Support effects on the structure and performance of ruthenium catalysts for the Fischer–Tropsch synthesis†
Juan María
González Carballo
a,
Elisabetta
Finocchio
b,
Sergio
García
a,
Sergio
Rojas
*a,
Manuel
Ojeda
a,
Guido
Busca
b and
José Luis García
Fierro
a
aGrupo de Energía y Química Sostenibles (EQS), Instituto de Catálisis y Petroleoquímica, CSIC, C/ Marie Curie 2, Cantoblanco, 28049 Madrid, Spain. E-mail: srojas@icp.csic.es; Fax: +34 91 585 4760
bDipartimento di Ingegneria Chimica e di Processo, Università di Genova, Fiera del Mare, Pad. D, I-16/129 Genova, Italy
First published on 5th July 2011
Abstract
The influence of support and metal precursor on Ru-based catalysts has been studied in the Fischer–Tropsch synthesis (FTS) combining flow reactor and quasi in situinfrared spectroscopy experiments. A series of supported ruthenium catalysts (3 wt.%) have been prepared using two different TiO2 (P25, 20% rutile and 80% anatase; Hombifine, 100% anatase) and SiO2·Al2O3 (28% Al2O3) as supports and RuCl3·nH2O as metal precursor. The catalysts were labeled as RuTi0.8, RuTi1 and RuSA respectively. Another catalyst (RuTi0.8N) has been synthesized with TiO2·P25 and Ru(NO)(NO3)3. After thermal treatments in air at 723 K and hydrogen at 443 K, ruthenium metal particles are agglomerated when pure anatase TiO2 and SiO2·Al2O3 are used as supports, leading to low active catalysts. In contrast, and despite the lower specific surface area of TiO2·P25 as compared to that of the other supports, well dispersed Ru particles are stabilized on titania P25. Remarkably, electronic microscopy studies demonstrate that Ru is deposited exclusively on the rutile phase of TiO2·P25. The catalytic performance shown by all these catalysts in FTS reactions follows the order: RuTi0.8 > RuTi0.8N > RuSA ≫ RuTi1. The same trend is observed during quasi in situ FTS experiments conducted in an infrared (IR) spectroscopy cell. The FTIR spectra of TiO2·P25 supported samples show that both samples behave similarly under the FTS reaction. This work shows that the structure of the support, rather than its specific surface area or the Ru precursor, is the parameter that determines the dispersion of Ru particles, hence their catalytic performance.
1. Introduction
Crude oil reserves are the main source of current transportation fuels, but environmental and political concerns require the exploration of other sources.1 The Fischer–Tropsch Synthesis (FTS) is a feasible and interesting process to produce sulfur, nitrogen and aromatics-free liquid fuels from syngas (H2/CO mixtures). The renewed interest in the FTS is related to the fact that this syngas can be obtained from renewable biomass. Iron- and cobalt-based catalysts are the archetypal and most studied systems for the FTS; however, ruthenium is known to be the most active metal for this reaction. Indeed, higher CO/H2 conversion levels, hydrocarbon productivities, and chain growth probabilities can be achieved when using Ru-based catalysts.1–3 This, in turn, results in a decreased cost of FTS fuels production, a specific requirement necessary for a wider implementation of the biomass-to-liquids (BTL) process.1 Furthermore, Ru catalysts can operate in the presence of high concentrations of water and other oxygenate-containing atmospheres, which is an important requisite to successfully convert biomass-derived syngas into hydrocarbons.4,5Previous studies have reported contradicting results concerning the influence of the support and metal particle size (dispersion) on the activity shown by supported ruthenium catalysts in the CO hydrogenation reaction. For instance, King6 reported a significant role of the support and the metal loading in the performance of ruthenium catalysts in FTS. In contrast, Vannice and Garten7 observed moderate modification of turnover frequencies and molecular weight product distribution with alumina- and silica-supported Ru catalysts, although a significant effect on titania was found,8 which was attributed to the development of strong metal support interactions.9 Tauster10 suggested that the increased activity (about one order of magnitude) with Ni/TiO2 and Pt/TiO2 catalysts was straightforward related to the formation of new active sites in the metal–TiOx contact perimeter.
Krishna and Bell11 investigated Ru/TiO2 catalysts and found that anatase-supported Ru clusters were more active than when deposited on rutile. The different performance was attributed, however, to the effect of chlorine impurities and different metal dispersion levels. Iglesia et al.12 showed that the catalytic performance of supported Ru in FTS is moderately affected by the support (silica, alumina, titania) or metal dispersion. Pérez-Zurita et al.13 studied the production of higher alcohols from syngas with Ru clusters deposited on different reducible supports, concluding that the catalytic performance depends on their reducibility. They found that the most active catalyst was obtained when Ru was supported on a non-reducible support, such as Al2O3. However, as the reducibility of the support increased, the selectivity to higher alcohols also increased.
Infrared spectroscopy (IR) is a powerful technique that may provide valuable information about the chemical nature, structure, and chemical bond strengths of adsorbed species during catalytic reactions.14 This technique has been previously used to determine different adsorbed species and mechanistic aspects in FTS with Fe15–17 and Co18,19catalysts. In the case of Ru catalysts, Ekerdt and Bell20 used IR spectroscopy to explore CO/H2 reactions with Ru/SiO2 catalysts and observed that neither CO pressure nor H2/CO ratio affected the position and intensity of the bands of CO adsorbed species. However, the intensity of this band decreased upon increasing the reaction temperature because of lower CO coverage. These authors assigned the IR bands at 2950 cm−1 to C–H stretching vibrations in methyl groups, whereas symmetric and asymmetric C–H vibrations in methylene groups were detected at 2910 and 2845 cm−1, respectively. They claimed that these species were not derived from reaction products adsorbed from the gas phase, but from reaction intermediates. Other authors have observed that, under FTS conditions, a minimum temperature or concentration of methylene groups on Ru/TiO2 is required to form methane. Moreover, these methylene moieties may be transformed into inactive hydrocarbon species upon thermal treatment or after significant chain growth.21 McQuire and Rochester22 detected different types of surface hydrocarbon on Ru/SiO2 catalysts, including ethoxy groups on silica originated via reaction of ethane with silanol groups, and methylene chains bonded to Ru atoms. They proposed that these hydrocarbons act as reaction intermediates in methane formation steps.
In order to clarify whether Ru–support interactions and metal precursor play a significant role on the structure and catalytic behavior of Ru clusters in FTS, IR measurements during CO adsorption and CO/H2 reactions at different temperatures were conducted. These investigations have been combined with catalytic studies with a laboratory-scale fixed bed reactor. Thus, a series of Ru-supported catalysts were prepared using different supports: TiO2 (anatase), TiO2 (anatase and rutile, P25), and SiO2·Al2O3. With the aim of studying the effect of the Ru precursor, two samples over TiO2·P25 were synthesized using RuCl3 and Ru(NO)(NO3)3. We have observed that after treating the catalytic precursors in air at 723 K, Ru clusters deposited on pure anatase or SiO2·Al2O3 do agglomerate, leading to low active FTS catalysts. In contrast, the rutile/anatase mixture (TiO2) anchors the ruthenium species more strongly and prevents, at least partially, ruthenium sintering. Consequently, more active catalysts are formed in this latter case. In addition, we have observed that Ru clusters size depends on the identity of the metal precursor, being smaller when Ru(NO)(NO3)3 is used. The IR spectra of adsorbed CO evidence a higher concentration of step-like defects as the mean Ru particles size decreases, which is a key factor to obtain active FTS catalysts.
