed methane, in which methane concentration is very high and oxygen concentration is low. Therefore, according to the basic requirements for a deoxidization of coal bed methane, we have developed the Er-doped LaCoO3 perovskite catalyst. The research results reveal that the presence of Er can remarkably improve the activity of the LaCoO3 catalyst for the deoxidization of CBM. To the best of our knowledge, there is little research work for the catalytic deoxidization of CBM, especially for the catalytic deoxidization of CBM over the Er-doped LaCoO3 perovskite catalyst, which has not been reported.
Herein, we synthesized a series of perovskite-type La1−xErxCoO3 (x = 0.0–0.4) catalysts by a co-precipitation method and the catalysts were characterized by X-ray diffraction (XRD), laser Raman spectroscopy (LRS), H2 and CH4 temperature-programmed reduction (H2, CH4-TPR), CO pulse and N2adsorption/desorption techniques and so on. And taking La0.8Er0.2CoO3 as a model catalyst, the influences of the operational parameters, such as methane concentration, oxygen concentration and space velocity, on the catalytic deoxygenation of CBM and its kinetics behaviours were investigated.
2. Experimental
2.1.
Catalyst preparation
The La1−xErxCoO3 (x = 0.0, 0.1, 0.2, 0.3 and 0.4) catalysts were prepared by the co-precipitation method. La(NO3)3·6H2O and Co(NO3)2·6H2O salts were dissolved in de-ionized water (0.3 mol L−1La3+ and 0.3 mol L−1Co2+). Then ammonium carbonate (1 mol L−1) aqueous solution was dropped in the above solution under stirring at 60 °C until pH ∼9, and the precipitate was obtained and aged in this matrix solution for 4 h at 60 °C under vigorous stirring. After filtering and washing with de-ionized water, the solid product was dried at 120 °C for 12 h and then calcined at 800 °C for 4 h in air.
2.2.
Catalyst characterization
Powder X-ray diffraction patterns were performed on a Rigaku D/max 2250VB/PC diffractometer with Cu Kα radiation (λ = 1.540656 Å, scanning step 0.02°) at scanning rate 6° min−1. The BET specific surface areas of catalysts were measured on a NOVA 4200e Surface Area and pore size analyser by N2 adsorption at −196 °C and Brunauer–Emmett–Teller method. The Raman spectra at 100–1000 cm−1 were obtained on a Renishaw Raman spectrometer equipped with a CCD detector at ambient temperature and moisture-free conditions, and the emission line at 514.5 nm from an Ar+ ion laser was used for excitation and the output laser power was 10 mW.
H2-temperature-programmed reduction (H2-TPR) of the sample was carried out in a conventional flow system equipped with a thermal conductivity detector (TCD). 50 mg sample was loaded in the U-shaped quartz tube reactor. 5% H2/N2 mixture gas at a flow rate of 45 mL min−1 was used and the heating rate was 10 °C min−1.
CH4-TPR and the CO pulse experiments were conducted in the U-shape quartz tube reactor and the effluent gas was monitored by on-line quadrupole mass spectrometer (MS, IPC 400, INFICON Co. Ltd.). The catalyst was pre-treated at 650 °C for 0.5 h in 20% O2/He of 50 mL min−1, followed by cooling down to room temperature at the same atmosphere. In the testing of CH4-TPR, 0.5% CH4/He of 50 mL min−1 was used as the reactant gas, and the heating rate was 10 °C min−1. In the CO pulse experiment, the reaction temperature was 410 °C, and the carrier gas was He with a flow rate of 50 mL min−1. After the system reaches equilibrium, 35.62% CO/Ar was pulsed into the reactor system at intervals of 1 min with a loop volume of 1 mL.
2.3. Testing of catalytic activity
The catalytic activities of La1−xErxCoO3catalysts for the catalytic deoxidization of CBM were tested in a stainless steel tube fixed-bed reactor (500 mm × Φ 21mm/inner diameter) at atmospheric pressure. The feed gas was consisted of 30% CH4 + 6% O2 + Ar (balance) with 50 mL min−1, and 0.5 g catalyst diluted with 15 g silica sands (20–40 mesh) was used. The weight hourly space velocity (WHSV) was 6000 mL h−1 g−1). The reactants and products were analyzed online by a gas chromatograph (GC) equipped with TCD. The catalyst activity was expressed by T10, T50 and T90 representing the reaction temperatures for the oxygen conversion of 10%, 50% and 90%, respectively.
