Carbon dioxide reforming of methane over ordered mesoporous NiO–Al2O3 composite oxides
Leilei
Xu
ab,
Huanling
Song
a and
Lingjun
Chou
*a
aState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, P. R. China. E-mail: ljchou@licp.cas.cn; Fax: +86 931 4968129; Tel: +86 931 4968066
bGraduate School of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 25th July 2011
Abstract
Ordered mesoporous NiO–Al2O3 composite oxides with different nickel content were facilely synthesized via an improved evaporation induced self-assembly (EISA) strategy with Pluronic P123 as template in absolute ethanol solvent. The catalytic properties of the obtained mesoporous materials were investigated for the carbon dioxide reforming of methane reaction. These materials were characterized by powder X-ray diffraction (XRD), transmission electron microscopy (TEM), N2 adsorption and desorption characterization, H2 temperature-programmed reduction (TPR), and thermogravimetry (TG). It was observed that these catalysts with mesostructure presented both high catalytic activity and long stability. The improved catalytic performance was suggested to be closely associated with both the amount of “accessible” active centers for the reactants on the mesopore wall surface and the stabilisation of the active sites by the alumina matrix due to the “confinement effect” of the mesopores. The “confinement effect” existing among the mesoporous structure of the materials contributed to preventing Ni particles from sintering under severe reduction and reaction conditions. The stabilized Ni nanoparticles had strong resistance to carbon deposition, accounting for no deactivation after a 100 h long-term stability test at 700 °C. Thus, the ordered mesoporous NiO–Al2O3 composite oxides promised a novel and stable series of catalyst candidates for the carbon dioxide reforming of methane reaction.
1. Introduction
As we all know, both carbon dioxide (CO2) and methane (CH4) are the contributors of the greenhouse effect. The significant and continuous rise of their concentrations in the atmosphere has become a major environmental problem resulting in global climate change. This situation heightened a worldwide interest in the control, conversion and utilization of these greenhouse gases.1–5 Hereby, carbon dioxide reforming of methane (CRM) provides an effective route to transform these two greenhouse gases simultaneously into valuable synthesis gas, namely, a mixture of CO and H2, which provides the basic building blocks for various compounds, such as hydrocarbon fuels, oxygenated chemicals, etc.5–10 Hence, the CRM reaction has received renewed interest due to the consideration of environmental protection as well as its potential industrial application.Based on the previous studies, it's well known that almost all of the transition metals of the VIII family are catalytically active catalysts towards the CRM reaction. Although the noble metals, such as Pt, Pd, Rh, Ru, Ir, possess perfect activity and ideal stability, they are not suitable for large-scale industrial applications owing to their high cost and limited availability.6,11–15 Therefore, nickel is the most appropriate candidate catalyst for this reaction due to its high initial activity almost equivalent to that of noble metals and low cost. Nevertheless, the main disadvantages for the Ni-based catalysts lie in its quick deactivation owing to carbon deposition and thermal sintering of the nickel particles during the process of reduction and reaction.7 How to effectively enhance the resistance to coke as well as the sintering of active sites for the Ni-based catalyst is the current research nodus and focus. Various strategies were employed to improve the stability of the Ni-based catalysts, such as altering the acid–base or redox properties of carriers,16,17 introducing a second active metal,8,10,18–20 controlling Ni particle size or distribution via special preparation methods,21–23 using various carriers,13,15,24–28etc. Most of these strategies were aimed at improving the dispersion of metal active components and inhibiting the thermal sintering of them. It is generally believed that smaller Ni particles possess enhanced capacity to suppress carbon deposition due to their contribution to the reaction for elimination of coke, leading to long time stability according to the pioneer studies. Carbon deposition is not susceptible to occur until the metal cluster is greater than a “critical size”.5,8,29,30 However, preventing Ni particles from sintering is a big challenge for the traditional metal supported catalysts, because the aggregation of Ni particles inevitably takes place under the typically severe reduction and reaction conditions of the CRM reaction.18
Since the ordered mesoporous silica material was firstly reported in 1992, the interests in this research field have expanded throughout the world due to their potential applications in the catalytic field.31–34 Mesoporous materials provide good opportunities to develop novel catalysts with improved catalytic performances. In the pioneer studies, Ni-containing mesoporous materials either mesoporous silica materials or mesoporous metal (or metal composite) oxides, such as Ni-incorporated MCM-41 (SBA-15, TUD-1), mesoporous Al2O3, mesoporous ZrO2, mesoporous Ni–CaO–ZrO2, mesoporous Ni–TiO2–SiO2, etc., all displayed excellent catalytic activity and stability as catalysts for the CRM reaction.12,16,17,22,35–38 The improved catalytic performances of these mesoporous catalysts might derive from the high dispersion and strong anti-sintering properties of the Ni active sites. The thermal agglomeration of the Ni nanoparticles is effectively prevented since the Ni atoms are stabilized by the “confinement effect” of the framework of the mesopores, which could suppress the growth of Ni nanoparticles during the reaction and promote catalytic stability.30,39,40 Moreover, the high dispersion of the Ni particles is able to provide more “accessible” Ni active sites for the reactants due to the predominant textural properties of the mesoporous materials, accounting for the good activity for these materials. Therefore, mesoporous materials promised ideal catalyst candidates for the CRM reaction.
Herein, a series of ordered mesoporous NiO–Al2O3 composite metal oxides with different Ni content were readily prepared and used as catalysts for the CRM reaction. All the catalysts showed excellent catalytic activity and stability. To the best of our knowledge, there has been almost no literature reporting similar materials as the catalysts for the CRM reaction. More details about the OM–xNiyAl materials as catalysts for the CRM reaction will be described extensively in the following text.