2. Experimental
2.1. Preparation of catalysts
The supported Ru catalysts (3 wt.%) were prepared by the incipient wetness impregnation technique. TiO2 (Degussa P25, 20% rutile, 80% anatase), TiO2 (Hombifine, 100% anatase) and SiO2·Al2O3 (Davison Chemical, 28% Al2O3) were used as supports. RuCl3·nH2O (40.49%, Johnson Matthey) and Ru(NO)(NO3)3 (31.30%, Alfa Aesar) were used as ruthenium precursors.Experimentally, an aqueous solution of the desired amount of the ruthenium salt precursor was added to the support. The solid was then dried at room temperature overnight and thermally treated in air (calcination) at 723 K (10 K min−1) for 3 h. This temperature was selected from the thermogravimetric analysis recorded in air (not shown). The solids thus obtained were denoted as RuM, where M is Ti0.8 (TiO2 80% anatase), Ti1 (TiO2 100% anatase) or SA (SiO2·AlO3). The catalyst prepared from nitrosyl nitrate of ruthenium was designated as RuTi0.8N.
2.2. Characterization techniques
Nitrogen adsorption–desorption isotherms were recorded at 77 K with a Micromeritics ASAP 2000 apparatus. The samples were degassed at 413 K for 16 h prior to the determination of the adsorption–desorption isotherm.Temperature programmed reduction (TPR) experiments were carried out in a Micromeritics TPR/TPD 2900 equipment. Experimentally, 30 mg of sample (0.25–0.30 mm pellet size) were loaded into a U-shaped quartz reactor and then thermally treated under flowing He at 393 K for 30 min. The TPR profiles were recorded by heating the sample from room temperature to 1173 K at a rate of 10 K min−1 under a 10 vol.% H2/Ar flow. H2 consumption was monitored with a thermal conductivity detector (TCD).
H2 chemisorption experiments were performed in a conventional glass vacuum apparatus. About 50 mg of the catalytic precursor were placed in a quartz cell, degassed at 523 K for 30 min, and then reduced in H2 (133 mbar) for 1 h and evacuated at 543 K for 20 min. The chemisorption temperature was conducted at 373 K23 and the equilibration time was 30 min. After recording the first isotherm, the cell was outgassed for 15 min and a second isotherm was subsequently measured. The amount of irreversibly adsorbed hydrogen was determined by subtraction of these two isotherms. Ru dispersion (D) was calculated as follows assuming a Ru/H2 adsorption stoichiometry equals to 2:
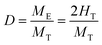 | (1) |
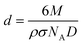 | (2) |
X-Ray photoelectron spectra (XPS) were acquired with a VG ESCALAB 200 R spectrometer in the pulse-count mode at a pass energy of 50 eV using a Mg Kα (hυ = 1253.6 eV) X-ray source. Kinetic energies of photoelectrons were measured using a hemispherical electron analyzer working in the constant pass energy mode. The background pressure in the analysis chamber was kept below 3 × 10−8 mbar during data acquisition. The powder samples were pressed into stainless steel holders and then mounted on a support rod placed in the pretreatment chamber. The XPS data were signal averaged for at least 200 scans and were taken in increments of 0.1 eV with dwell times of 50 ms. Since the binding energy of the C 1s core level (usually taken as reference) overlaps with the binding energy of the Ru 3d core level, binding energies were calibrated relative to the main lines of the supports;25Ti 2p peak at 458.6 eV in the case of TiO2 samples and Si 2p peak at 103.4 eV in the case of SiO2·Al2O3 sample to correct the contact potential differences between the sample and the spectrometer. High-resolution spectral envelopes were obtained by curve fitting using the XPS peak software.
A JEM-2100F 200 kV transmission electron microscope (JEOL Ltd.) equipped with an Oxford INCAx-Sight EDS detector (Oxford Instruments Ltd.) was used. High angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) images were obtained upon operating the microscope in scanning mode with an electron probe size of 1 nm and the signal was recorded with an annular dark field detector with an inner collection angle of 59 mrad (Z-contrast) and a maximum point resolution of 0.1 nm.
2.3. Quasi in situinfrared spectroscopy studies
Fourier Transform Infrared (FTIR) spectra were recorded with a Nicolet Nexus instrument, using conventional IR cells connected to a gas handling system, under static conditions. Pressed disks of pure catalysts and support powders (∼20 mg) were thermally treated within the IR cell at 673 K under vacuum (10−4 mbar). The catalysts were reduced in situ with pure H2 (800 mbar) at 673 K. The reducing process was conducted in two cycles of 30 min each followed by degassing at the same temperature. CO adsorption (8 mbar) experiments were performed at the temperature of liquid nitrogen, and spectra were recorded in the range 133–298 K while outgassing. In another set of experiments, CO was adsorbed (13 mbar) at room temperature and then evacuated at increasing temperatures. In the so-called quasi in situ FTS experiments, after reducing the sample, a mixture of H2/CO = 6 (365 mbar total pressure) was admitted into the IR cell. Spectra of the catalyst and the gas phase were recorded at 298, 473, 523, and 573 K.2.4. Catalytic experiments
Catalysts were tested in the CO hydrogenation reaction using a fixed-bed microreactor (300 mm long and 9 mm i.d.). The reactor temperature was measured with a K-type thermocouple directly placed in the catalytic bed. Flow rates were controlled with TOHO TTM 005 mass flow controllers. The catalytic bed consisted of 0.1 g of the catalyst precursor diluted with 2.0 g of quartz (0.25–0.30 mm pellet size) to avoid hot spots. The catalysts were subjected to a thermal treatment in situ under H2 at 443 K (10 K min−1). The temperature was afterwards raised to 523 K (10 K min−1) under an N2 atmosphere and immediately, the atmosphere was switched to H2/CO/N2 (62/31/7, GHSV = 1250 h−1) flow and the pressure was increased to 4.04 MPa. Analysis of reactant gases and products was performed on line with a gas chromatograph (Varian CP-3800) equipped with a cryogenic unit which allowed the gas chromatograph oven to be cooled down to 203 K. A Hayesep Q (2 m × 0.32 cm) column connected to a TCD detector was used to identify and quantify inorganic gases (H2, N2, CO and CO2). Hydrocarbon compounds were identified and quantified with an Rtx-1 column (60 m × 0.25 mm i.d. × 0.25 μm) connected to a flame ionization detector (FID). N2 was used as internal standard for chromatographic analyses.3. Results and discussion
3.1. Characterization
The supported Ru samples display N2 adsorption–desorption isotherms classified as type IV (not shown), typical of mesoporous materials.26 A moderate decrease of the specific surface area of Ti1 and SA supports is observed after Ru incorporation and subsequent calcination (Table 1). However, neither the impregnation with the Ru solution nor the calcination process affects the surface area of the titania P25 support (Ti0.8 samples, Table 1).| Sample | BET surface area/m2 g−1 | H2 chemisorption | ||
|---|---|---|---|---|
| Support | Catalyst | Dispersion (%) | Particle size/nm | |
| RuTi0.8 | 45 | 44 | 11 | 8.7 |
| RuTi0.8N | 45 | 44 | 14 | 7.0 |
| RuTi1 | 120 | 101 | — | — |
| RuSA | 340 | 317 | 1 | >50 |
The TPR-H2 profiles shown by the different supports and catalytic precursors are depicted in Fig. 1. Each sample presents a characteristic hydrogen consumption profile with maximum reduction rates appearing at different temperatures. The broad peaks observed in all cases might indicate the presence of ruthenium species with different oxidation states.27 Furthermore, the profile of the hydrogen consumption process depends also on the Ru precursor (RuTi0.8Nvs.RuTi0.8), suggesting that its identity may influence significantly the final Ru state.