3. Results and discussion
3.1.
Catalyst characterization
The XRD patterns of La1−xErxCoO3 (x = 0–0.4) samples with different Er contents are shown in Fig. 1, and their BET surface areas and crystallite sizes are listed in Table 1. In the XRD pattern of LaCoO3, there are the typical diffraction peaks of rhombohedral LaCoO3 perovskite (JCPDS card: 48–0123), and a very weak diffraction peak of La2O3 at 2θ = 27–28° (JCPDS card: 54-0213). Replacing La3+ (ionic radius 1.06 Å) with Er3+ (0.88 Å) of a lower cation radius LaCoO3 would decrease the tolerance factor,14 resulting in the aberration of the perovskite structure and a combine of doublet peaks at around 33°, 41°, 59°, 70°, and 79°. Hence, the diffractograms of the substituted Er for La samples (La1−xErxCoO3) are similar to that of Ce, Cu, or Ni partial-substitution onto LaCoO3 perovskite.14,15 The intensities of the diffraction peaks decrease gradually with an increase in Er amount, which indicates that Er ions enters into the perovskite crystalline lattice and distort the crystalline structure. However, for the La1−xErxCoO3 (x = 0.3 and 0.4) samples a weak signal of Er2O3 at 2θ ≈ 29° (JCPDS card: 02–0930) can also be observed, that is to say, with an increase in Er amount some Er ions cannot enter into the lattices. The results in Table 1 show that, the presence of erbium in La1−xErxCoO3 increases its BET surface area and decreases the crystallite sizes. The results above indicate that there may bring lattice distortion and crystalline structure change of perovskite when part La3+ ions in LaCoO3 are substituted by smaller Er3+ ion.
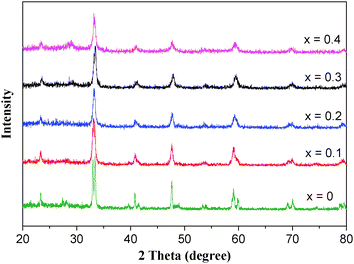 |
| | Fig. 1
XRD patterns of La1−xErxCoO3catalysts. | |
Table 1 BET surface areas (SA), crystallite sizes (d110) of La1−xErxCoO3catalysts and their T10, T50 and T90
| La1−xErxCoO3 |
T
10 (°C) |
T
50 (°C) |
T
90 (°C) |
d
110 (nm) |
SA (m2 g−1) |
|
x = 0 |
388 |
463 |
500 |
21.7 |
9.5 |
|
x = 0.1 |
454 |
418 |
440 |
18.7 |
13.4 |
|
x = 0.2 |
324 |
389 |
419 |
18.5 |
11.9 |
|
x = 0.3 |
337 |
412 |
446 |
18.8 |
14.1 |
|
x = 0.4 |
350 |
415 |
465 |
19.7 |
15.2 |
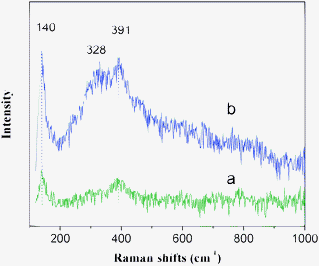
Comparing with X-ray diffraction technique, Raman spectroscopy is very sensitive to structure distortion and oxygen motion, and can be used to investigate an electron excitation for the perovskite-type structure materials. The Raman spectra of LaCoO3 and La0.8Er0.2CoO3 at room temperature are shown in Fig. 2, and there are two similar bands at ∼140 and 391 cm−1 with different intensities, which are similar to that reported by Popa.16 The bands intensities of La0.8Er0.2CoO3 are much larger than that of LaCoO3, due to disorder induced by erbium substitution for La and a distortion of the lattice structure. According to the data reported,16,17 we can assign the band at ∼391 and ∼140 cm−1 to Eg mode symmetry. Compared with the Raman spectrum of LaCoO3, there is a new band at ∼328 cm−1 in the Raman spectrum of La0.8Er0.2CoO3, which was not found in the Raman spectrum of LaCoO3 and further indicates that erbium substitution leads to structure distortion or oxygen motion of perovskite.
3.2.