2. Experimental
2.1 Preparation of ordered mesoporous catalysts
Aluminum isopropoxide (C9H21AlO3, Sinopharm Chemical Reagent Co. Ltd.), (EO)20(PO)70(EO)20 triblock copolymer (Pluronic P123, Sigma-Aldrich), 67 wt% nitric acid (HNO3, local vendor), nickel nitrate hexahydrate (Ni(NO3)2·6H2O, Shanghai No. 2 Reagent Factory, China), and absolute ethanol (C2H5OH, local vendor) employed in this study were all A.R. grade. All the chemicals were used without further purification.Ordered mesoporous x mol% NiO − y mol% AlO3/2 (x mol% = nNi/(nNi + nAl) × 100%, 1 ≤ x ≤ 10, x + y = 100, denoted as OM–xNiyAl in the following text) composite metal oxides were synthesized via an improved one pot evaporation-induced self-assembly (EISA) by fine control of the volatile process.41,42 In a typical synthesis procedure, approximately 1.0 g of (EO)20(PO)70(EO)20 triblock copolymer was dissolved in 20.0 mL anhydrous ethanol with vigorous stirring. Then, 1.6 mL of 67 wt% nitric acid, approximately A mmol aluminum isopropoxide, B mmol nickel nitrate hexahydrate (A + B = 10 mmol) were sequentially added into the above solution with vigorous stirring. The final mixture was covered with PE film and stirred at room temperature for at least 5 h. Finally, the mixture was transferred to a Petri dish, the Petri dish was covered with punctured PE film, and finally the covered Petri dish was put into a 60 °C drying oven to undergo the slow EISA process for 48 h. A light green solid gel (with the increase of the Ni content, the color of the gel would deepen) was obtained after 48 h via a precisely controlled EISA process. The final gel was calcined by slowly increasing the temperature (1 °C min−1 ramping rate) to 600 °C and maintaining this final temperature for 5 h. Finally, the OM–xNiyAl composite metal oxide powders with large BET specific surface areas, big pore volumes, and narrow pore size distributions were obtained. The as-prepared mesoporous materials were pressed, crushed, and sieved through 20–40 meshes.
2.2 Catalysts characterization
Power X-ray diffraction (PXRD) measurements were performed using an X'Pert Pro multipurpose diffractometer (PANalytical, Inc.) with Ni-filtered Cu-Kα radiation (0.15046 nm) at room temperature from 10.0° to 80.0° (wide angle range) and 0.6° to 5.0° (small angle range). Measurements were conducted using a voltage of 40 kV, current setting of 40 mA, step size of 0.02°, and count time of 4 s. Crystallite sizes of Ni particles were calculated using Scherrer' equation: D(hkl) = Kλ/(βcosθ), where K was the shape factor of the average crystalline, λ, the wavelength (1.54056 Å for Cu-Kα), β, defined by a relationship between the full wave at half maximum (FWHM) of the sample and that of a quartz standard, and θ was the peak position.The nitrogen adsorption and desorption isotherms at −196 °C were recorded on an Autosorb-iQ analyzer (Quantachrome Instruments U.S.). Prior to the tests, samples were degassed at 200 °C for 4 h. The specific surface areas were calculated via the BET method in the relative pressure range of 0.05–0.3; the single-point pore volume was calculated from the adsorption isotherm at a relative pressure of 0.990; pore size distributions were calculated using adsorption branches of nitrogen adsorption-desorption isotherms by BJH method.
TEM images were taken on the JEM-2010 high-resolution transmission electron microscopy under a working voltage of 200 kV. The samples were dispersed in absolute ethanol by moderate sonication at a concentration of 5 wt% solid. A Lacy carbon-coated 200 mesh TEM micro-grid was dipped into the sample suspension and then dried under vacuum at given temperature for a while prior to analysis.
Thermogravimetry–differential scanning calorimetry (TG–DSC) measurements were carried out on a NETZSCH STA 449F3 thermogravimetric analyzer from room temperature to 800 °C with the rate of 10 °C min−1 under air atmosphere.
H2 temperature programmed reduction (TPR) measurements were performed on an AMI-100 (Zeton-Altamira instrument) multifunctional catalyst characterization instrument employing hydrogen as reducing agent. The samples (350 mg) were loaded in a U-shaped quartz reactor. Prior to the TPR measurements, samples were pre-treated at 300 °C for 0.5 h in flowing He (50 mL min−1) to remove any moisture and other impurities. After cooling the reactor to room temperature, a 5 vol% H2–He (50 mL min−1) gas mixture was introduced. The catalyst was heated to 950 °C at a rate of 10 °C min−1 and the hydrogen consumption was measured using an AMETEK (LC-D-200 Dycor AMETEK) mass spectrum.
2.3 Catalysts evaluation
The CRM reaction was performed in a vertical fixed bed quartz tubular reactor (8 mm i.d.) under atmospheric pressure. The flow of all gases was precisely controlled by mass flow meters (MT50-4J METRON Instruments). The catalyst bed was constituted by 100 mg OM–xNiyAl diluted with 300 mg quartz sand (20–40 mesh). Prior to the reaction, the catalyst was reduced in situ in a mixed flow of H2 and N2 (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 mL min−1) with a heating rate of 1.5 °C min−1 rate to 800 °C, and maintained at 800 °C for 120 min. Before introducing in the reaction gases, the catalyst bed was purged with N2 for half an hour to remove the absorbed hydrogen. The effluent mixed gases were cooled in an ice-water trap and analyzed by online gas chromatography (SP-6800A GC) equipped with thermal conductivity detector (TCD) using a TDX-01 packed column.
:20 mL min−1) with a heating rate of 1.5 °C min−1 rate to 800 °C, and maintained at 800 °C for 120 min. Before introducing in the reaction gases, the catalyst bed was purged with N2 for half an hour to remove the absorbed hydrogen. The effluent mixed gases were cooled in an ice-water trap and analyzed by online gas chromatography (SP-6800A GC) equipped with thermal conductivity detector (TCD) using a TDX-01 packed column.
The conversion of methane and carbon dioxide were calculated by means of eqn (1) and (2), respectively:
| CH4 conversion, vol% = 100 × [(H2)out/2]/[(CH4)out + (H2)out/2] | (1) |
| CO2 conversion, vol% = 100 × [(CO)out/2]/[(CH4)out + (CO)out/2] | (2) |
3. Results and discussion
3.1 Phase structure of materials
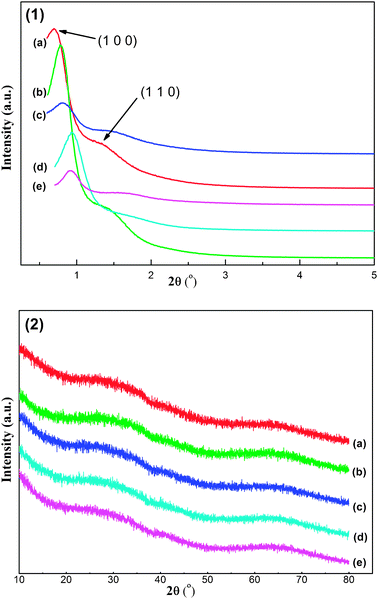 | ||
| Fig. 1 Small-angle X-ray diffraction (1) and wide-angle X-ray diffraction (2) patterns of as-synthesized OM–xNiyAl materials calcined at 600 °C: (a) OM–10Ni90Al, (b) OM–7Ni93Al, (c) OM–5Ni95Al, (d) OM–3Ni97Al, (e) OM–1Ni99Al. | ||
The WXRD analysis was carried out to determine the crystalline phase of the as-prepared OM–xNiyAl calcined at 600 °C. Unlike the SXRD patterns, the WXRD patterns in Fig. 1 (2) showed no evident diffraction peaks of crystalline metal oxides, suggesting the high dispersion of NiO nanoparticles among the framework of the amorphous alumina. In the OM–xNiyAl materials, Ni atoms were inlaid among the mesoporous frameworks of the catalysts and segregated by the Al atoms due to the special traits of this one pot preparation method. Hereby, the thermal conglomeration of the NiO particles during the progress of the calcination was avoided to some extent, accounting for the high dispersion of the NiO particles.