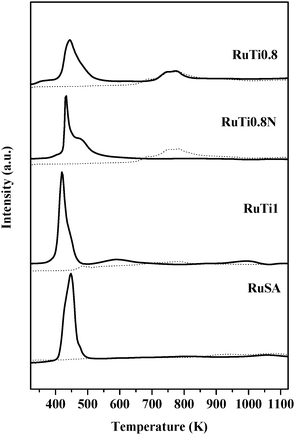 | ||
| Fig. 1 TPR-H2 profiles of the supports (dotted line) and catalytic precursors (solid line). | ||
Binding energy values and relative abundances of the different surface species after treatment in H2 at 443 K are shown in Table 2. The Ru 3d region exhibits Ru 3d5/2 peaks at 279.3–280.4 eV and 281.2–282.1 eV, attributed, respectively, to Ru0 and oxidized ruthenium species. The actual oxidation state of the latter species is not well defined, being widely admitted that several Ru species, such as RuOx, RuClx, RuO2·xH2O or RuOxHy, may coexist.28–32 The Ru/M atomic ratio values are also shown in Table 2. The three TiO2-based samples (RuTi1, RuTi0.8 and RuTi0.8N) display similar Ru/M values (0.07–0.08), while that for the RuSA sample is significantly higher (1.24).
| Sample | Region | B.E./eV | Species | M/supporta |
|---|---|---|---|---|
| a Ru or Cl or S surface atoms/Ti or (Si + Al) surface atoms from the corresponding support. Values in parenthesis show the relative concentration of Ru species. | ||||
| RuTi0.8 | Ru 3d5/2 | 279.3 (85) | Ru0 | 0.066 |
| 281.2 (15) | Ruδ+ | 0.012 | ||
| Ti 2p3/2 | 458.6 | TiO2 | ||
| Cl 2p3/2 | 198.0 | 0.030 | ||
| RuTi0.8N | Ru 3d5/2 | 280.1 (63) | Ru0 | 0.041 |
| 281.9 (37) | Ruδ+ | 0.024 | ||
| Ti 2p3/2 | 458.6 | TiO2 | ||
| Cl 2p3/2 | 198.3 | 0.014 | ||
| RuTi1 | Ru 3d5/2 | 280.4 (70) | Ru0 | 0.054 |
| 282.0 (30) | Ruδ+ | 0.024 | ||
| Ti 2p3/2 | 458.6 | TiO2 | ||
| Cl 2p3/2 | 199.7 | 0.014 | ||
| S 2p3/2 | 168.5 | SO42− | 0.013 | |
| RuSA | Ru 3d5/2 | 279.8 (88) | Ru0 | 1.090 |
| 282.1 (12) | Ruδ+ | 0.150 | ||
| Si 2p3/2 | 103.4 | SiO2 | ||
| Al 2p3/2 | 74.9 | Al2O3 | ||
| Cl 2p3/2 | — |
The XPS analyses reveal the presence of chlorine species on all titania-based samples. Remarkably, chlorine species are also detected in RuTi0.8N, even though this solid was, in principle, prepared with a Cl-free Ru precursor. Chen and Goodwin24 have also reported the presence of residual Cl species on commercial Ru(NO)(NO3)3 prepared from RuCl3. Furthermore, sulfur species were detected on the surface of RuTi1. These species may cause catalyst poisoning by avoiding the adsorption of the reactants and/or leading to the formation of catalytically inactive Ru–S species.33
Fig. 2 shows HAADF-STEM images of all H2-reduced samples. Clearly, Ru species are deposited as large aggregates on Ti1 (RuTi1, Fig. 2a) and particularly, in the case of RuSA (Fig. 2b), where large Ru islands (>50 nm) are deposited on some silica–alumina particles. HAADF-STEM images of the H2-reduced Ti0.8-based samples (Fig. 2c and d for RuTi0.8 and RuTi0.8N, respectively) show that TiO2 particles range from 10 to 60 nm. Since atomic numbers of Ru and Ti are very different (44 and 22, respectively), HAADF-STEM images allow identifying the Ru clusters as the bright spots on the border of the TiO2 particles.
 | ||
| Fig. 2 HAADF-STEM images of RuTi1 (a), RuSA (b), RuTi0.8 (c) and RuTi0.8N (d) samples previously treated in H2 at 443 K. | ||
This is because the contrast in the HAADF-STEM technique is a function of ∼Z2. Therefore, the brighter spots of the image correspond directly to areas of higher mean atomic number, provided that the thickness of these areas is identical.34,35
The Ru particles in RuTi0.8 and RuTi0.8N samples (arrowed in Fig. 2c and d) can be described as epitaxially grown particles of 1–2 nm thickness forming islands over the TiO2 surface. These islands cover areas from 1 × 1 nm to 20 × 20 nm. Substantial differences between the morphology of the Ru particles on RuTi0.8 and RuTi0.8N samples are not found.
A positive effect of anionic oxygen vacancies on rutile for avoiding the agglomeration of metallic particles has been previously reported.36 The appearance of O2− ions is related to the formation of Ti3+ species on the surface of rutile particles during anatase–rutile transformation, being the rutile particles enriched in these Ti3+ ions and oxygen vacancies.36–38 It is likely that the anionic oxygen vacancies on rutile particles may act as anchor sites for cationic species of ruthenium. It is also well documented that in water solution, RuCl3 species evolve with time to form RuClxδ+ cationic species.39–41 Similarly, Ru(NO)3+ cationic species can be stabilized from the nitrosyl precursor of Ru.41 On anatase (Ti1), the Ru-precursor species are poorly dispersed, and consequently, large Ru particles are formed after the thermal treatment step in H2. In contrast, the same species are highly dispersed on the rutile surface because of the interaction with O2− ions, thus forming stable metallic ruthenium particles.
Remarkably, a heterogeneous distribution of the Ru particles on Ti0.8 is found. As a matter of fact, Ru is exclusively deposited on the rutile phase of the TiO2·P25 substrate. This feature is clearly illustrated in Fig. 3. The squared area corresponds to a TiO2 particle in which the rutile and anatase phases are identified (R and A, respectively). It can be seen that the Ru particles are deposited exclusively on the rutile component of titania. The assignment of the TiO2 structure to either rutile or anatase phases is corroborated by indexing the digital diffraction patterns (DDP) obtained after applying the Fast Fourier Transform (FFT) to the high resolution images of the particles, which is equivalent to indexing the electron diffraction pattern42 (Fig. S1 in ESI†) or simply by the identification of the d-spacing of the {110} planes of rutile, d = 3.247 Å, or {101} planes of anatase, d = 3.520 Å43 (Fig. S1 in ESI†). The structure of more than 20 individual Ru/TiO2 particles was analyzed, in all of them the only TiO2 phase observed was the rutile one.
 | ||
| Fig. 3 TEM images of the RuTi0.8N sample treated at 773 K in H2. R: rutile; A: anatase. | ||
On the other hand, the migration process of TiO2 crystals over Ru particles has been studied. The strong metal–support interaction effect for group 8–10 metals on TiO2 is well documented.10 Such an interaction has been studied thoroughly by Bernal44 and explained in terms of structural changes and decorating microcrystals observed by high resolution-transmission electronic microscopy (HR-TEM) for Rh and Pt supported on CeO2. The interplanar spacings and angles observed in the digital diffraction pattern (DDP) of RuTi0.8N (Fig. S2 in ESI†) correspond to a particle with the rutile structure oriented along the [121] zone axis. The coherence between the crystal structure of the epitaxial Ru islands and the structure of TiO2 impedes the identification of the Ru particles by HR-TEM. Therefore, information about metal–support interactions was obtained by an energy dispersive X-ray spectroscopy (EDS) line scan.45
Fig. 4 shows the EDS profile recorded across one individual TiO2 particle with Ru particles on its surface. The profiles clearly show that Ru particles are deposited on the periphery of the TiO2 particles with no evidence of coverage of those Ru particles by TiO2 layers. Some small Ru islands are also identified. The absence of TiO2 on the outmost layer of the Ru particles indicates that no decoration process has occurred under our experimental conditions, i.e.hydrogen flow at 443 K.