H2-TPR
The H2-TPR profiles of La1−xErxCoO3catalysts are shown in Fig. 3, and there are two H2 consumption peaks at 100–800 °C, which correspond to two successive reduction steps. For the perovskite composite oxide, the first step is ascribed to the reduction of M3+ to M2+ at 100–500 °C,18,19 and the second step is attributed to the reduction of M2+ to metallic state M0 at 500–800 °C, resulting in a destruction of perovskite structures.20 Herein, the first step may be corresponding to the reduction of Co3+ to Co2+ at 300–460 °C and the second reduction step is to the reduction of Co2+ to Co0 at 550–750 °C with a destruction of the perovskite structure.21–23
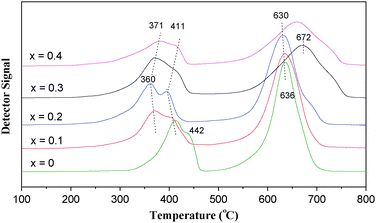 |
| | Fig. 3
H2-TPR profiles of La1−xErxCoO3catalysts. | |
After Er is add in La1−xErxCoO3 (x = 0.1–0.4), the reduction temperature of the first step declines obviously, and when x = 0.2 the reduction peak temperature for the La0.8Er0.2CoO3 sample was down by ∼50 °C compared with pure LaCoO3; for the high temperature reduction peak at 600–700 °C, La0.8Er0.2CoO3 is lowest (630 °C) among all samples. The results above show that Er-doped catalysts are reduced more easily by H2 than pure LaCoO3, that is to say, the lattice oxygen of Er-doped catalysts are more active than pure LaCoO3, and an appropriate doping amount of Er is x = ∼0.2.
3.3.
CH4-TPR
The CH4-TPR profiles of La1−xErxCoO3catalysts are shown in Fig. 4, and CO2 (m/z = 44) signal was detected by MS. During the CH4-TPR on La1−xErxCoO3catalysts, the most reactive oxygen species can be removed at low temperatures, and the less reactive oxygen species are only removed at higher temperatures, in which the deep oxidation product CO2 can be obtained at both stage temperatures.24
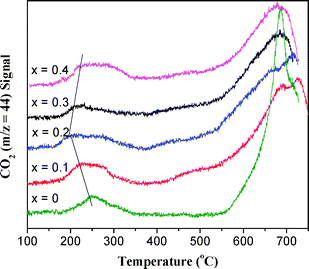 |
| | Fig. 4
CO2 curves in CH4-TPR on La1−xErxCoO3catalysts. | |
For the first reaction peak at 150–350 °C, the peak temperature (Tp) of CO2 desorption over LaCoO3 catalyst is 246 °C, and the presence of Er in LaCoO3 obviously lowers this peak temperature, for instance, Tp on La0.8Er0.2CoO3 is lowest (194 °C) and Tp on La0.6Er0.4CoO3 is 222 °C, which should be attributed to the reaction of methane with surface oxygen on the catalyst. At 370–530 °C, there is no CO2 peak on LaCoO3, while there are smaller CO2 peaks on the Er-doped samples. At >530 °C, the CO2 peak over Er-doped samples is wider than that on LaCoO3, which is attributed to the reaction of methane with bulk lattice oxygen of the catalyst. The results above indicate that La0.8Er0.2CoO3 has more active surface oxygen and higher amounts of reactive lattice oxygen than LaCoO3. This situation is similar to the results of H2-TPR.
3.4.
CO pulse experiment
The oxygen migrations in LaCoO3 and La0.8Er0.2CoO3catalysts were investigated by the CO pulse experiment, and the results (CO signal, m/z = 28) are shown in Fig. 5. It can be seen that the unreacted CO (m/z = 28) amount detected on LaCoO3 is much more than that on La0.8Er0.2CoO3, that is, more CO have been converted to CO2 on La0.8Er0.2CoO3. As the surface oxygen amount is less, pulsed CO is mainly converted by lattice oxygen from the sub-surface and bulk oxygen of the catalyst after the first pulse. The slope of the change in CO signals on LaCoO3 is milder than that on La0.8Er0.2CoO3. These different slopes may be induced by different amounts of the active lattice oxygen, and different reactivities and migration rates of lattice oxygen. The results in Fig. 5 show that the presence of Er in La0.8Er0.2CoO3 increases the amount of the active lattice oxygen and improves obviously the reactivity and migration rate of lattice oxygen in LaCoO3, which is consistent with the results in H2-TPR and CH4-TPR.