000 mL g−1 h−1, 1 atm). Compared with the as-reduced OM–10Ni90Al, the grain size of Ni particles of the spent OM–10Ni90Al suffered a slight increase. However, the average D(200) (D is the crystallite size) of the Ni particles was only 5.74 nm, as calculated by the Scherrer equation, illuminating that the Ni nanoparticles still maintained a highly dispersed state. The “confinement effect” of the mesostructure contributed to the stabilization of the metallic nanoparticles.30,39,40 These phenomena suggested that this sort of mesostructured material possessed excellent thermal stability. Besides, one diffraction peak with very low intensity of graphic carbon (JCPDS Card No. 12-0212) was found in the spent catalyst.43 The graphic carbon phase could be ascribed to the carbon deposition on the surface of the catalyst after a catalytic stability test for 100 h. Generally speaking, this kind of mesoporous material showed extraordinary stability after undergoing these severe reduction and reaction conditions.
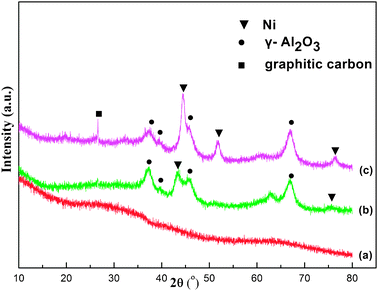 | ||
| Fig. 2 Wide-angle X-ray diffraction patterns of OM–xNiyAl: (a) as-prepared OM–10Ni90Al, (b) as-reduced OM–10Ni90Al, (c) spent OM–10Ni90Al. | ||
3.2 Morphology analysis of as-prepared OM–xNiyAl
TEM images of OM–5Ni95Al and OM–10Ni90Al materials calcined at 600 °C are displayed in Fig. 3. The ordered hexagonal arrangement of pores along the [001] direction (Fig. 3(b)) and the alignment of cylindrical pores along the [110] direction (Fig. 3(a)) for OM–5Ni95Al were distinctly observed. Furthermore, the images of OM–10Ni90Al (Fig. 3(c) and (d)) also showed a similar well-ordered pore network. These TEM images further confirmed the presence of ordered mesopores with p6mm hexagonal symmetry. The characterization results of the TEM experiments were in good agreement with the deduction based on the SXRD characterization results. | ||
| Fig. 3 High resolution TEM images of as-synthesized OM–xNiyAl calcined at 600 °C: (a) and (b) OM–5Ni95Al, (c) and (d) OM–10Ni90Al. | ||
3.3 Textural properties of the materials
The textural properties of these mesoporous materials were characterized by nitrogen adsorption and desorption at −196 °C. Fig. 4 exhibited the nitrogen adsorption and desorption isotherms as well as the pore size distributions of the as-prepared OM–xNiyAl composite metal oxides. Almost all the isotherms presented in Fig. 4 (1) were IV type isotherms with H1 shaped hysteresis loops characteristic for ordered mesoporous materials. Besides, nearly all the capillary condensation steps of the hysteresis loops were very steep, indicating the presence of the mesopores with uniformity among the frameworks of the OM–xNiyAl materials. The H1 shaped hysteresis loops also illuminated that all the mesopores were “cylindrical-shaped” channels. The characteristic results of TEM (refer to Fig. 3) had further confirmed the deduction. In addition, the pore size distributions of all the materials shown in Fig. 4 (2) were extremely narrow. The curves of pore size distributions were all extremely sharp and the position of the peaks were located in the range of “mesopore” according to the definition of the IUPAC (2–50 nm).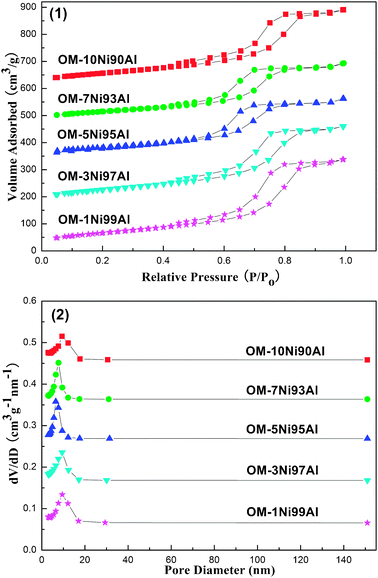 | ||
| Fig. 4 Isotherms (1) and pore size distributions (2) of as-synthesized OM–xNiyAl materials calcined at 600 °C. | ||
In order to investigate the thermal stability of this kind of materials, the OM–10Ni90Al material was selected as an example. As presented in Fig. 5, the structural properties of as-reduced OM–10Ni90Al (reduced at 800 °C for 2 h) and spent OM–10Ni90Al (after 100 h long term stability test at 700 °C, GHSV = 15000 mL g−1 h−1, CH4 = 1 atm, CO2 = 1 atm) were also carefully studied. As for the as-reduced OM–10Ni90Al, the isotherms displayed IV type isotherm with H2 shaped hysteresis loop, which was the typical characteristic for the mesoporous materials with “ink-bottle” shaped mesopores. Compared with the as-synthesized OM–10Ni90Al with H1 shaped hysteresis loop, the H2 shaped of as-reduced OM–10Ni90Al indicated that the uniform “cylindrical-shaped” mesopores were deformed to “ink-bottle” shaped pores after being reduced at temperature as high as 800 °C for 2 h. But the pore size distribution for this as-reduced OM–10Ni90Al material was still very narrow around 6.51 nm. Hence, the mesoporous structure of as-reduced OM–10Ni90Al was retained after reduction. Similar to the as-reduced OM–10Ni90Al, the spent OM–10Ni90Al also possessed IV typed isotherms affiliated with H2 shaped hysteresis loop and narrow pore size distribution (also centered around 6.51 nm). Therefore, the damage of the framework of as-reduced OM–10Ni90Al during the 100 h long term catalytic stability test was avoided to some extent. In a word, all the evidence mentioned above suggested that the OM–10Ni90Al composite metal oxides were equipped with enhanced thermal stability to withstand the severe reduction and reaction conditions.