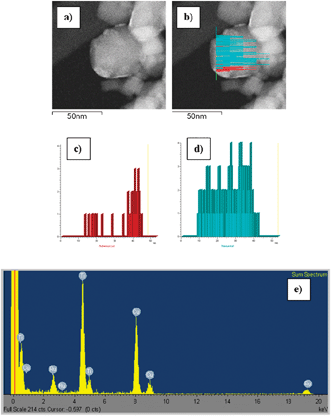 | ||
| Fig. 4 EDS line scans recorded for the RuNTi0.8N sample reduced at 443 K: (a) HAADF-STEM image, (b) HAADF-STEM image with the EDS intensity profile of Ru–Lα1 (red) and Ti–Kα1 (blue) energy lines overlapping, (c) Ru–Lα1 and (d) Ti–Kα1 profiles, (e) sum up spectra. | ||
3.2. FTIR characterization of the catalysts
Fig. 5a shows the FTIR spectra of RuSA after outgassing, as well as after reduction in hydrogen at 673 K and subsequent outgassing at the same temperature. The spectrum of the silica–alumina support (SA) recorded after outgassing at 673 K is also shown as a reference. The IR spectrum of the SA support is consistent with the literature.46 The IR spectrum of the outgassed RuSA sample shows an increased absorption baseline, with a significantly reduced absorbance of the band near 3745 cm−1 (detailed in the inset) associated to free surface silanol groups. The broad absorptions in the 2100–1500 cm−1 region, due to overtones of bulk vibrations, are also reduced. This observation would be in line with the exchange of Ru species with the protons of surface silanol groups. It is also possible that the overtones in the 2100–1500 cm−1 region have a predominant surface character and may be relaxed due to the presence of surface Ru species. Both these effects are attenuated after the reduction treatment, suggesting that Ru metal atoms coalesce into large particles, thus regenerating the surface hydroxyl groups and decreasing the Ru surface coverage. The sample reduced in H2 shows a further increased absorbance baseline, possibly because of the continuous absorption of Ru metal particles. This behavior is consistent with our previous characterization data (STEM and H2 chemisorption) showing large ruthenium clusters in RuSA, see also XPS results.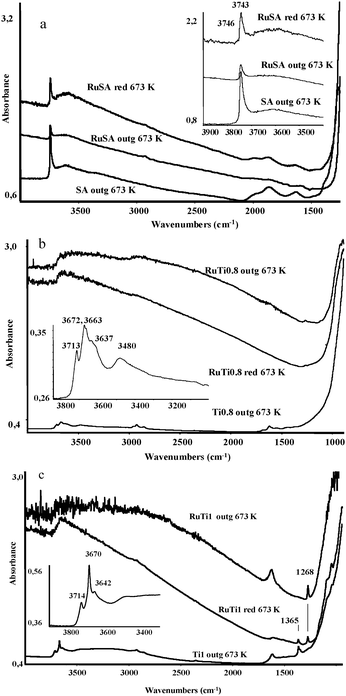 | ||
| Fig. 5 (a) FTIR spectra of silica–alumina (SA) and RuSA catalyst disks after outgassing and reduction. Inset: OH stretching region. (b) FTIR spectra of TiO2 (Ti0.8) and RuTi0.8 catalyst disks after outgassing and reduction. Inset: OH stretching region. (c) FTIR spectra of TiO2 (Ti1) and RuTi1 catalyst disks after outgassing and reduction. Inset: OH stretching region. | ||
The spectra of TiO2 and Ru–TiO2 samples are shown in Fig. 5b for Ti0.8 and Fig. 5c for Ti1. Irrespective of the support used, a pronounced increase of the absorbance is observed in the spectra of the outgassed RuTi1 and RuTi0.8 samples. Upon thermal treatment in hydrogen at 673 K, spectra with decreasing absorbance are recorded. Altogether, these facts point to a strong electronic interaction between Ru species and bulk TiO2. It is interesting to remark that RuO2, which displays high conductivity, is stable in the rutile structure, probably because both phases share the same crystal structure (see discussion below). Furthermore, RuxTi1−xO2rutile-type solid solutions may be formed, which also display high conductivity and light absorption properties.47 Our data suggest that after impregnation, a strong interaction occurs between the bulk titania and the supported Ru oxide phase, which is responsible for the increased absorption of the IR radiation. After reduction, the Ru centers would coalesce into metal particles, thus decreasing the electronic interaction with titania.48 In fact, our microscopy study (see above) shows no evidence of any decoration of the Ru particles by TiO2 layers after thermal treatment in hydrogen.
The region of the FTIR spectra corresponding to the surface hydroxyl groups of the two titania powders (insets in Fig. 5b and c) are akin to those reported in the literature for similar systems.49,50 The addition of ruthenium causes the disappearance of the sharp bands of the surface hydroxyl groups, which are now apparent as a broad absorption in the 3700–3500 cm−1 region. This provides evidence of the perturbation of the surface by ruthenium oxide species, which persists after reduction in hydrogen.
It is important to remark the presence of a sharp band at 1365 cm−1 in the spectrum of Ti1, which is characteristic of surface sulfate species, a typical contaminant in some anatase preparations.49 This band seems to evolve into another sharp one centered at 1268 cm−1 after ruthenium addition, which we tentatively assign to ruthenium sulfate species.
The catalysts were further characterized by FTIR after CO adsorption at low temperature, which allows CO to interact with both metallic Ru sites and support. Moreover, the oxidation of the ruthenium particles by CO is impeded at low temperatures.51 The spectra of CO adsorbed (COad) at low temperature on reduced RuSA after different evacuation temperatures are shown in Fig. 6. The high frequency bands at 2228 and 2157 cm−1 (strong) are due to CO adsorbed on the support and disappear almost completely after outgassing. The most intense band due to the CO stretching mode of Ru-carbonyls appears at 2035 cm−1 with a shoulder at 1995 cm−1 (Fig. 6, inset). Upon outgassing, the main band is shifted to 2014 cm−1. Other broad and less intense bands appear at 1885 and 1845 cm−1. Similar bands have been reported after CO adsorption on reduced Ru/alumina, Ru/ZSM5 zeolite and Ru/silica catalysts. These bands are associated to weakly adsorbed species, more likely bridging carbonyl species,58 which disappear after outgassing at room temperature. On the other hand, the band centered at 1995 cm−1 is associated to a second, more labile, on-top carbonyl species.
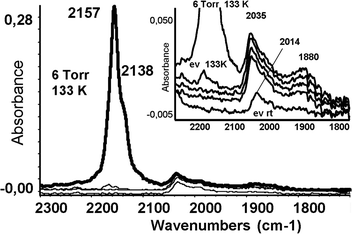 | ||
| Fig. 6 FTIR spectra of CO adsorbed on reduced RuSA at 133 K and upon warming under outgassing up to 293 K. Inset: enlargement of the metal-carbonyl region. | ||
CO species adsorbed on Ru particles are more stable than CO adsorbed on the support, as revealed by the evolution of the COad bands with the outgassing temperature shown in Fig. 6. This is a typical behavior of on-top carbonyls on extended metal particles, due to vibrational coupling effects.59
Fig. 7a shows the spectra of CO adsorbed at low temperature on the reduced RuTi0.8 sample. The sharp strong band observed at 2178 cm−1 is assigned to CO adsorbed on Ti4+ sites.49 Upon outgassing, the position of the bands shifts to 2193 cm−1, and it eventually appears at 2207 cm−1. Another component at 2156 cm−1 is due to CO weakly interacting with the support through H-bonds. The broader band at 2033 cm−1 reveals CO species linearly coordinated to metallic Ru particles. This band shifts to a lower frequency (2012 cm−1; Δν = 21 cm−1) with decreasing the CO coverage. The spectrum of CO adsorbed on RuTi0.8N follows a similar pattern to that recorded for RuTi0.8, as shown in Fig. 7b. Nonetheless, certain differences are appreciable. In this sample, the features of CO adsorbed on the strongest anatase Ti4+ sites at 2206 cm−1 are very weak, if any. The presence of bands accounting for CO adsorbed on anatase is in good agreement with the preferential deposition of Ru on the rutile phase. It also suggests that the amount of anatase sites exposed on the surface of RuTi0.8 is higher than on RuTi0.8N. Additionally, the broad band assigned to on-top carbonyls adsorbed on Ru metallic particles is recorded between 2010–1990 cm−1; a significantly lower frequency compared to that on RuTi0.8.