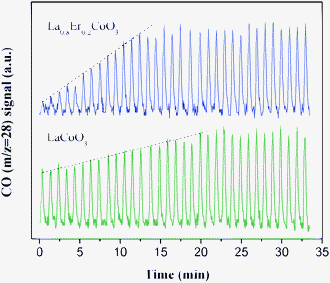 |
| | Fig. 5
CO pulse experiment at 410 °C on LaCoO3 and La0.8Er0.2CoO3. | |
3.5.
Catalytic performances of La1−xErxCoO3catalysts for deoxidization of CBM
The effect of Er content (x) on the catalytic performance of La1−xErxCoO3 for CBM deoxidization is shown in Fig. 6, in which CO2 and H2O are the only products. The results show that the activities of Er-doped catalysts are apparently superior to that of the LaCoO3 catalyst, and when x = 0.2 the La0.8Er0.2CoO3catalyst presents the maximum activity, for instance, T90 (the reaction temperature for 90% O2 conversion) over La0.8Er0.2CoO3 is ∼420 °C, and T90 over LaCoO3 is ∼500 °C.
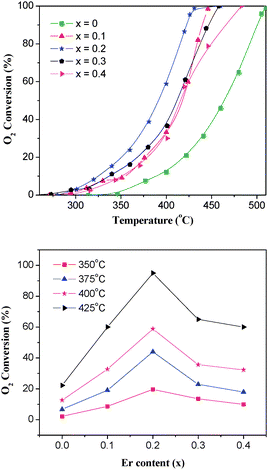 |
| | Fig. 6 Effect of Er content on the catalytic activity of La1−xErxCoO3 for the deoxidization of CBM. (30% CH4–6% O2/Ar balance, WHSV = 6000 mL h−1 g−1). | |
The effects of CH4 and O2 concentrations on the conversion of O2 over La0.8Er0.2CoO3 are shown in Fig. 7 and 8. As shown in Fig. 7, increasing methane concentration can increase the O2 conversion at the same space velocity; contrarily, increasing oxygen concentration decreases the O2 conversion under the same reaction conditions (Fig. 8).
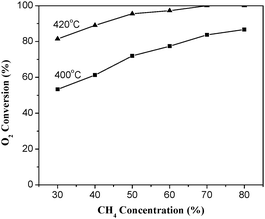 |
| | Fig. 7 Effect of CH4 concentration on the O2 conversion over La0.8Er0.2CoO3. (6% O2/Ar balance, WHSV = 6000 mL h−1 g−1). | |
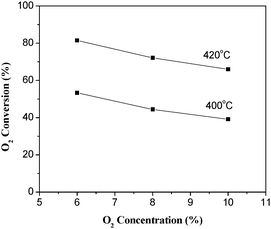 |
| | Fig. 8 Effect of O2 concentration on the O2 conversion over La0.8Er0.2CoO3. (30% CH4/Ar balance, WHSV = 6000 mL h−1 g−1). | |
Methane
combustion over the metal oxides catalysts or Pd supported on oxide catalysts is known to follow a redox mechanism, and a variety of kinetic models for the catalytic combustion of methane were reported,25–30 which is related to the reaction mechanisms of Rideal–Eley, or Langmuir–Hinshelwood bi-molecules, or Mars–van Krevelen, or purely empirical expression. In general, in studying the kinetic models for the catalytic combustion of methane, a low concentration methane and excess oxygen were adopted, which is as a model of VOCs (volatile organic compounds) purification, that is to say, it is a process of de-methane. Herein we focus on a deoxygenation of CBM, so that in the condition of high concentration and excess methane the kinetic behaviours for the catalytic combustion of methane over the La0.8Er0.2CoO3catalyst were investigated. Although the general equation for methane combustion is relatively complex, by the help of the empirical kinetic expression the rate equation of removing oxygen by the catalytic combustion of methane can be obtained as follows:
| | 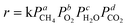 | (1) |
Based on the facts that CO
2 and H
2O are the products in the methane combustion, their concentrations in the feed gases were almost zero and the oxygen conversion at 320 °C is <13%,
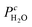
and
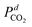
can be supposed to be approximately constant, while
eqn (1) can be simplified as:
| | 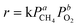 | (2) |
Taking the logarithm of
eqn (2),
eqn (3) can be obtained:
| | | ln r = a ln PCH4 + b ln PO2 + ln k | (3) |
In the conditions of 30–80% CH
4 and 6% O
2 at a space velocity (WHSV) of 6000 mL h
−1 g
−1) and 320 °C,
eqn (3) can be simplified to ln
r =
a ln
PCH4 +
C; ln
r against ln
PCH4 over the La
0.8Er
0.2CoO
3catalyst at 320 °C has been plotted in
Fig. 9. The slope of the straight line,
a is ∼0.9. In the conditions of 6–10% O
2 and 30% CH
4 at a space velocity (WHSV) of 6000 mL h
−1 g
−1) and 320 °C,
eqn (3) can be simplified to ln
r =
b ln
PO2 +
C; ln
r against ln
PO2 over the La
0.8Er
0.2CoO
3catalyst at 320 °C has been plotted in
Fig. 10. The slope of the straight line,
b is ∼−0.6. It is found that the order of removing O
2 for methane concentration is 0.9, in agreement with the first order found in literatures;
30–33 and the order for oxygen concentration is −0.6, unlike zero order for oxygen reported in the literature.