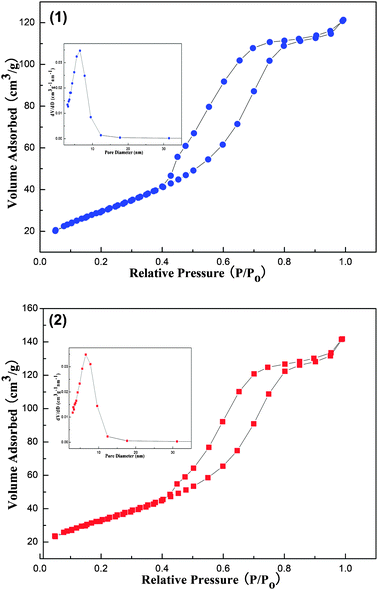 | ||
| Fig. 5 Isotherms and pore size distributions of as-reduced (1) and spent (2) OM–10Ni90Al materials. | ||
Furthermore, the characterization results of the textural properties of the as-prepared OM–xNiyAl, as-reduced OM–10Ni90Al, and spent OM–10Ni90Al were summarized in Table 1. It could be observed that all the as-prepared OM–xNi90Al samples possessed large specific surface areas up to 241.24 m2 g−1 and big pore volumes up to 0.504 cm3 g−1. The average pore diameters of the OM–xNiyAl were all in the range from 6.5–9.5 nm. It was worth mentioning that the specific surface areas and pore volumes suffered from a little decline as the Ni content of OM–xNiyAl increased. The reason for these phenomena might derive from relatively larger ionic radius of Ni2+ (0.69 Å) compared to that of Al3+ (0.51 Å). Ni2+ ions usually took up a much larger area and bigger space in contrast with those of Al3+. The as-reduced OM–10Ni90Al took on a much smaller specific surface area as well as pore volume compared with those of as-prepared OM–10Ni90Al. The shrinkage of the skeleton of material and the deformation from “cylindrical-shaped” to “ink-bottle” shaped mesopores under the severe reduction condition (800 °C for 2 h) could account for this phenomenon. Whereas, as for the spent OM–10Ni90Al, its specific surface area and pore volume were only a little smaller than those of the as-reduced OM–10Ni90Al, implying that the carbon deposition on the surface of the catalyst after 100 h long time stability test was negligible. Furthermore, the average pore diameters of the as-reduced as well as spent OM–10Ni90Al were almost identical at around 6.51 nm. Thus, the blockade of the mesopores was effectively inhibited due to the reinforced ability of eliminating coke. The evidence mentioned above all, once again, demonstrated that the mesostructures of the materials were not damaged under the harsh reduction and reaction conditions. Generally speaking, these OM–xNiyAl mesostructured materials possessed eminent thermal stability.
| Samples | Specific surface area (m2 g−1) | Pore volume (m3 g−1) | Average pore diameter (nm) | Isotherm type |
|---|---|---|---|---|
|
a The OM–10Ni90Al was in situ reduced under H2/N2 (H2:N2 = 10:20 mL min−1) atmosphere at 800 °C for 2 h.
b The OM–10Ni90Al was used as catalyst of carbon dioxide reforming of methane reaction for the 100 h long term stability test. Reaction conditions: CH4/CO2 = 1, GHSV = 15000 mL g−1 h−1, 700 °C, 1 atm.
|
||||
| OM–10Ni90Al | 188.571 | 0.347 | 9.418 | IV H1 |
| OM–7Ni93Al | 189.990 | 0.358 | 7.756 | IV H1 |
| OM–5Ni95Al | 222.246 | 0.437 | 6.474 | IV H1 |
| OM–3Ni97Al | 223.003 | 0.443 | 9.448 | IV H1 |
| OM–1Ni99Al | 241.240 | 0.504 | 9.488 | IV H1 |
| As-reduced OM–10Ni90Ala | 123.438 | 0.213 | 6.509 | IV H2 |
| Spent OM–10Ni90Alb | 112.430 | 0.184 | 6.511 | IV H2 |
3.4 Reducibility of the OM–xNiyAl catalysts
The TPR profiles of the OM–xNiyAl mesostructured materials were presented in Fig. 6. All the samples except for the OM–1Ni99Al presented similar profiles of hydrogen reduction, displaying only one distinct reduction peak in the high temperature region irrespective of the Ni loading. For all the OM–xNiyAl catalysts, the locations of the reduction peaks gradually migrated towards the higher temperature region until they disappeared at 1% with Ni loading decreasing, in the range of 720–830 °C, which was contrary to the results of Ni grafted on SBA-15 and MCM-41 materials reported in the precursory reference.36 No obvious reduction peaks located at the range of 300–400 °C could be observed. This phenomenon suggested the absence of dissociated or free nickel oxide among the framework, which was not interacting with the alumina mesoporous framework. This was in good agreement with the above-mentioned WXRD results (refer to Fig. 1 (2)). On the other hand, these reduction peaks in a higher temperature region (720–830 °C) were attributed to the reduction of NiO having strong interaction with the alumina mesostructured skeleton. 27Al NMR spectra of OM–10Ni90Al given by the pioneer work showed that the nickel replaced tetrahedrally coordinated aluminum (also observed in pure alumina samples), forming nickel aluminate-like species.41,42 Therefore, Ni atoms were embedded among the alumina skeleton, explaining the relatively high reduction temperature. Regardless of the Ni loading, all the OM–xNiyAl catalysts possessed higher reduction temperature than that of the pure NiO sample, indicating the formation of strong metal–support interactions between Ni and the alumina framework. It was this strong metal–support reaction that partly contributed to preventing the nickel nanoparticles from sintering into large clusters, in general, the accumulation of nickel nanoparticles would eventually result in the loss of active sites and catalyze the carbon deposition.5,8,22,30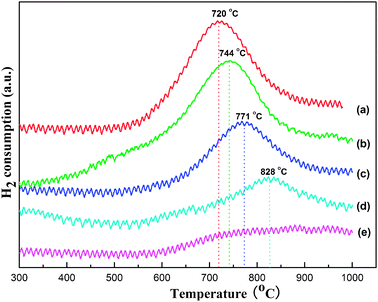 | ||
| Fig. 6 TPR profiles of various as-synthesized OM–xNiyAl catalysts: (a) OM–10Ni90Al, (b) OM–7Ni93Al, (c) OM–5Ni95Al, (d) OM–3Ni97Al, (e) OM–1Ni99Al. | ||
3.5 Catalytic performances of carbon dioxide reforming of methane (CRM) reaction
:1) of the CRM reaction, there was always an excess of CO with respect to H2 for all temperatures investigated regardless of the catalyst. It was also considered that the RWGS reaction could be responsible for the phenomenon. H2, one of the two main products of the CRM reaction, was consumed in the RWGS reaction, leading to the relatively lower H2/CO ratio compared to the stoichimetric ratio (1:1) of the CRM reaction. Whereas, the H2/CO ratio was found to be elevated with the increase of the reaction temperature according to Fig. 7 (3), which agreed well with the thermodynamics tendency caused by the RWGS side reaction.