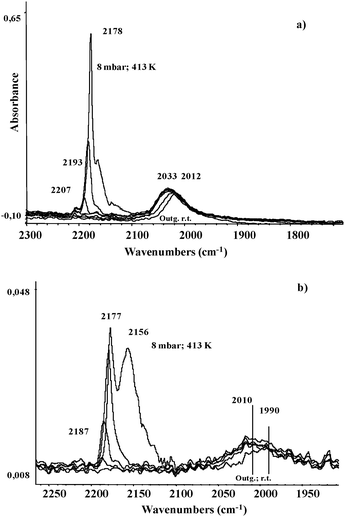 | ||
| Fig. 7 FTIR spectra of CO adsorbed on reduced RuTi0.8 (a) and RuTi0.8N (b) upon warming under outgassing up to 293 K. | ||
The FTIR spectra recorded after room temperature CO adsorption on the reduced Ru catalysts show similar bands to those recorded after low temperature CO adsorption (see Fig. S3 in the ESI†). The most intense band of COad species on metallic Ru appears at similar frequencies than those reported for the low temperature experiments. However, two main differences are observed: (i) the spectra lack COad bands on the support, and (ii) a further set of less intense bands can be detected at the higher frequency side of the most intense COad band. These bands account for CO adsorbed on oxidized Ru species, which are formed during CO adsorption at room temperature.14 Accordingly, these species are not recorded for RuTi1, where the dispersion of ruthenium is very low.
3.3. FTS studies by quasi in situFTIR
The performance of the Ru catalysts in CO hydrogenation (FTS) has been followed by quasi in situFTIR experiments. First, samples are reduced in situ under a H2 atmosphere at 673 K. Then, a CO/H2 mixture (365 mbar total pressure) is admitted into the FTIR cell at the desired reaction temperature for 5 min. Three reaction temperatures for the CO hydrogenation reaction are studied, 473, 523 and 573 K; the FTIR spectra were recorded at room temperature.RuTi0.8 and RuTi0.8N samples display very similar FTIR spectra during FTS at different temperatures (Fig. 8). The most intense COad band appears at 2027 cm−1, which shifts to lower frequency and its intensity decreases with the increasing reaction temperature. The next most intense band appears in the range 1930–1910 cm−1, and their intensity is found to be independent of the reaction temperature. Additionally, two weaker components are also observed at 2132 and 2068 cm−1 and assigned to CO adsorbed on oxidized Ru species (see above).
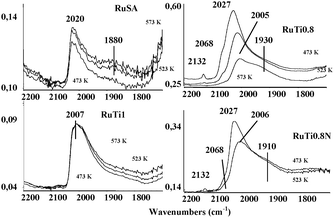 | ||
| Fig. 8 FTIR spectra of CO adsorbed on the four reduced catalysts after contact with CO + H2 at different temperatures. | ||
Again, the shape of the COad bands recorded on RuSA and RuTi1 is similar, although different to those observed for RuTi0.8 and RuTi0.8N. The main COad band in RuSA and RuTi1 appears at 2020 cm−1 and 2007 cm−1, respectively; only faint components at the lower and higher frequency to the main COad band are recorded.
Fig. 9 compares the spectra of the adsorbed species formed during the CO hydrogenation reaction at 523 K with the most active catalyst, RuTi0.8, and the less active catalyst, RuTi1. For comparison, both spectra have been normalized to show the same intensity of the COad band at ca. 2007 cm−1. It is evident that on the RuTi0.8 catalyst, strong bands due to C–H stretching of polymethylene chains at 2924 and 2853 cm−1 are formed, consistent with its higher activity in the FTS (Table 3). On the almost inactive RuTi1 catalyst, such bands are very weak, and actually are only observed after spectrum magnification. In both cases, other C–H stretching modes appear, evidencing the presence of other CHx species. The analysis of the IR spectrum of the gas phase after CO hydrogenation on RuTi0.8 (see inset in Fig. 9) reveals a number of additional bands due to the presence of methane, CO2, water, ethylene (H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) wagging mode at 949 cm−1), propene (H2C wagging mode at 912 cm−1) and butenes (H2C wagging mode 889 at cm−1). The appearance of these species confirms that even under these mild conditions of pressure and temperature, this catalyst shows a detectable FTS activity by FTIR and that this technique is suitable to follow both the reaction products and the evolution of Ru centers during FTS. The results obtained with RuTi0.8N (Fig. 4 in ESI†) are similar to those obtained with RuTi0.8, in good agreement with their comparable FTS activity recorded in a fixed bed reactor under more realistic FTS conditions.
wagging mode at 949 cm−1), propene (H2C wagging mode at 912 cm−1) and butenes (H2C wagging mode 889 at cm−1). The appearance of these species confirms that even under these mild conditions of pressure and temperature, this catalyst shows a detectable FTS activity by FTIR and that this technique is suitable to follow both the reaction products and the evolution of Ru centers during FTS. The results obtained with RuTi0.8N (Fig. 4 in ESI†) are similar to those obtained with RuTi0.8, in good agreement with their comparable FTS activity recorded in a fixed bed reactor under more realistic FTS conditions.
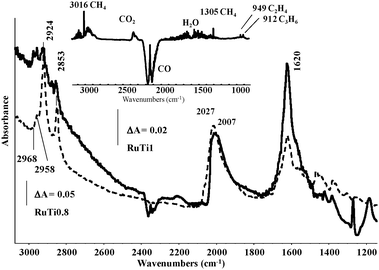 | ||
| Fig. 9 FTIR spectra of the adsorbed species formed on the active catalyst RuTi0.8 and on the inactive catalyst RuTi1 after contact with CO + H2 at 523 K for 5 min. Inset: gas phase species detected in the same conditions over the RuTi0.8 catalyst. | ||
3.4. Catalytic activity
All supported Ru catalysts display a high initial activity (not shown), reaching the steady-state after 10–12 h on stream. Table 3 summarizes the catalytic performance of Ru-based samples in FTS at CO conversion of 36%. The CO conversion rate follows the order: RuTi0.8 ≈ RuTi0.8N > RuSA ≫ RuTi1.The identity of the support is of extreme importance to obtain active Ru-based catalysts. Those prepared with titania P25 (RuTi0.8 and RuTi0.8N) show the best FTS performance in terms of CO conversion rate and selectivity to long chain hydrocarbons, as deduced from the chain growth probability values (α) followed by RuSA. In contrast, RuTi1 is inactive under typical FTS reaction conditions.
We note that RuTi0.8 shows a similar reaction rate compared to RuTi0.8N, even though smaller Ru clusters are found in this latter case. In contrast, reaction selectivity to the different hydrocarbons and olefins/paraffins distribution is not affected by the identity of the Ru precursor.
4. Discussion
H2 chemisorption and electron microscopy experiments show that Ru dispersion, or particle size, varies with the support. In principle, solids with high specific surface area should lead to higher metallic dispersion when used as support; however, the dispersion of the Ru particles derived from H2 chemisorption analysis (see Table 1) records the highest value for the catalysts prepared on TiO2·P25, the support showing the lower specific surface area value in the series; 45 m2 g−1vs. 120 m2 g−1 and 340 m2 g−1 for Ti1 and SiO2·Al2O3, respectively. The particle size of Ru calculated from H2 chemisorption data is 8.7 nm for RuTi0.8 and 7.0 nm for RuTi0.8N. HAADF-STEM images show larger Ru clusters on RuTi1, and especially on RuSA, consistent with H2 chemisorption data. These results indicate that the effects of both the specific surface area of the support and that of the Ru precursor on the particle size is negligible as compared to the effect of the structure of the support itself (see below). The XPS results are in good agreement with the aforementioned tendencies, except for RuSA, where the surface atomic abundance of Ru is higher than for the other catalysts (Table 2). In principle, the high Ru/support atomic ratio derived from the XPS analysis of RuSA would suggest that this sample consists of highly dispersed Ru particles. However, H2 chemisorption data point otherwise, a very low dispersion of ∼1% (Table 1). The microscopy images of RuSA show that this sample consists of very large Ru particles deposited on some silica–alumina particles. This explains the low Ru dispersion value found by H2 chemisorption. Moreover, since Ru islands actually cover the whole surface of the support particles, those would not be detected by a surface sensitive technique such as XPS, hence recording a surface enriched in Ru atoms.The dispersion of the Ru particles is improved on Ti0.8 irrespectively of the Ru precursor used to prepare the catalysts, consistent with previous studies for other metals. Thus, Jongsomjit et al.60 have recently reported that the highest degree of metallic Co dispersion is achieved when TiO2 (anatase/rutile ratio of 81/19) is used as a support. These authors claimed that this effect is the result of the higher reducibility of the cobalt species deposited on the rutile phase.