30,33 This can be explained that methane is adsorbed on oxygen vacancies on the surface. These vacancies can be occupied by molecular oxygen in a linear way of O
2(g) + * → O
2*, and then by atomic oxygen of O
2* + * → 2O*, resulting in the number of vacancies being dependent on the pressure
PO2 of the gas phase, that is, the reaction rate of removing O
2 is dependent on
PO2. As
PO2 pressure increases, the number of vacancies decreases and the order with respect to
PO2 is negative. This is the pseudo-Langmuir–Hinshelwood mechanism proposed by Iglesia
et al.34,35 It is interesting that based on the pseudo-Langmuir–Hinshelwood mechanism, the results calculated by a Monte Carlo simulation for the catalytic oxidation of methane shows that the order for oxygen is −0.62,
30 being the same as the order of oxygen here. It is understandable that, for the bi-molecules reaction followed Langmuir–Hinshelwood mechanism, the reaction would be subjected to limitation if one of the reactants adsorbed very strongly on the active sites competitively against another reactant, and hence we can deduce that an adsorption of methane should be the rate-determining step, which should be attributed to the catalytic combustion kinetics behaviours for high concentration methane (deoxygenation) being different from that for very low concentration methane (de-methane).
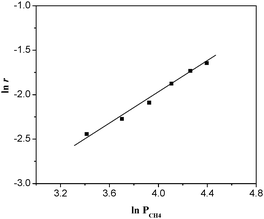 |
| | Fig. 9 ln r as a function of ln PCH4 over La0.8Er0.2CoO3 at 320 °C. (The feed gas consisted of 30–80% CH4 and 6% O2 at a space velocity (WHSV) of 6000 mL g−1 h−1), the conversion of O2 was adjusted to below 13%). | |
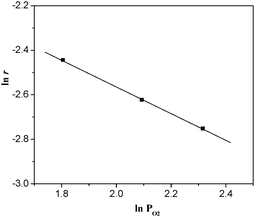 |
| | Fig. 10 ln r as a function of ln PO2 over La0.8Er0.2CoO3 at 320 °C. (The feed gas consisted of 6–10% O2 and 30% CH4 at a space velocity (WHSV) of 6000 mL h−1 g−1), the conversion of O2 was adjusted to below 6%). | |
The effect of space velocity on the O2 conversion over La0.8Er0.2CoO3catalyst is shown in Fig. 11. It indicates that, at the same reaction conditions a lower space velocity results in higher O2 conversion. This is because lower space velocity gives longer residence times of reactant gases in the catalyst bed.
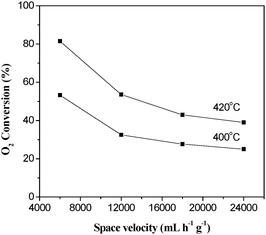 |
| | Fig. 11 Effect of space velocity on the O2 conversion over La0.8Er0.2CoO3. (30% CH4–6% O2/Ar balance). | |
4. Conclusions
For medium concentration drained gas (30–80% CH4) mixed with some oxygen, oxygen should be removed before using it to avoid an explosion. For the catalytic deoxygenation of CBM over perovskite catalysts, the effective La1−xErxCoO3 (x = 0–0.4) perovskite catalysts were prepared successfully. The results show that the partial substitution of La by Er modifies the rhombohedral structure of the LaCoO3 perovskite and promotes its activity of lattice oxygen, thereby, improving its reactivity for deoxidization of CBM. When x = 0.2, the La0.8Er0.2CoO3 perovskite catalyst possesses higher amounts of active lattice oxygen and higher migration capability of lattice oxygen, resulting in the highest activity for CBM deoxidization among all La1−xErxCoO3catalysts.
The influences of operation parameters, such as methane concentration, oxygen concentration and space velocity, on the activity of La0.8Er0.2CoO3catalyst for CBM deoxygenation were investigated. The results show that increasing methane concentration can increase the oxygen conversion, and increasing oxygen concentration conversely decreases the oxygen conversion. The study on the kinetics for CBM deoxygenation over La0.8Er0.2CoO3catalyst indicates that the reaction order of removing oxygen is 0.9 for methane partial pressure and −0.6 for oxygen partial pressure.
Acknowledgements
This project was supported financially by the National Basic Research Program of China (2010CB732300), and the “Shu Guang” Project (10GG23) and Leading Academic Discipline Project (J51503) of Shanghai Municipal Education Commission and Shanghai Education Development Foundation.
Notes and references
- G. J. Li, Procedia Earth and Planetary Science, 2009, 1, 94 Search PubMed.
- B. Stasiñska and A. Machocki, Pol. J. Chem. Technol., 2007, 9, 29 Search PubMed.
- Goaf (pl. Goafs or Goaves): That part of a mine from which the mineral has been partially or wholly removed; the waste left in old workings;—called also {gob}. [1913 Webster] http://www.theenglishdictionary.org/definition/goafs.
- S. Su and J. Agnew, Fuel, 2006, 85, 1201 CrossRef CAS.
- A. Olajossy, A. Gawdzlk, Z. Budner and J. Dula, Chem. Eng. Res. Des., 2003, 81, 474 Search PubMed.
- G. S. Gallego, J. G. Marín, C. B. Dupeyrat, J. Barrault and F. Mondragón, Appl. Catal., A, 2009, 369, 97 Search PubMed.
- D. S. Qiao, G. Z. Lu, X. H. Liu, Y. Guo, Y. Q. Wang and Y. L. Guo, J. Mater. Sci., 2011, 46, 3500 CrossRef CAS; L. L. Zhu, G. Z. Lu, Y. Q. Wang, Y. Guo and Y. L. Guo, Chin. J. Catal., 2010, 31, 1006 Search PubMed.
- N. A. Merino, B. P. Barbero, P. Grange and L. E. Cadús, J. Catal., 2005, 231, 232 CrossRef CAS.
- S. Khan, R. J. Oldman, S. A. French and S. A. Axon, J. Phys. Chem. C, 2008, 112, 12310 Search PubMed.
- J. Li, L. Zhao and G. Z. Lu, Ind. Eng. Chem. Res., 2009, 48, 641 Search PubMed.
- L. Fabbrini, I. Rossetti and L. Forni, Appl. Catal., B, 2006, 63, 131 Search PubMed.
- C. Q. Chen, W. Li, C. Y. Cao and W. G. Song, J. Mater. Chem., 2010, 20, 6968 RSC; I. Rossetti, C. Bif and L. Forni, Chem. Eng. J., 2010, 162, 768 Search PubMed.
- M. S. Cui, M. L. Li, S. L. Zhang, Z. Q. Long, D. L. Cui and X. W. Huang, J. Rare Earths, 2004, 22, 623 Search PubMed; H. Taguchi, S. Matsuoka, M. Kato and K. Hirota, J. Mater. Sci., 2009, 44, 5732 Search PubMed.
- M. Ghasdi, H. Alamdari, S. Royer and A. Adnot, Sens. Actuators, B, 2011 DOI:10.1016/j.snb.2011.04.003.
- B. Białobok, J. Trawczynski, W. Mista and M. Zawadzki, Appl. Catal., B, 2007, 72, 395 CrossRef CAS; N. Tien-Thaoa, H. Alamdarib and S. Kaliaguine, J. Solid State Chem., 2008, 181, 2006 Search PubMed; X. D. Yang, S. Li, Y. Liu, X. Z. Wei and Y. N. Liu, J. Power Sources, 2011, 196, 4992 Search PubMed.
- M. Popa, J. Frantti and M. Kakihana, Solid State Ionics, 2002, 154–155, 135 Search PubMed; M. Popa and J. M. C. Moreno, Thin Solid Films, 2009, 517, 1530 Search PubMed.
- E. Granado, N. O. Moreno, A. Garcia, J. A. Sanjurjo, C. Rettori, I. Torriani, S. B. Oseroff, J. J. Neumeier, K. J. McClellan, S. W. Cheong and Y. Tokura, Phys. Rev. B: Condens. Matter, 1998, 58, 11435 CrossRef CAS.
- B. Levasseur and S. Kaliaguine, Appl. Catal., B, 2009, 88, 305 Search PubMed; S. Royer, F. Berube and S. Kaliaguine, Appl. Catal., A, 2005, 282, 273 CrossRef.
- J. L. G. Fierro, M. A. Pena and L. G. Tejuca, J. Mater. Sci., 1988, 23, 1018 Search PubMed; A. Baiker, P. E. Martin, P. Keusch, E. Fritsch and A. Reller, J. Catal., 1994, 146, 268 CrossRef.
- M. Crespin and W. K. Hall, J. Catal., 1981, 69, 359 Search PubMed.
- Y. G. Wang, J. W. Ren, Y. Q. Wang, F. Y. Zhang, X. H. Liu, Y. Guo and G. Z. Lu, J. Phys. Chem. C, 2008, 112, 15293 CrossRef CAS.
- N. A. Merino, B. P. Barbero, C. Cellier, J. A. Gamboa and L. E. Cadus, Catal. Lett., 2007, 113, 130 Search PubMed.
- S. Royer, H. Alamdari, D. Duprezc and S. Kaliaguine, Appl. Catal., B, 2005, 58, 237 Search PubMed.
- V. A. Sadykov, T. G. Kuznetsova, G. M. Alikina, Y. V. Frolova, A. I. Lukashevich, Y. V. Potapova, V. S. Muzykantov, V. A. Rogov, V. V. Kriventsov, D. I. Kochubei, E. M. Moroz, D. I. Zyuzin, V. I. Zaikovskii, V. N. Kolomiichuk, E. A. Paukshtis, E. B. Burgina, V. V. Zyryanov, N. F. Uvarov, S. Neophytides and E. Kemnitz, Catal. Today, 2004, 93–95, 45 Search PubMed.
- V. C. Belessi, A. K. Ladavos and P. J. Pomonis, Appl. Catal., B, 2001, 31, 183 CrossRef CAS.
- V. C. Belessi, A. K. Ladavos, G. S. Armatas and P. J. Pomonis, Phys. Chem. Chem. Phys., 2001, 3, 3856 RSC.
- S. Specchia, F. Conti and V. Specchia, Ind. Eng. Chem. Res., 2010, 49, 11101 Search PubMed.
- N. Bahlawane, Appl. Catal., B, 2006, 67, 168 CrossRef CAS.
- T. N. Angelidis and V. Tzitzios, Stud. Surf. Sci. Catal., 1999, 122, 341 Search PubMed.
- J. Cortes, E. Valencia and P. Araya, Catal. Lett., 2006, 112, 121 Search PubMed.
- J. G. Deng, L. Zhang, H. X. Dai, H. He and C. T. Au, Appl. Catal., B, 2009, 89, 87 Search PubMed.
- M. V. Bukhtiyarova, A. S. Ivanova, E. M. Slavinskaya, L. M. Plyasova, V. A. Rogov, V. V. Kaichev and A. S. Noskov, Fuel, 2011, 90, 1245 Search PubMed.
- W. Sheng, D. N. Gao, C. X. Zhang, Z. S. Yuan, P. Zhang and S. D. Wang, Prog. Chem., 2008, 26, 789 Search PubMed.
- K. Fujimoto, F. H. Ribeiro, M. Avalos-Borja and E. Iglesia, J. Catal., 1998, 179, 431 CrossRef CAS.
- J. Au-Yeung, K. Chen, A. T. Bell and E. Iglesia, J. Catal., 1999, 188, 132 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.