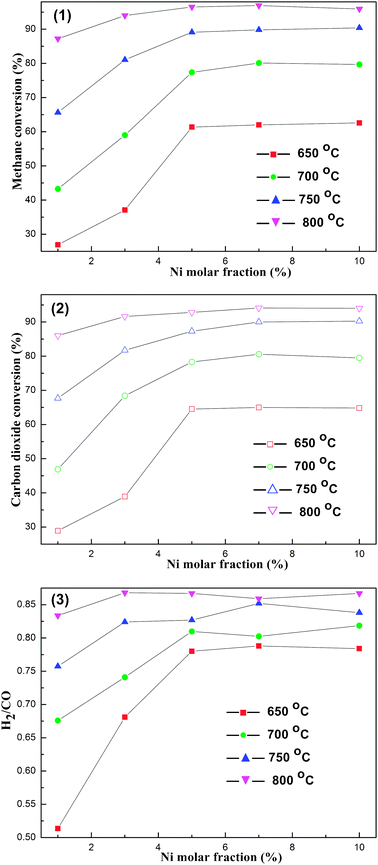 | ||
| Fig. 7 The curves of average (1) CH4 conversion, (2) CO2 conversion, (3) H2/CO ratio, versusNi molar fraction at different reaction temperatures. Reaction conditions: CH4/CO2 = 1, GHSV = 15000 mL g−1 h−1, 1 atm. | ||
In addition to the relationship between the initial catalytic activity and reaction temperature, the connections of the catalytic activity and Ni molar content in OM–xNiyAl also could be observed in Fig. 7 (1) and (2). As the Ni content mounted up, the conversions of CH4 and CO2 increased rapidly and monotonously until they reached a plateau at 5% for all the temperature stages investigated. With further increase in the Ni content (above 5%), minor improvement in CH4, CO2 conversions could be observed. In other words, as the Ni molar content was greater than or equal to 5%, the conversions of CH4, CO2 over different OM–xNiyAl catalysts were similar to each other, respectively. It was considered that their conversions were close to and even exceeded their respective theoretical equilibrium conversions (650 °C, 62.86%; 700 °C, 78.10%; 750 °C, 89.52%; 800 °C, 95.24%, it was supposed that the CRM reaction was not affected by other side reactions and the conversions of CH4 and CO2 were equal).5 It was the thermodynamic equilibrium that limited the further improvement of CH4 and CO2 conversions. Therefore, the OM–5Ni95Al catalyst had been able to provide sufficient nickel active sites to make this reaction achieve thermodynamic equilibrium under the given conditions (CH4/CO2 = 1, GHSV = 15000 mL g−1 h−1, 1 atm) as the nickel content was as low as 5%. The catalytic activity of the OM–5Ni95Al was much higher than that of other Ni based catalysts, such as Ni supported on mesoporous ZrO2 (5%), mesoporous Ni–CaO–ZrO2 (above 10%), mesoporous Ni–TiO2–SiO2 (10%), and so on, with similar or even higher Ni content under the given conditions.16,17,22,38 The reason for the OM–xNiyAl catalysts having such excellent catalytic activity might be due to their relatively high specific surface areas and large pore volumes. The nickel atoms were studded in the skeleton of the alumina due to the special features of this one pot preparation approach. Hence, in the CRM reaction, the OM–xNiyAl catalysts were able to provide more “exposed” (or “accessible”) Ni active sites for the reactants because of their above-mentioned textural properties, making for much higher catalytic activities. Therefore, the mesoporous structure played a critical role in determining the catalytic performances of OM–xNiyAl catalysts. The relations between H2/CO ratio and nickel content are also shown in Fig. 7 (3). As a whole, the H2/CO ratio initially increased as the Ni content mounted up until reached 5% Ni content. Whereas, their connections became quite complicated and no law could be abided by with further increase in Ni content.
000 to 60600 mL g−1 h−1 led to rapid decreases in the CH4, CO2 conversions (e.g. decreased from 92.28% to 73.57% for the CH4 conversion of OM–7Ni93Al) over all the catalysts. Whereas, the specific reasons for the rapid decline in catalytic activities with the increase of the GHSV were not quite sure. A pioneer paper presumed that the reduction in the residence time of reactants on the surface of the catalyst and the drop in the temperature of the catalyst bed might account for these encountered phenomena.44 However, we concluded that the decrease in the residence time on the surface of the catalyst and the limited active sites for the increasing reactants might be the main reasons. As the GHSV increased, the velocity for the reactants through the catalyst bed became faster; therefore, their contact time on the surface of the catalyst bed became shorter. Thus, the thermodynamic equilibrium conversions for the reactants were not achieved due to insufficient contact time. In addition, with the increase of GHSV, the number of the reactants through the catalyst bed became larger; consequently, the catalysts were not able to provide sufficient active sites for redundant reactants. Those factors aforementioned might make clear the phenomenon that the increase of the GHSV brought on the decline in CH4, CO2 conversions.