STEM images of RuTi0.8N clearly show (Fig. 3) that the preferred anchor site for the Ru particles is the rutile phase of Ti0.8. In line with our results, the preferential location of IrO2 on the rutile phase of TiO2·P25 has been previously reported.61,62 It should be recalled that both rutile and anatase phases coexist in Ti0.8 and according to XRD calculations, rutile accounts for ca. 20% of the crystalline particles in P25.42 The preferential deposition of Ru on the rutile phase can be explained by taking into account that both RuO2 and rutile crystallize in the same space groupP42/mnm, whereas anatase space group is I41/amd, see Scheme 1. That is, rutile can accommodate the Ru oxide particles within its structure. This also explains the epitaxial growth of the Ru particles on Ti0.8 as discussed above. This feature explains the predominant role of the structure of the support in the dispersion of the Ru particles as compared to other features such as specific surface area of the support or the nature of the Ru precursor.
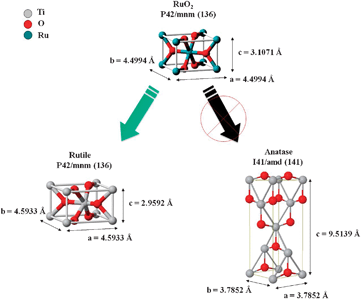 | ||
| Scheme 1 Cartoon type illustration on how RuO2 can accommodate on the crystal structure of the rutile phase. | ||
After thermal treatment in hydrogen, the Ir particles referred above become re-distributed homogeneously on both the rutile and anatase phase of TiO2·P25.62 This is not the case, however, for our Ru samples, where the preferential location of Ru on the rutile crystals is maintained even after thermal treatment in hydrogen (Fig. 3b). The high stability of the Ru particles towards thermal treatment in H2 could be explained by the formation of RuxTi1−xO2rutile type solid solutions. As a matter of fact, the high absorbance recorded for the degassed spectrum of RuTi0.8 (Fig. 5b) could be indicative of the presence of such species.
The FTIR spectra recorded after either low temperature or room temperature CO adsorption show a single COad broad band, as expected for Ru-based samples. As summarized recently by Payne et al.,63CO adsorption on Ru(0001) single crystal surfaces gives rise essentially to a single IRAS and EELS band at any coverage shifting from about 1980 cm−1 to about 2060 cm−1 with the increasing CO loading from zero coverage to saturation, due to static and dynamic coupling effects. This band is due to on-top CO species forming the (√3 × √3)R30° pattern evident in LEED experiments. A similar situation was found on some stepped surfaces such as Ru(109), where terminal carbonyls absorb at 2063 cm−1 at the highest coverage, and at 1973 cm−1 at the lowest coverage, where CO has been also found to dissociate producing Ru–C and Ru–O species at the step sites and recombine near 530 K.58 On the Ru(11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 0) face, terminal carbonyls are observed to shift from 1937 to 2058 cm−1 by increasing coverage. Another COad species characterized by unusually low stretching frequency (1558 cm−1) together with an unusually high deformation frequency (694 cm−1) has been identified as the precursor of CO dissociation.64 This species is supposed to be tilted, bonded to a kind of fourfold hollow site. On Ru(10
0) face, terminal carbonyls are observed to shift from 1937 to 2058 cm−1 by increasing coverage. Another COad species characterized by unusually low stretching frequency (1558 cm−1) together with an unusually high deformation frequency (694 cm−1) has been identified as the precursor of CO dissociation.64 This species is supposed to be tilted, bonded to a kind of fourfold hollow site. On Ru(10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0), terminal carbonyls near 2000 cm−1 were found together with a second species absorbing at 1810 cm−1.65
0), terminal carbonyls near 2000 cm−1 were found together with a second species absorbing at 1810 cm−1.65
The position of the COad band recorded at high CO coverage for the Ru-supported catalysts in the present study ranges between 2039 and 2007 cm−1, which is a significantly lower value than that observed on Ru single crystal faces (typically near 2060 cm−1). This shifting to lower frequencies suggests that the surfaces of the supported Ru metal particles are more electron rich than that of the extended Ru crystal faces, hence being able to back-donate more electrons into the π* orbital of CO, thus recording a lower νCO frequency. This behavior is usually associated to the small crystal size of the Ru particles in the supported catalyst and/or to the co-presence of adsorbed hydrogen species in our experiments. It is not possible, however, to establish a direct correlation between the COad band position and the size of the Ru particles on the support. This is because the presence of ions such as Cl− and/or sulfate species (as detected by XPS) can affect the electron donating ability of the Ru particles, masking thus pure size effects.
Concerning the support effects, the FTIR spectra shown in Fig. 6 and 7 illustrate that COad species on the support are detected only after low temperature CO adsorption. After outgassing, these COad species disappear and only those ascribed to COad on Ru remain. COad species on the support were not detected in the high temperature experiments.
The FTIR spectra after FTS reaction at 473 K in the IR cell (Fig. 8) show broad COad bands centered at 2007 cm−1 (RuTi1), 2020 cm−1 (RuSA), and 2027 cm−1 (RuTi0.8, and RuTi0.8N). Noticeably, the position of these bands shifts to lower frequency values with the increasing reaction temperature, except for RuTi1 and RuSA. Furthermore, the intensity of this band also decreases with the reaction temperature with the exception of RuTi1. On the other hand, the catalytic experiments conducted in a fixed bed reactor show that both RuTi0.8 and RuTi0.8N render the most active catalysts in FTS, whereas RuTi1 is almost inactive for the synthesis of hydrocarbons. While the FTIR spectra of COad on the Ti0.8-based catalysts show certain differences (not shown) as discussed above, identical spectra are recorded for both samples during quasi in situ FTS experiments at every reaction temperature. In addition to the main COad band at 2027 cm−1, COad bands at 2132 and 2068 cm−1, along with a broad absorption ∼1900 cm−1 are found. The latter band is assigned to νCO of bridging carbonyls, which are usually not found on Ru single crystal surfaces. The COad bands in the high frequency region are usually ascribed to COad on partially oxidized Ru species. Over an oxidized Ru(0001) surface, the C–O stretching tends to increase with respect to the clean surface. According to Jakob and Schiffer,66 the observed frequency for the Ru(0001)–(√3 × √3)R30° CO pattern is 2030.8 cm−1, shifting to 2050.7 cm−1 for the Ru(0001)–(2 × 2)–(CO–O) mode, and to 2090.3 cm−1 for the Ru(0001)–(2 × 2)–(CO + 2O) mode. Instead, for the Ru(0001)–(2 × 2)–(2CO + O) mode, the frequency of terminal carbonyls is observed at 2068.6 cm−1, while a second species considered to be triply bridging CO on fcc-type sites is observed at 1849.7 cm−1. Two groups reported the adsorption and reaction of CO on the RuO2(110) single crystal surface. Using HREELS, Wang et al.67 found that CO adsorbs on-top on coordinatively unsaturated Ru species absorbing at 2115 cm−1, while after reduction of the surface by CO (forming CO2), new bands appeared at 1975 and 1895 cm−1, associated to species adsorbed on centers similar to reduced ruthenium. Using IRAS, Farkas et al.68 found on the stoichiometric RuO2(110) surface CO adsorbed on-top shifting from 2110 to 2123 cm−1 by increasing coverage, with an additional weaker feature at 2001 cm−1 attributed to asymmetrically bridging CO. On the mildly reduced RuO2(110) surface, the main band shifts from 2016 to 2086 cm−1, with the weaker feature at 2001–1994 cm−1. A symmetrically bridging species is also observed in some conditions at 1860–1880 cm−1. Both groups also showed several complex effects associated to the different oxygen coverage of the surface during oxidation or reduction treatments.69,70 Indeed, multiple bands in the region 2150–2080 cm−1 have been frequently observed on supported Ru catalysts and attributed to polycarbonyls on incompletely reduced ruthenium.51–57
Interestingly, the shape and intensity of the COad band at ∼1900 cm−1 remains invariable during the FTS reaction. In contrast, the bands at the higher frequency values disappear with the increasing FTS temperature. This is because the partially oxidized Ru species are reduced within the reducing atmosphere of the FT reaction.