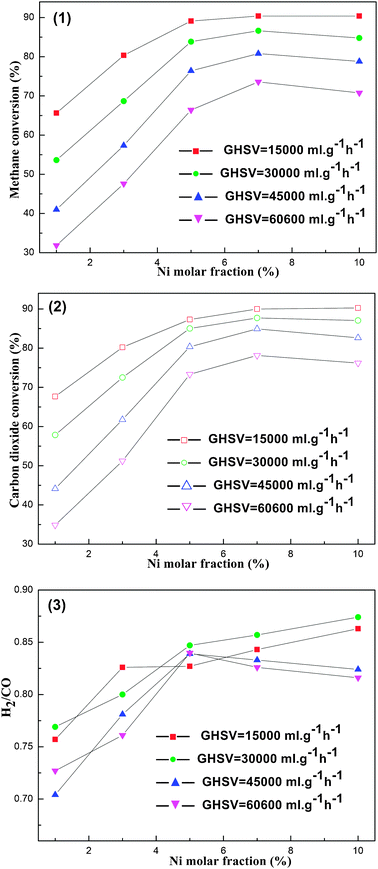 | ||
| Fig. 8 The curves of (1) CH4 conversion, (2) CO2 conversion, (3) H2/CO ratio versusNi molar fraction at different gas hourly space velocity (GHSV). Reaction conditions: CH4/CO2 = 1, 750 °C, 1 atm. | ||
Similar to Fig. 8 (1), (2), the effect of Ni molar content on the catalytic activity for the OM–xNiyAl catalysts under various GHSV was also studied in detailed. When the GHSV was kept at a constant value, the conversions of CH4 and CO2 increased and reached a maximum at 7% as the Ni content increased. As the GHSV increased from 15000 to 60600 mL g−1 h−1, the advantage of the OM–7Ni93Al catalyst in catalytic activity became more and more remarkable. The reason for the outstanding performances of OM–7Ni93Al compared to other OM–xNiyAl catalysts might derive from its more adequate active centers. Compared to other OM–xNiyAl catalysts with Ni molar content less than 7%, the OM–7Ni93Al was originally able to provide more active sites than those catalysts excluding the occurrence of sintering. As for the OM–10Ni90Al sample, slight sintering of the metallic active sites might take place during the severe reduction and reaction processes because of its relatively high levels of Ni active ingredients. Furthermore, the connections between the H2/CO ratio and the GHSV as well as the Ni content were also investigated in Fig. 8 (3). Nevertheless, their relationships were very intricate and required further study.
000 mL g−1 h−1, 1 atm. The OM–10Ni90Al material was chosen as the catalyst for the long lifetime test, because it was more inclined to suffer the sintering of the nickel active sites and collapse of the mesostructure during the reaction due to its higher Ni content than that of the others. As shown in Fig. 9, the catalyst exhibited highly efficient catalytic activity and stability throughout the 100 h long term stability test, and provided over 78.0% and 81.0% conversions of methane and carbon dioxide, respectively, both of which were close to or even exceeded the thermodynamic equilibrium level (700 °C, 78.10%).5 It was also observed that the conversions of CH4 and CO2 firstly experienced a minor rise in the first 5 h from 77.0% to 78.5% for CH4, and from 79.0% to 81.0% for CO2, respectively. Thereafter, the conversions of CH4, CO2 remained stable and no deactivation was observed during the 100 h time on stream, showing prominent catalytic stability over this catalyst. The H2/CO ratio also remained steady and performed as the periodic oscillation around 0.80 from 0.77 to 0.83 throughout the stability test.
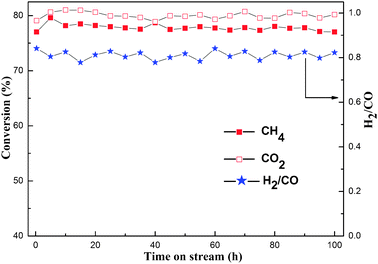 | ||
| Fig. 9 Long term stability test over as-synthesized OM–10Ni90Al catalyst; Reaction conditions: CH4/CO2 = 1, GHSV = 15000 mL g−1 h−1, 700 °C, 1 atm. | ||
It was well known that the deactivation of Ni-based catalysts for the CRM reaction was mainly caused by carbon deposition on the surface of the active sites and the sintering of Ni active components. Moreover, it was reported that smaller Ni particles had an enhanced ability to suppress carbon deposition due to the “size effect”.5,8,29 As observed in the WXRD pattern of as-reduced OM–10Ni90Al (see Fig. 2), the Ni active component maintained a highly dispersed state among the framework of the catalyst. The metal nanoparticles loaded on mesoporous materials were found perform a unique catalytic performance in the previous studies.17,30,35,38 This phenomenon occurring on the mesoporous catalyst could be due to the “confinement effect” of the mesopores and strong metal–support interaction (see Fig. 6), which would limit the growth of Ni particles during the reaction and ultimately promote catalytic stability. Therefore, the mesoporous framework of the OM–10Ni90Al not only provided the catalyst with large specific surface area, big pore volume for its high activity, but was also able to limit the growth of the Ni nanoparticlesvia the “confinement effect” and strong metal–support interaction during the reaction. Consequently, the Ni nanoparticles were stabilized and the coke was suppressed. Therefore, the excellent catalytic activity of OM–10Ni90Al for long term reaction was preserved because of the high coke resistance of its smaller Ni nanoparticles.
3.6 TG analysis
The properties of carbon deposition over the OM–10Ni90Al catalyst after reaction at 700 °C for 10 h and 100 h are displayed in Fig. 10. Overall, the two TG curves in the figure were identical in shape, indicating that similar physical and chemical processes occurred during the TG analysis process. As the temperature increased, the general trends of the curves were downward. However, these two curves both experienced a slight rise in the region from 250 °C to 400 °C. It was assumed that the oxidation of elemental Ni during the process of TG analysis contributed to it. The curve for 100 h in Fig. 10 showed that the weight loss over the catalyst was only 3.31%. Compared with the traditional catalysts reported in previous work,22 this catalyst displayed an excellent ability to resist carbon deposition. As mentioned above, smaller Ni particles had a better ability to suppress carbon deposition.5,8,29,30 The peak broadening of the WXRD pattern for the spent OM–10Ni90Al (700 °C, 100 h) in Fig. 2 had indicated that the sizes of the Ni particles were still preserved nanocrystalline state (5.74 nm) after the 100 h lifetime test. The eminent performance of this catalyst further confirmed the pioneer hypothesis that smaller Ni particles possessed reinforced the ability to inhibit carbon deposition.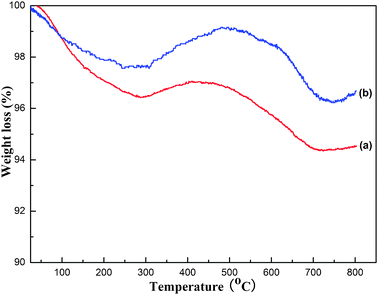 | ||
| Fig. 10
TG analysis of the spent OM–10Ni90Al catalysts for different reaction time: (a) 10 h, (b) 100 h. Reaction conditions: CH4/CO2 = 1, GHSV = 15000 mL g−1 h−1, 700 °C, 1 atm. | ||
As shown in Fig. 10, the weight loss of coke over the spent catalyst for 10 h was as high as 5.46%. This phenomenon was amazing and barely met with in the previous works. It was commonly believed that catalyst with long reaction time had more coke amount than the one with short reaction time under the identical reaction conditions. Generally speaking, the coke formation reactions in the CRM reaction are mainly derived from the methane decomposition reaction (CH4 = C + 2H2) and the Boudouard reaction (2CO = CO2 + C). Grounded on the thermodynamic analysis, the Boudouard reaction could be completely inhibited at 700 °C, therefore, this reaction barely contributed to the carbon deposition under these reaction conditions. On the contrary, the methane decomposition reaction could be triggered at temperature as low as 557 °C. Therefore, the methane decomposition reaction was the main coke reaction for the CRM reaction at 700 °C. Meanwhile, the elimination of the carbon deposition on the surface of the catalyst was ongoing due to the presence of these reactions: C + CO2 = 2CO and C + H2O = H2 + CO. Furthermore, it was amusing to notice that the H2/CO ratio (see Fig. 9) varied in an oscillating way between 0.77 and 0.83 over OM–10Ni90Al catalyst, illuminating a periodic cycle of carbon deposition and elimination reaction.44 The coke previously accumulated on the surface of the catalyst could be eliminated by the above-mentioned reactions in the subsequent process. The reason for the unique phenomenon met with in the TG analysis was that the carbon deposition reaction and elimination over the spent catalyst of 10 h had not reached the final equilibrium and carbon deposition reaction gained the upper hand compared to the coke elimination reaction. However, it could be confidently affirmed that equilibrium between carbon deposition and elimination reaction would be finally achieved so long as the reaction time was long enough. In summary, the OM–10Ni90Al catalyst displayed excellent catalytic activity, long lifetime stability, and good anti-coking performance.
4. Conclusion
Ordered mesoporous NiO–Al2O3 composite oxides with various Ni molar contents were readily prepared and used as catalysts for the CRM reaction. This series of materials possessed large specific surface areas, big pore volumes, narrow pore size distributions, and good thermal stability. The catalytic performances of OM–xNiyAl for the CRM reaction were excellent, presenting high catalytic activity and long catalytic stability. They were ideal candidates as catalysts for the CRM reaction. The ordered mesostructure of OM–xNiyAl catalysts played a significant role to provide high activity and long stability, which provided much more “accessible” (or “exposed”) Ni active sites to the reactants and stabilized Ni nanoparticlesvia the “confinement effect” during the reaction. Strong metal–support interactions had been also formed between the Ni atom and the mesoporous framework, which likewise helped to prevent the nickel nanoparticles from sintering. As a result, the OM–10Ni90Al catalyst had strong a anti-coking ability due to the nano-sized Ni particles existing among the framework of the material, which contributed to suppressing the carbon deposition due to the “size effect” of the Ni active sites. Due to these favorable properties, the OM–xNiyAl series of materials could be considered as promising catalysts for the CRM reaction.Acknowledgements
The authors sincerely acknowledge the financial support from the National Basic Research Program of PR China (No. 2011CB201404).References
- K. M. Walter, S. A. Zimov, J. P. Chanton, D. Verbyla and F. S. Chapin, Methane bubbling from Siberian thaw lakes as a positive feedback to climate warming, Nature, 2006, 443, 71–75 CrossRef CAS.
- C. S. Song, Global challenges and strategies for control, conversion and utilization of CO2 for sustainable development involving energy, catalysis, adsorption and chemical processing, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- H. Rodhe, A comparison of the contribution of various gases to the greenhouse-effect, Science, 1990, 248, 1217–1219 CAS.
- J. M. Melillo, A. D. McGuire, D. W. Kicklighter, B. Moore, C. J. Vorosmarty and A. L. Schloss, global climate-change and terrestrial net primary production, Nature, 1993, 363, 234–240 CrossRef CAS.
- M. C. J. Bradford and M. A. Vannice, CO2 reforming of CH4, Catal. Rev.-Sci. Eng., 1999, 41, 1–42 CAS.
- A. T. Ashcroft, A. K. Cheetham, M. L. H. Green and P. D. F. Vernon, partial oxidation of methane to synthesis gas-using carbon-dioxide, Nature, 1991, 352, 225–226 CrossRef CAS.
- A. M. Gadalla and M. E. Sommer, carbon-dioxide reforming of methane on nickel-catalysts, Chem. Eng. Sci., 1989, 44, 2825–2829 CrossRef CAS.
- Y. H. Hu and E. Ruckenstein, Binary MgO-based solid solution catalysts for methane conversion to syngas, Catal. Rev.-Sci. Eng., 2002, 44, 423–453 CAS.
- J. H. Lunsford, Catalytic conversion of methane to more useful chemicals and fuels: a challenge for the 21st century, Catal. Today, 2000, 63, 165–174 CrossRef CAS.
- J. G. Zhang, H. Wang and A. K. Dalai, Development of stable bimetallic catalysts for carbon dioxide reforming of methane, J. Catal., 2007, 249, 300–310 CrossRef CAS.
- M. C. J. Bradford and M. A. Vannice, CO2 reforming of CH4 over supported Ru catalysts, J. Catal., 1999, 183, 69–75 CrossRef CAS.
- S. Damyanova, B. Pawelec, K. Arishtirova, J. L. G. Fierro, C. Sener and T. Dogu, MCM-41 supported PdNi catalysts for dry reforming of methane, Appl. Catal. B-Envir., 2009, 92, 250–261 CrossRef CAS.
- S. Damyanova, B. Pawelec, K. Arishtirova, M. V. M. Huerta and J. L. G. Fierro, The effect of CeO2 on the surface and catalytic properties of Pt/CeO2–ZrO2 catalysts for methane dry reforming, Appl. Catal. B-Envir., 2009, 89, 149–159 CrossRef CAS.
- J. Nakamura, K. Aikawa, K. Sato and T. Uchijima, role of support in reforming of CH4 with CO2 over rh catalysts, Catal. Lett., 1994, 25, 265–270 CrossRef CAS.
- M. Sigl, M. C. J. Bradford, H. Knozinger and M. A. Vannice, CO2 reforming of methane over vanadia-promoted Rh/SiO2 catalysts, Top. Catal., 1999, 8, 211–222 CrossRef CAS.
- S. G. Liu, L. X. Guan, J. P. Li, N. Zhao, W. Wei and Y. H. Sun, CO2 reforming of CH4 over stabilized mesoporous Ni–CaO–ZrO2 composites, Fuel, 2008, 87, 2477–2481 CrossRef CAS.
- M. Rezaei, S. M. Alavi, S. Sahebdelfar and Z. F. Yan, Effects of K2O promoter on the activity and stability of nickel catalysts supported on mesoporous nanocrystalline zirconia in CH4 reforming with CO2, Energy Fuels, 2008, 22, 2195–2202 CrossRef CAS.