On the other hand, strong bands of polymethylene chains are observed on the spectra of RuTi0.8 and RuTi0.8N. Faint bands of those species are observed in the spectra of RuTi1 or RuSA. This result is in good agreement with the actual FTS performance of the catalysts in the fixed bed reactor. In addition, a strong broad band is observed at ca. 1620 cm−1 due to water molecules produced during FTS. This broad band would overlap with those of CO tilted species, proposed as intermediate species in the FT process.
Early FTS literature suggested that larger Ru particles yield more active catalysts. It is also proposed that the surface carbon species formed by hydrogen-assisted CO dissociation participate in the FT synthesis reaction and as precursor for the species that causes catalyst deactivation.11,71,72 Recent theoretical studies suggest, however, that the CO dissociation ability is favored on couples of atoms on monoatomic steps being the key requirement for Ru to develop a good FTS catalyst,73,74 followed by hydrogenation and coupling of CHx fragments75 or CO insertion.74 Experimental studies have shown that water presents a positive effect on the reaction rate and selectivity, in part due to its ability to remove surface carbon atoms,4 and in part because Ru particles around 8 nm perform better than smaller particles for FTS.76
This study shows that Ru particles are accommodated and stabilized on the rutile phase of TiO2 showing a size between 7–8 nm. Both Ti0.8 supported catalysts show higher activity for the FTS reaction than those supported on Ti1 and SA, due to the smaller size of the Ru clusters. Also, the catalytic performance of RuTi1 is affected negatively by the formation of Ru-sulfate like species, which are known to be inactive for the FT synthesis reaction. Quasi in situ FTS studies by FTIR demonstrate that both RuTi0.8 and RuTi0.8N yield similar Ru species within the reaction atmosphere, hence their comparable performance in FTS.
5. Conclusions
The dispersion of Ru particles is dominated by the structure of the support rather than by features such as metal precursor or specific surface area of the support. Remarkably, Ru deposition occurs preferentially on the rutile phase of TiO2·P25, rendering particles of ca. 7–8 nm which are stable after successive thermal treatments in air and hydrogen. This behavior has been ascribed to the similar crystal phase of rutile and Ru which favors the epitaxial growth of the Ru particles, impeding their agglomeration. Furthermore, irrespectively of the Ru precursor, when supported on TiO2·P25 Ru is an active FTS catalyst. On the other hand, when supported on pure anatase or SiO2·Al2O3, the catalytic performance drops dramatically. The performance of the catalysts in FTS follows the order: RuTi0.8 > RuTi0.8N > RuSA ≫ RuTi1. The same trend was observed during the quasi in situ FTS experiments recorded in an IR cell at 473–573 K and 667 mbar. The formation of surface polymethylene chains is evident on the Ti0.8 catalyst. Methane, ethylene and propylene were detected in the spectra of the gas phase. The FTIR spectra of CO adsorbed on the different Ru catalysts, characteristic of each sample, are mostly determined by the Ru size and the presence of other ions. However, after quasi in situ FTS, both RuTi0.8 and RuTi0.8N display similar spectra, in line with their catalytic performance in FTS.Acknowledgements
J. M. González-Carballo acknowledges the Ministerio de Educación of Spain through the Formación de Profesorado Universitario program (FPU) for financial support. The authors also acknowledge projects ENE2007-67533-C02-02/ALT from Ministerio de Ciencia e Innovación and Project S2009ENE-1743 from Comunidad de Madrid. Programa de Actividades de I + D entre Grupos de Investigación en Tecnologías for funding this work.Notes and references
- C. N. Hamelink, A. P. C. Faaij and H. den Uil, Energy, 2004, 29, 1743–1771 CrossRef CAS
.
- J. P. Hindermann, G. J. Hutchings and A. Kiennemann, Catal. Rev., 1993, 35, 1–127 CrossRef CAS
- G. P. van der Laan and A. A. C. M. Beenackers, Catal. Rev. Sci. Eng., 1999, 41, 255–318 CrossRef
- M. Claeys and M. van Steen, Catal. Today, 2002, 71, 419–427 CrossRef CAS
- J. Kang, S. Zhang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2009, 48, 2565–2568 CrossRef CAS
- D. L. King, J. Catal., 1978, 51, 386–397 CrossRef CAS
- M. A. Vannice and R. L. Garten, J. Catal., 1980, 63, 255–260 CrossRef
- E. Kikuchi, M. Matsumoto, T. Takahashi, A. Machino and Y. Morita, Appl. Catal., 1984, 10, 251–260 Search PubMed
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389–394 CrossRef CAS
- K. R. Krishna and A. T. Bell, J. Catal., 1991, 130, 597–610 CrossRef CAS
- E. Iglesia, S. L. Soled and R. A. Fiato, J. Catal., 1992, 137, 212–224 CrossRef CAS
- M. J. Pérez-Zurita, M. Dufour, Y. Halluin, A. Griboval, L. Leclercq, G. Leclercq, M. Goldwasser, M. L. Cubeiro and G. Bond, Appl. Catal., A, 2004, 274, 295–301 CrossRef
- K. I. Hadjiivanov and G. N. Vayssilov, Adv. Catal., 2002, 47, 307–511 CAS
- G. Bian, A. Oonuki, Y. Kobayashi, N. Koizumi and M. Yamada, Appl. Catal., A, 2001, 219, 13–24 CrossRef CAS
- E. Boellaard, A. M. van der Kraan and J. W. Geus, Appl. Catal., A, 1996, 147, 229–245 CrossRef CAS
- M. Jiang, N. Koizumi and M. Yamada, Appl. Catal., A, 2000, 204, 49–58 CrossRef CAS
- V. Sanchez-Escribano, M. A. Larrubia Vargas, E. Finocchio and G. Busca, Appl. Catal., A, 2007, 316, 68–74 CrossRef CAS
- C. G. Visconti, L. Lietti, E. Tronconi, P. Forzatti, R. Zennaro and E. Finocchio, Appl. Catal., A, 2009, 355, 61–68 CrossRef CAS
- J. G. Ekerdt and A. T. Bell, J. Catal., 1978, 58, 170–187
- N. M. Gupta, V. S. Kamble, R. M. Iyer, K. Ravindranathan Thampi and M. Gratzel, J. Catal., 1992, 137, 473–486 CrossRef CAS
- M. W. McQuire and C. H. Rochester, J. Catal., 1993, 141, 355–367 CrossRef CAS
- J. Okal, M. Zawadzki, L. Kepinski, L. Krajczyk and W. Tylus, Appl. Catal., A, 2007, 319, 202–209 CrossRef CAS
- B. Chen and J. G. Goodwin, Jr., J. Catal., 1996, 158, 228–235 CrossRef CAS
- C. Elmasides, D. I. Kondarides, W. Grünert and X. E. Verykios, J. Phys. Chem. B, 1999, 103, 5227–5239 CrossRef CAS
- K. S. W. Sing, K. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS
- L. Li, L. Qu, J. Cheng, J. Li and Z. Hao, Appl. Catal., B, 2009, 88, 224–231 CrossRef CAS
- M. G. Cattania, F. Parmigiani and V. Ragaini, Surf. Sci., 1989, 211–212, 1097–1105 CrossRef
- H. Y. H. Chan, G. Takoudis Ch and M. J. Weaver, J. Catal., 1997, 172, 336–345 CrossRef CAS
- J. L. Gómez de la Fuente, M. V. Martínez-Huerta, S. Rojas, P. Hernández-Fernández, P. Terreros, J. L. G. Fierro and M. A. Peña, Appl. Catal., B, 2009, 88, 505–514 CrossRef CAS
- K. S. Kim and N. Winograd, J. Catal., 1974, 35, 66–72 CrossRef CAS
- D. R. Rolison, P. L. Hagans, K. E. Swider and J. W. Long, Langmuir, 1999, 15, 774–779 CrossRef
- C. H. Bartholomew, P. K. Agrawal and J. R. Katzer, Adv. Catal., 1982, 31, 135–242 CAS
- E. M. James, N. D. Browning, A. W. Nicholls, M. Kawasaki, Y. Xin and S. Stemmer, J. Electron Microsc., 1998, 47, 561–574 CAS
- J. Liu, J. Electron Microsc. Tech., 2005, 54, 251–278 Search PubMed
- A. Nobile, Jr. and M. W. Davis, Jr., J. Catal., 1989, 116, 383–398 CrossRef
- C. Di Valentin, G. Pacchioni and A. Selloni, J. Phys. Chem. C, 2009, 113, 20543–20552 CrossRef CAS
- T. Ohno, K. Sarukawa, K. Tokieda and M. Matsumura, J. Catal., 2001, 203, 82–86 CrossRef CAS
- H. H. Cady and R. E. Connick, J. Am. Chem. Soc., 1958, 80, 2646–2652 CrossRef CAS
- R. E. Connick and D. A. Fine, J. Am. Chem. Soc., 1960, 82, 4187–4191 CrossRef CAS
- J. A. Rard, Chem. Rev., 1985, 85, 1–39 CrossRef CAS
- R. I. Bickley, T. González-Carreno, J. S. Lees, L. Palmisano and R. J. D. Tilley, J. Solid State Chem., 1991, 92, 178–190 CrossRef CAS
- V. Puddu, H. Choi, D. D. Dionysiou and G. L. Puma, Appl. Catal., B, 2010, 94, 211–218 CrossRef CAS
- S. Bernal, J. J. Calvino, M. A. Cauqui, J. M. Gatica, C. Larese, J. A. Pérez Omil and J. M. Pintado, Catal. Today, 1999, 50, 175–206 CrossRef CAS
- A. A. Herzing, M. Watanabe, J. K. Edwards, M. Conte, Z. Tang, G. J. Hutchings and C. J. Kiely, Faraday Discuss., 2008, 138, 337–351 RSC
- M. Bevilacqua, T. Montanari, E. Finocchio and G. Busca, Catal. Today, 2006, 116, 132–142 CrossRef CAS
- S. Saito, K. Okano, T. Hayqashi and Y. Nakahashi, J. Ceram. Soc. Jpn., 1992, 100, 663–667 CAS
- H. Lin, S. Kumon, H. Kozuka and T. Yoko, Thin Solid Films, 1998, 315, 266–272 CrossRef CAS
- G. Busca, H. Saussey, O. Saur, J. C. Lavalley and V. Lorenzelli, Appl. Catal., 1985, 14, 245–260 Search PubMed
- J. A. Toledo-Antonio, M. A. Cortés-Jácome, J. Navarrete, C. Angeles-Chavez, E. López-Salinas and A. Rendon-Rivera, Catal. Today, 2010, 155, 247–254 CrossRef CAS
- K. Hadjiivanov, J.-C. Lavalley, J. Lamotte, F. Maugé, J. Saint-Just and M. Che, J. Catal., 1998, 176, 415–425 CrossRef CAS
- S. Y. Chin, C. T. Williams and M. D. Amiridis, J. Phys. Chem. B, 2006, 110, 871–882 CrossRef CAS
- C. Crisafulli, S. Scirè, S. Minicò, R. Maggiore and S. Galvagno, Appl. Surf. Sci., 1996, 99, 401–409 CrossRef CAS
- O. Dulaurent, M. Nawdali, A. Bourane and D. Bianchi, Appl. Catal., A, 2000, 201, 271–279 CrossRef CAS
- C. Elmasides, D. I. Kondarides, W. Grunert and X. E. Verikios, J. Phys. Chem. B, 1999, 103, 5227–5239 CrossRef CAS
- A. Maroto-Valiente, M. Cerro-Alarcón, A. Guerrero-Ruiz and I. Rodríguez-Ramos, Appl. Catal., A, 2005, 283, 23–32 CrossRef CAS
- G. H. Yokomizo, C. Louis and A. T. Bell, J. Catal., 1989, 120, 1–14 CrossRef CAS
- T. Zubkov, G. A. Morgan, Jr., J. T. Yates, Jr., O. Kühlert, M. Lisowski, R. Schillinger, D. Fick and H. J. Jänsch, Surf. Sci., 2003, 526, 57–71 CrossRef CAS
-
M. A. Vannice, in Catalysis-Science and Technology, ed. J. R. Anderson and M. Boudart, Springer-Verlag, Berlin, 1982, vol. 3, pp. 140–149 Search PubMed
-
B. Jongsomjit, T. Wongsalee and P. Praserthdam, in Studies in Surf. Sci. and Catalysis New Development and Application in Chemical Reaction Engineering, ed. I.-S. N. Hyun-Ku Rhee and P. Jong Moon, Elsevier, 2006, vol. 159, pp. 285–288 Search PubMed
- T. Akita, M. Okumura, K. Tanaka and S. Tsubota, J. Electron Microsc. Tech., 2004, 53, 29–35 Search PubMed
- A. Gómez-Cortés, G. Díaz, R. Zanella, H. Ramírez, P. Santiago and J. M. Saniger, J. Phys. Chem. C, 2009, 113, 9710–9720 CrossRef CAS
- S. H. Payne, J. S. McEwen, H. J. Kreuzer and D. Menzel, Surf. Sci., 2005, 594, 240–262 CrossRef CAS
- J. Wang, Y. Wang and K. Jacobi, Surf. Sci., 2001, 488, 83–89 CrossRef CAS
- G. Lauth, T. Solomon, W. Hirschwald and K. Christmann, Surf. Sci., 1989, 210, 201–224 CrossRef CAS
- P. Jakob and A. Schiffer, Surf. Sci., 2009, 603, 1135–1144 CrossRef CAS
- J. Wang, C. Y. Fan, K. Jacobi and G. Ertl, Surf. Sci., 2001, 481, 113–118 CrossRef CAS
- A. Farkas, G. C. Mellau and H. Over, J. Phys. Chem. C, 2009, 113, 14341–14355 CrossRef CAS
- F. Gao, Y. Wang, Y. Cai and D. W. Goodman, Surf. Sci., 2009, 603, 1126–1134 CrossRef CAS
- K. Jacobi, J. Wang and G. Ertl, J. Phys. Chem. B, 2006, 110, 6115–6122 CrossRef CAS
- J. A. Mieth and J. A. Schwarz, J. Catal., 1989, 118, 203–217 CrossRef
- S. Mukkavilli, C. V. Wittmann and L. L. Taviarides, Ind. Eng. Chem. Process Des. Dev., 1986, 25, 487–494 CrossRef CAS
- J. Chen and Z. P. Liu, J. Am. Chem. Soc., 2008, 130, 7929–7937 CrossRef CAS
- C. Welker, N. S. Phala, J. R. Moss, M. Claeys and M. van Steen, J. Mol. Catal. A: Chem., 2008, 288, 75–82 CrossRef CAS
- J. Cheng, P. Hu, P. Ellis, S. French, G. Kelly and C. M. Lok, J. Phys. Chem. C, 2008, 112, 6082–6086 CrossRef CAS
- M. Nurunnabi, K. Murata, K. Okabe, M. Inaba and I. Takahara, Appl. Catal., A, 2008, 340, 203–211 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1, digital diffraction patterns of the areas shown in Fig. 3 in the manuscript used to identify the rutile and anatase phases in P25; Fig. S2, HR-TEM images of RuTi0.8 illustrating the deposition of Ru on the rutile phase of P25; Fig. S3, FTIR spectra of CO adsorbed on the reduced catalysts at room temperature; Fig. S4, FT-IR spectra of surface and gas phase species formed with RuTi0.8N and RuSA catalysts after contact with CO + H2 at 523 K. See DOI: 10.1039/c1cy00136a |
| This journal is © The Royal Society of Chemistry 2011 |