- H. W. Chen, C. Y. Wang, C. H. Yu, L. T. Tseng and P. H. Liao, Carbon dioxide reforming of methane reaction catalyzed by stable nickel copper catalysts, Catal. Today, 2004, 97, 173–180 CrossRef CAS.
- C. Crisafulli, S. Scire, S. Minico and L. Solarino, Ni-Ru bimetallic catalysts for the CO2 reforming of methane, Appl. Catal. A, 2002, 225, 1–9 CrossRef CAS.
- S. Irusta, L. M. Cornaglia and E. A. Lombardo, Hydrogen production using Ni–Rh on ZrO2 as potential low-temperature catalysts for membrane reactors, J. Catal., 2002, 210, 263–272 CrossRef CAS.
- S. Y. Shang, G. H. Liu, X. Y. Chai, X. M. Tao, X. Li, M. G. Bai, W. Chu, X. Y. Dai, Y. X. Zhao and Y. X. Yin, Research on Ni/gamma-Al2O3 catalyst for CO2 reforming of CH4 prepared by atmospheric pressure glow discharge plasma jet, Catal. Today, 2009, 148, 268–274 CrossRef CAS.
- S. B. Zhang, J. K. Wang, H. T. Liu and X. L. Wang, One-pot synthesis of Ni-nanoparticle-embedded mesoporous titania/silica catalyst and its application for CO2-reforming of methane, Catal. Commun., 2008, 9, 995–1000 CrossRef CAS.
- B. Fidalgo, L. Zubizarreta, J. M. Bermudez, A. Arenillas and J. A. Menendez, Synthesis of carbon-supported nickel catalysts for the dry reforming of CH4, Fuel Proc. Tech., 2010, 91, 765–769 CrossRef CAS.
- N. Laosiripojana and S. Assabumrungrat, Methane steam reforming over Ni/Ce–ZrO2 catalyst: Influences of Ce–ZrO2 support on reactivity, resistance toward carbon formation, and intrinsic reaction kinetics, Appl. Catal. A, 2005, 290, 200–211 CAS.
- K. Nagaoka, K. Takanabe and K. Aika, Influence of the phase composition of titania on catalytic behavior of Co/TiO2 for the dry reforming of methane, Chem. Commun., 2002, 1006–1007 RSC.
- D. Qin and J. Lapszewicz, Study of mixed steam and CO2 reforming of CH4 to syngas on mgo-supported metals, Catal. Today, 1994, 21, 551–560 CrossRef CAS.
- B. Q. Xu and W. M. H. Sachtler, Rh/NaY: A selective catalyst for direct synthesis of acetic acid from syngas, J. Catal., 1998, 180, 194–206 CrossRef CAS.
- Q. H. Zhang, L. Yan and B. Q. Xu, Reforming of methane and coalbed methane over nanocomposite Ni/ZrO2 catalyst, Catal. Today, 2004, 98, 601–605 CrossRef CAS.
- Y. H. Hu, Solid-solution catalysts for CO2 reforming of methane, Catal. Today, 2009, 148, 206–211 CrossRef CAS.
- N. N. Sun, X. Wen, F. Wang, W. Wei and Y. H. Sun, Effect of pore structure on Ni catalyst for CO2 reforming of CH4, Energy Environ. Sci., 2010, 3, 366–369 CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, A new family of mesoporous molecular-sieves prepared with liquid-crystal templates, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- A. Corma, From microporous to mesoporous molecular sieve materials and their use in catalysis, Chem. Rev., 1997, 97, 2373–2419 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, ordered mesoporous molecular-sieves synthesized by a liquid-crystal template mechanism, Nature, 1992, 359, 710–712 CrossRef CAS.
- J. C. Vartuli, S. S. Shih, C. T. Kresge and J. S. Beck, Potential applications for M41S type mesoporous molecular sieves, Mesoporous Molecular Sieves, 1998, 117, 13–21 CrossRef CAS.
- D. P. Liu, R. Lau, A. Borgna and Y. H. Yang, Carbon dioxide reforming of methane to synthesis gas over Ni-MCM-41 catalysts, Appl. Catal., A, 2009, 358, 110–118 CrossRef CAS.
- D. P. Liu, X. Y. Quek, H. H. A. Wah, G. M. Zeng, Y. D. Li and Y. H. Yang, Carbon dioxide reforming of methane over nickel-grafted SBA-15 and MCM-41 catalysts, Catal. Today, 2009, 148, 243–250 CrossRef CAS.
- X. Y. Quek, D. P. Liu, W. N. E. Cheo, H. Wang, Y. Chen and Y. H. Yang, Nickel-grafted TUD-1 mesoporous catalysts for carbon dioxide reforming of methane, Appl. Catal., B, 2010, 95, 374–382 CrossRef CAS.
- M. Rezaei, S. M. Alavi, S. Sahebdelfar, X. M. Liu, L. Qian and Z. F. Yan, CO2–CH4 reforming over nickel catalysts supported on mesoporous nanocrystalline zirconia with high surface area, Energy Fuels, 2007, 21, 581–589 CrossRef CAS.
- W. Chen, Z. L. Fan, X. L. Pan and X. H. Bao, Effect of confinement in carbon nanotubes on the activity of Fischer–Tropsch iron catalyst, J. Am. Chem. Soc., 2008, 130, 9414–9419 CrossRef CAS.
- Z. X. Li, F. B. Shi, L. L. Li, T. Zhang and C. H. Yan, A facile route to ordered mesoporous-alumina-supported catalysts, and their catalytic activities for CO oxidation, Phys. Chem. Chem. Phys., 2011, 13, 2488–2491 RSC.
- Q. Yuan, A. X. Yin, C. Luo, L. D. Sun, Y. W. Zhang, W. T. Duan, H. C. Liu and C. H. Yan, Facile synthesis for ordered mesoporous gamma-aluminas with high thermal stability, J. Am. Chem. Soc., 2008, 130, 3465–3472 CrossRef CAS.
- S. M. Morris, P. F. Fulvio and M. Jaroniec, Ordered Mesoporous Alumina-Supported Metal Oxides, J. Am. Chem. Soc., 2008, 130, 15210–15216 CrossRef CAS.
- W. H. Shen, K. Komatsubara, T. Hagiyama, A. Yoshida and S. Naito, Steam reforming of methane over ordered mesoporous Ni–Mg–Al oxides, Chem. Commun., 2009, 6490–6492 RSC.
- J. M. Wei, B. Q. Xu, J. L. Li, Z. X. Cheng and Q. M. Zhu, Highly active and stable Ni/ZrO2 catalyst for syngas production by CO2 reforming of methane, Appl. Catal., A, 2000, 196, L167–L172 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |