Model Ag/HOPG catalysts: preparation and STM/XPS study
Demid V.
Demidov
*,
Igor P.
Prosvirin
,
Alexei M.
Sorokin
and
Valerii I.
Bukhtiyarov
Boreskov Institute of Catalysis SB RAS, Lavrentieva Ave. 5, Novosibirsk, 630090, Russia. E-mail: demidoff@catalysis.ru
First published on 22nd August 2011
Abstract
Model catalysts—Ag on highly oriented pyrolytic graphite (Ag/HOPG)—have been studied using scanning tunneling microscopy (STM), scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS) and X-ray induced Auger electron spectroscopy (XAES). Two types of catalysts were compared: Ag nanoparticles supported on sputtered and non-sputtered HOPG, and the influence of graphite surface defects on the stabilization of Ag nano-sized particles was discussed. A procedure for the preparation of stable (up to 250 °C under a submillibar oxygen/ethylene pressure) silver nanoparticles is suggested. The disappearance of Ag particles in STM images after a sample treatment in the ambient conditions is explained in terms of silver penetration into graphite and the loss of conductivity due to adsorption of contaminants.
1. Introduction
Metallic particles dispersed on porous supports, such as alumina, silica or carbon, represent one of the most important types of heterogeneous catalysts.1 They are used for the abatement of car exhaust gases,2 for increasing the octane number of straight-run gasoline,3 for electro-oxidation of H2 and electro-reduction of O2 in fuel cells with the proton electrolyte membrane,4etc. Practical importance of the supported metal catalysts explains the great interest in the studies of the nature of active centers in these catalysts. Such studies are a part of the fundamental approach to the development of improved catalysts, in which a study of mechanisms of catalytic reactions precedes the molecular design of an optimal catalyst surface.Unfortunately, low loading of a catalytically active metal in real catalysts that use high-surface area supports limits the application of many physical methods in such studies. In addition, intermediates and adsorbed surface species often can not be identified due to masking of their spectroscopic characteristics by corresponding characteristics of the support. For example, an intense O1s signal in the X-ray photoelectron spectroscopy (XPS) from oxide supports (Al2O3, SiO2, TiO2, etc.) overlaps the corresponding signals of oxygen-containing species adsorbed on the surface of supported metal particles in the oxidation catalysts.
One common way of solving these problems is the application of model catalysts in which metal particles are deposited on the surface of planar supports. Indeed, in this case the surface concentration of the particles of a catalytically active metal can be increased above the sensitivity limit of most physical methods. Besides, the oxide support can be exchanged with carbon, e.g. with highly oriented pyrolytic graphite (HOPG), which is substantially free from oxygen contaminations.5–7 Variation of the metal surface concentration allows one to also change the particle sizes, which makes it possible to investigate the size effects in adsorption and catalysis by supported metal particles.5,8,9
This situation is quite typical of silver catalysts for ethylene epoxidation in which Ag/α-Al2O3 catalysts promoted with cesium, chlorine, rheniumetc. are used. The principal challenge associated with the epoxidation process is the prevention of CO2 formation due to the total oxidation of ethylene and secondary oxidation of ethylene oxide,10 which reduce the process selectivity. Practical importance of the process continues to attract great attention to the study of both ethylene epoxidation and oxygen adsorption over various silver samples.11–15 One of the most frequent opinions originating from these numerous studies is the conclusion that the routes of ethylene oxidation are affected by the nature of the oxygen species. The epoxidation route is commonly associated with atomically adsorbed oxygen in an electrophilic state Oδ−, whereas nucleophilic oxygen O2− is supposed to oxidize ethylene to CO2 and H2O. However, this statement, which has been formulated on the basis of single crystal studies performed with XPS,16,17 can not be confirmed for commercial catalysts. Indeed, a huge O1s signal at 531.6 eV from oxygen in alumina completely masks O1s signals both from the electrophilic (Eb(O1s) ∼ 530 eV) and the nucleophilic (Eb(O1s) ∼ 528.4 eV) oxygen species, in spite of the lower binding energies of the latter species. This masking effect also makes it impossible to study the size effects in activity and selectivity reported in a number of papers.8,14,18 It is evident that single crystals are not suitable for these purposes.
As mentioned above, application of HOPG, which is widely used as a support for metallic particles,19–24 could be a good solution for the problem. The main advantage of HOPG is the very small background signals in the O1s region.7 Furthermore, since HOPG is a conductor, scanning tunneling microscopy (STM) and scanning electron microscopy (SEM) could be applied to characterize the surface morphology of the catalysts. Conducting properties of HOPG also simplify analysis of XPS data due to the absence of charging effects, which, in some cases, do not allow measurement of XPS spectra from oxide-based catalysts. Finally, HOPG is quite stable and presumably can withstand the oxidation under reaction conditions (T = 200–250 °C, excess of oxygen).
There are a number of papers devoted to the preparation of metallic particles on HOPG in vacuum.19–24 However most of them have not paid attention to the stability of particles in real catalytic conditions: at high temperatures and elevated pressures of reactants (if compared to the standard surface science pressures). This problem is aggravated by the weak adhesion of metallic nanoparticles to the regular graphite surface.19,21 Instability of nanoparticles against agglomeration and sintering could affect the results of adsorption and catalytic studies. A possible solution to the problem is a formation of artificial defects on the regular HOPG surface. As shown earlier,6,19–23 preliminary bombardment of the HOPG surface with accelerated Ar+ ions improves the metal–support adhesion and stabilizes silver particles. Furthermore, the sticking probability of silver particles was significantly enhanced on the sputtered graphite surface as compared to well-annealed HOPG.21 Still these effects have not been studied in detail. In particular, the stability of silver particles against sintering in these conditions and the stability of HOPG against oxidation in the presence of silver particles have not been analyzed.
In this paper, we present a comparative investigation of silver nanoparticles deposited on sputtered and non-sputtered HOPG surfaces. The results show that preliminary sputtering of the HOPG surface increases the stability of Ag particles against sintering. The reasons for the stabilization are discussed. In addition, the upper limit of support stability under conditions of oxidative reaction has been studied as well.
2. Experimental
Model Ag/HOPG catalysts were prepared inside a preparation chamber of a SPECS photoelectron spectrometer. Before loading to the spectrometer, each sample of HOPG (SPI, Inc.) was cleaned by removing the upper layers with a double-side adhesive scotch-tape. Then, the sample was annealed at 500 °C for 1 h inside the spectrometer at vacuum better than 5 × 10−9 mbar. Since the goal of the present study was to compare the behavior of silver particles on annealed and defect pyrolytic graphite surfaces, samples of HOPG in the latter case were briefly sputtered using an argon ion source (acceleration voltage was 0.5 kV, sputter time was 2–3 s, Ar pressure was 3.5 × 10−6 mbar, current density was estimated to be 5 × 1013 ions cm−2s−1). Silver particles on HOPG were prepared via electron beam evaporation of a silver rod using an OMICRON EFM3 evaporator. The HOPG surface during the evaporation was located perpendicularly to the Ag beam. An amount of deposited silver, which was controlled by the ratio of Ag3d to C1s XPS peak areas between evaporation steps (Ag/C), was varied by changing the deposition time. To increase the resolution, XPS and X-ray induced Auger electron spectra (XAES) of the samples were recorded using a SPECS spectrometer with a PHOIBOS-150-MCD-9 hemispherical energy analyzer and a FOCUS-500 X-ray monochromator (Al-Kα irradiation, hν = 1486.74 eV, 200 W). STM experiments were performed using an ultra high vacuum (UHV) scanning tunneling microscope GPI-300.02 that allows one to record atomically resolved images of a conductive surface at room temperature. A Pt wire (0.25 mm) truncated with nippers was used as a tip. STM images were processed using the WSxM software.25SEM investigation was carried out with a Hitachi S4800 microscope (Fritz-Haber-Institute, Berlin). In situXPS measurements were performed using a VG ESCALAB High Pressure photoelectron spectrometer (Mg-Kα irradiation, hν = 1253.6 eV).263. Results
Fig. 1 shows STM images of annealed (a) and sputtered (b) HOPG surfaces. | ||
| Fig. 1 10 × 10 nm STM images of non-sputtered (a) and sputtered (b) HOPG surfaces. Tunneling parameters: (a) −0.3 nA, −200 mV; (b) 0.7 nA, −100 mV. | ||
Atomically resolved images, which show an ordered structure of the graphite surface, could be obtained only for the flat surface of non-sputtered graphite (Fig. 1a). From Fig. 1b one can not see the atomic structure of graphite, because Ar+ bombardment disordered the surface layers of HOPG and formed the defects.
Images of Ag/HOPG model catalysts prepared using these two graphite surfaces (Ag/C was about 0.4) are presented in Fig. 2a and 3a. As one can see, the surface of non-sputtered graphite is covered by silver nanoparticles homogeneously distributed over the surface (Fig. 2a).
 | ||
| Fig. 2 100 × 100 nm STM image from Ag/HOPG sample prepared on non-sputtered HOPG surface (a) and corresponding particles size distribution (b). Tunneling parameters: −0.5 nA, −100 mV. | ||
 | ||
| Fig. 3 100 × 100 nm STM images from Ag/HOPG samples prepared on sputtered HOPG: after loading in STM (a) and after heating inside STM 1 h, 250 °C, UHV (b). Tunneling parameters: 0.3 nA, −50 mV (a), 0.3 nA, 2000 mV (b). | ||
The mean diameter of the particles is about 2.6 nm (Fig. 2b). In contrast, STM images of the sample on the sputtered HOPG have no indication of silver particles: images of this surface are similar to the images of clean sputtered HOPG (Fig. 3a). This observation disagrees with XPS data which clearly indicate the presence of silver in the XPS analysis depth (∼2 nm). Silver particles on the surface of the sputtered HOPG appear in STM images after annealing of the sample in UHV at 250 °C for 1 h (Fig. 3b). Neighboring silver particles are in tight contact with each other.
Annealing at high temperatures in UHV also causes significant changes in STM images of the Ag/HOPG (non-sputtered) sample: silver particles are sintered into agglomerates that are concentrated on the steps of the HOPG surface (Fig. 4a). The particle size distribution becomes quite broad (Fig. 4c) indicating high mobility of silver particles on the ordered HOPG surface. The STM image of the Ag/HOPG (sputtered) sample taken with the same resolution shows a much more homogeneous distribution of the silver particles on the sputtered HOPG surface (Fig. 4b). Besides, this sample is characterized by much narrower particle size distribution with a mean particle size of ∼8 nm (Fig. 4d), which indicates higher stability of the silver particles on the sputtered HOPG surface against sintering. It should be noted that STM images shown in Fig. 3b and 4b were measured after cooling down the Ag/HOPG (sputtered) sample, followed by a transfer to the STM chamber without contact with the atmosphere. In the previous case (Fig. 3a), this sample was characterized by STM after transfer from the XPS spectrometer, where it was prepared, i.e. after contact with the air at atmospheric pressure. To check the influence of the atmospheric pressure on STM images of the Ag/HOPG (sputtered) sample, three experiments were performed.
 | ||
| Fig. 4 500 × 500 nm STM images (a, b) and particle size distributions (c, d) from Ag/HOPG samples on non-sputtered (a, c) and sputtered (b, d) HOPG surfaces after 1 h annealing at 250 °C in UHV. Tunneling parameters: (a) −1.0 nA, −1500 mV and (b) −1.0 nA, −30 mV. | ||
The experiments included (i) admission of various gases at 1 bar (air in the 1st cycle, nitrogen in the 2nd cycle, argon in the 3rd cycle) into the STM microscope, (ii) evacuation of the chamber followed by measurement of STM images, (iii) annealing the sample at 250 °C inside the microscope chamber for 1 h followed by cooling to room temperature in UHV with a subsequent measurement of STM images at room temperature. Fig. 5, for example, shows the corresponding STM images taken from the Ag/HOPG (sputtered) sample after the 3rd cycle of the gas (argon) admission (Fig. 5a) followed by evacuation and 1 h annealing at 250 °C in UHV (Fig. 5b), as well as the silver particle size distribution (Fig. 5c) after annealing. Admission of the gases, irrespective of their nature, resulted each time in the disappearance of silver particles from the STM images (Fig. 5a), whereas the subsequent heating in UHV restored the images with the clear indication of three-dimensional metallic particles (Fig. 5b).
 | ||
| Fig. 5 100 × 100 nm STM images (a, b) and particle size distribution (c) from Ag/HOPG (sputtered) sample after exposure with argon at 1 bar (a) and subsequent annealing for 1 h in UHV at 250 °C (b, c). Tunneling parameters: (a) −0.3 nA, −30 mV, (b) −0.5 nA, −1500 mV. | ||
It should also be noted that in spite of the higher stability of silver particles on the sputtered HOPG surface, repetition of admission/annealing procedures decreases the mean particle size (compare Fig. 4d and 5c). Since additional analysis of the STM images reveals the permanency of the surface density of the silver particles (∼6 × 103 particles per μm2 before and after cycles of atmosphere exposure), a partial removal of silver from the sample surface could be supposed. This assumption is in agreement with an XPS observation that the ratio Ag/C reduces from the initial 0.40 to 0.25 after the third procedure.
Increasing the annealing temperatures up to 300 °C was found to finally stabilize the sample, i.e.silver particles did not disappear under 1 bar. Moreover, final stabilization of the sample preserves silver from removal from the surface.
This conclusion follows, for example, from scanning electron microscopy (SEM) images shown in Fig. 6. The first SEM image (Fig. 6a) was taken after the preparation of the Ag/HOPG (sputtered) sample with the final stabilization at 300 °C and storage of the sample for 1 month in air, while the second image (Fig. 6b) was taken after repetitive annealing for 1 h at 300 °C in UHV. As one can see, the images have almost no differences in particle morphology, size distribution or location on the HOPG surface. The only difference (by image contrast) is related to measuring and focusing conditions. This sample was used to confirm the possibility of distinguishing the different oxygen species adsorbed on the silver and graphite. Fig. 7 shows O1s spectra from the Ag/HOPG sample measured in situ in an oxygen/ethylene reaction mixture at two different temperatures: 80 and 200 °C.
 | ||
| Fig. 6 SEM images of the same area of the Ag/HOPG (sputtered) sample after two annealing processes at 300 °C in UHV (see the text). | ||
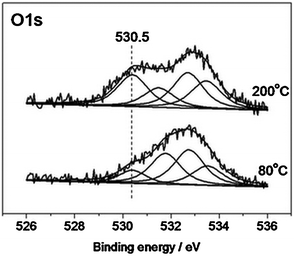 | ||
Fig. 7 O1s spectra from Ag/HOPG (sputtered) sample measured in reaction mixture: O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C2H4 = 1:1, P = 0.25 mbar, at different temperatures. :C2H4 = 1:1, P = 0.25 mbar, at different temperatures. | ||
One can see that both spectra are characterized by several oxygen species with the relative population being dependent on temperature. The species at 530.5 eV, which increases its intensity at 200 °C, can be attributed to oxygen in the electrophilic state adsorbed on silver,5,16,17 while oxygen species with higher binding energies are related to different oxygen groups of HOPG: with BE = 531.5–532.2 eV related to the C=O bond in carbonyls, carboxyls and esters; with BE > 533 eV related to the C–O bond in hydroxyls, carboxyls and ethers.5,7 The data indicate that these model samples can be used to study oxygen species on catalytically active metallic particles.
This sample was also used to check the upper temperature limit of the HOPG stability against oxidation. Fig. 8 shows SEM images from the Ag/HOPG catalyst measured after catalytic experiments at different temperatures. These SEM images indicate that silver particles can promote the oxidation of the HOPG surface at temperatures higher than 250 °C, but at typical temperatures of ethylene epoxidation (180–230 °C) the Ag/HOPG sample remains stable against combustion of the HOPG. Burning of graphite at the contact points with silver particles is accompanied by the migration of the metallic particles on the HOPG surface.
 | ||
| Fig. 8 SEM images from the Ag/HOPG (sputtered) sample measured after experiments on ethylene oxidation at 230 °C (a) and 250 °C (b), P = 0.25 mbar. | ||
4. Discussion
The detailed STM/XPS investigation of preparation of the Ag/HOPG model catalysts and their thermal stability indicates great differences in the behavior of silver particles deposited on annealed and sputtered surfaces of pyrolytic graphite. In the former case, silver particles homogeneously distributed on the HOPG surface are clearly seen immediately after silver deposition and transfer of the sample from the photoelectron spectrometer to the microscope (Fig. 2a).Subsequent annealing of the sample at T > 200 °C in UHV causes a coarsening of the silver particles (Fig. 4a). This effect is in line with the well-known fact of weak adhesion of metal particles to the well-ordered HOPG surface.19,27 In full agreement with this explanation, silver particles located on the steps are much more stable than the particles on terraces and practically do not change their size during the annealing (Fig. 4a). As a consequence, the size distribution of silver particles becomes rather broad (Fig. 4c). It is evident that these samples can not be used for in situ studies of the size effects in adsorption and catalytic reactions. In contrast to the Ag/HOPG (non-sputtered) sample, silver particles deposited on the sputtered HOPG surface are not observed in STM images after transfer of the Ag/HOPG (sputtered) sample from the spectrometer to the microscope (Fig. 3a), and only subsequent annealing of the sample at T > 200 °C in UHV makes them visible in STM (Fig. 3b and 4b). However, the distribution of silver particles by size is much narrower in this case (Fig. 4d), indicating higher stability of the silver particles against sintering on the sputtered HOPG surface. This result has already been reported in literature and was explained by the fact that ion sputtering creates vacancy defects on the HOPG surface that serve as sites for nucleation and stabilization of silver particles.19,22,27
It is more difficult to explain the disappearance of silver particles from STM images of the Ag/HOPG (sputtered) samples after their contact with different gases at atmospheric pressure (Fig. 2a and 5a). We did not find any reports of this effect in the literature. There are two possible explanations. On one hand, contact of the samples with the atmosphere can cause loss of conductivity due to adsorption of some impurities on their surface. As a consequence, a good STM image from the catalyst surface could not be measured. In this case, the restoration of STM images after annealing at T > 200 °C could be caused by desorption of the impurities at high temperatures. O1s photoelectron and Ag MNN Auger spectra measured from the Ag/HOPG (sputtered) sample before and after final annealing at T = 300 °C (shown in Fig. 9) seem to confirm this assumption. The O1s spectrum measured after the exposure of the sample to the atmosphere is characterized by the rather intense signal at ∼531.8 eV (Fig. 9a). This signal is strongly reduced after annealing which indicates desorption of oxygen-containing species. The use of the Ag MNN spectrum instead of the Ag3d photoelectron line is justified by its higher sensitivity to the chemical state of silver.28,29Annealing at high temperatures also shifts the Ag MNN Auger spectrum to direction of metallic silver.
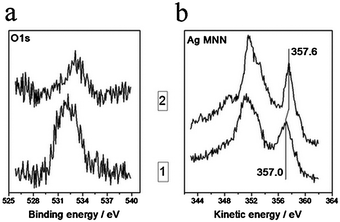 | ||
| Fig. 9 Variations of O1s photoelectron (a) and Ag MNN Auger (b) spectra from Ag/HOPG (sputtered) sample after contact with atmosphere (curve 1) and after heating up to 300 °C (curve 2). | ||
To exclude the influence of final state effects on the shift of Ag MNN line, we have calculated the so-called modified Auger parameter (α′):28,29
| α′ = BE(Ag3d5/2) + KE(Ag MNN), |
At the same time, there are two observations that can not be explained by the adsorption of atmospheric impurities on silver particles. The first is the possibility of seeing silver particles on the non-sputtered HOPG after the same contact with the atmosphere (Fig. 2a). The second is that the disappearance of silver particles from STM images after treatment of the Ag/HOPG (sputtered) samples at P = 1 bar does not depend on the nature of the gas phase above the sample: argon, nitrogen, or a mixture of nitrogen with oxygen.
Another possible explanation of the particle disappearance from STM images is a penetration of the silver atoms into the upper graphite layers distorted by Ar+ sputtering, under the influence of atmospheric pressure. Then, the subsequent appearance of silver particles in STM images after annealing can be explained by wedging out of silver atoms. This mechanism can be realized if several upper layers of graphite are distorted, i.e. due to penetration of argon ions (according to SRIM simulation,30 the penetration depth of argon is about 5–6 graphite layers). The presence of Ar on the surface has been confirmed by the Ar2p peak in XPS spectra.
In spite of the earlier reported fact that surface defects of graphite cause changes in C1s spectra,31,32 we have observed only insignificant broadening of the C1s peak after Ar+ sputtering of HOPG. This fact can be explained by weak distortion of a surface after mild Ar+ sputtering and still high contribution to C1s spectra from non-damaged graphite. Therefore, C1s photoelectron spectra are not suitable to use in our case as opposed to the case of higher extent of defectiveness. Much more dramatic changes were observed in the C KVV Auger spectra of HOPG (Fig. 10), which are more surface sensitive (2–3 layers of graphite)33 than C1s ones. As a consequence, the Auger spectra were used as a support for STM data to explain the processes on the surface, notwithstanding its complexity and some limitations in use.
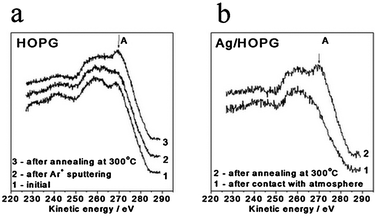 | ||
| Fig. 10 C KVV Auger spectra from clean HOPG (a) and Ag/HOPG (sputtered) (b) samples after different treatments. For (a): 1. initially annealed at 500 °C HOPG, 2. after Ar+ sputtering, 3. after annealing at 300 °C, 1 h. For (b): 1. after contact with atmosphere, 2. after annealing at 300 °C, 1 h. | ||
Distortion of the upper graphite layers can be characterized via analysis of a C KVV Auger spectrum, the line shape of which in a first approximation can be written as:34
| I(C KVV) ∼ (σ + πint + πs) × (σ + πint + πs), |
In accordance with HOPG valence band density of states (DOS), the high kinetic energy feature (A) is attributed to πint × πs and πint × πint terms, i.e. it characterizes π–π interaction between neighboring graphene sheets.33–35 Therefore, reduction of intensity of feature A after sputtering reflects the damaging of the upper surface layers of graphite. Argon ions with energy of 0.5 keV can produce two types of defects: carbon single/multi atom vacancy defects created by displacement of carbon atoms from graphene sheets (displacement threshold for one C atom < 50 eV) and interlayer defects caused by penetration of noble gas atoms into graphite.22,36–39 A decrease in feature A intensity suggests partial exfoliation of several upper layers of HOPG as a result of Ar penetration. The further annealing of graphite at 300 °C causes a healing of interlayer defects and resumption of π–π interactions between upper graphene sheets. Contact of the HOPG sample with atmosphere leads to a decrease in feature A as well (spectrum not shown). It means that feature A is sensitive also to the presence of atmospheric adsorbates on the HOPG surface.
Returning to the Ag/HOPG model catalysts, Fig. 10b shows C KVV spectra from the Ag/HOPG (sputtered) sample before (curve 1) and after (curve 2) final annealing at 300 °C. One can see that in agreement with the concept of creation of interlayer defects by Ar+ sputtering, the spectrum from the sample after exposure to the atmosphere does not show any indication of the feature A, even in the presence of silver atoms penetrated in the upper distorted layers of HOPG (note that Ag deposition is not able to produce the defects due to low condensation energy of silver). This indicates that π–π interactions between neighboring graphene sheets are absent in this case. Restoration of π–π interactions in the upper HOPG layers occurs after annealing of the sample at 300 °C. As a consequence, feature A appears in the spectra. This result is also in agreement with the observation that this procedure finally stabilizes the Ag/HOPG samples and silver particles do not disappear even after the gas exposure at atmospheric pressure (Fig. 6).
There are some additional facts in favor of the “silver penetration” concept. First, dependencies of C1s (Fig. 11a) and Ag3d (Fig. 11b) spectral intensities on polar angles of photoelectron escape (take-off angle) were measured for three Ag/HOPG (sputtered) samples: (i) from freshly prepared sample (just after silver deposition on the sputtered HOPG); (ii) after three treatments of the sample with air, nitrogen and argon at atmospheric pressure and RT followed by annealing in vacuum at 250 °C (see Results section) and transfer through the atmosphere from STM microscope into the photoelectron spectrometer and (iii) after final annealing in vacuum at 300 °C. It is easy to see that an increase of take-off angle (Θ) measured from perpendicular to the HOPG surface decreases the corresponding intensities due to decrease in depth of analysis (d):
| d = λ × cos Θ, |
 | ||
| Fig. 11 Angle-resolved C1s (a) and Ag3d (b) spectral intensities and Ag/C ratio (c) from Ag/HOPG (sputtered) sample after different treatment: silver deposition, 3 cycles of contact with atmosphere and final 1 h annealing at 300 °C in UHV. | ||
The higher intensity of Ag3d spectra for the freshly prepared sample is in agreement with our notification (see Results) that repetition of admission/annealing procedures decreases the mean size of silver particles. To estimate the location of silver in all three samples the Ag/C ratios have been calculated for three different take-off angles (see Fig. 11c). The Ag/C behavior for the freshly prepared sample clearly indicates the surface location of silver when the ratio of intensity of surface located component (Ag) to the bulk support (HOPG) is increased with decrease of analysis depth.40 Opposite Ag/C behavior, which is observed for two other samples, means the location of the great number of silver atoms in the subsurface layer of graphite in these cases. Annealing of the sample leads to an increase in the Ag/C ratio at lower depth of analysis, while the Ag/C ratio for the high analysis depth is almost constant (∼0.25). This result indicates partial wedging out of silver to the HOPG surface as a result of annealing.
Another important observation in favor of silver penetration into graphite has been obtained in an experiment with fast annealing of the sample. If the Ag/HOPG (sputtered) sample after silver deposition was heated up to 300 °C for several minutes, but not for several hours as usual, XPS did not show any signals in the Ag3d spectrum. Since a temperature of 300 °C is not high enough to evaporate silver, penetration of silver in HOPG layers deeper than XPS analysis depth should be proposed in this case.
We suppose that there are several simultaneous processes that take place on the surface of the sputtered HOPG with Ag nanoparticles as a result of sample heating: (1) sintering of metallic nanoparticles in the lateral direction; (2) penetration of silver into the distorted graphite layers; and (3) annealing of the defects (mainly interlayer defects). In our opinion, the relative rates of these processes control the formation of silver particles on the sputtered HOPG surface. In other words, the ability of silver to penetrate into surface layers of graphite depends on the degree of the interlayer defectiveness of HOPG. To shed light on the penetration effect, some additional experiments should be carried out in the future. For example, atomic force microscopy, which does not depend on the sample conductivity, can be used for this goal. To sum up, we propose the following mechanism of the processes occurring on the sputtered HOPG surface during the catalyst preparation. Ar+ sputtering of the HOPG leads to a formation of crater-like defects and to distortion and exfoliation of the upper graphite layers with the partial loss of conductivity in the direction perpendicular to the surface plane. After the Ag evaporation, atoms or small clusters of silver are able to penetrate into the interlayer space between exfoliated areas of the surface. Contact with the atmosphere results in the adsorption of contaminants on the defective graphite surface. In this case, Ag particles can not be obtained at STM. The following annealing at < 250 °C leads to adsorbate desorption from the surface and to a partial restoration of conductivity. In this case, STM images of Ag nanoparticles could be obtained. However, after the subsequent contact with the atmosphere, the surface of the sample is covered by adsorbates. Only annealing at higher temperatures (300 °C) results in appreciable healing of defects and finally stabilizes Ag nanoparticles. In spite of complexity of C KVV spectra, a good correlation between changes in high energy component of the C KVV Auger spectra (feature A) and STM data allows us to use this component as a reliable qualitative indicator for examination of the surface during the preparation of model samples.
The procedure of preparation of stable metallic nanoparticles on HOPG can be formulated as follows. First, sputtering of the annealed HOPG is necessary to create the defects (vacancy and interlayer defects). A second step is evaporation of a metal on the defective HOPG surface. After these stages, silver particles are stable against movements and sintering in lateral direction and are also capable of penetrating into the upper HOPG layers. Final stabilization of silver particles requires a complete annealing of interlayer defects of HOPG. It can be reached by slow heating of the sample up to temperatures of about 300 °C in UHV. This preparation technique will be applied for the synthesis of the model Ag/HOPG catalysts for further investigation of the size effects in ethylene and propylene oxidation reactions using in situXPS. Both reactions proceed at temperatures (150–230 °C) below the upper limit of the system stability in oxidation atmosphere (250 °C at 0.25 mbar of the ethylene/oxygen reaction mixture).
5. Summary
Surface defects play a crucial role in the stabilization of silver nanoparticles on a HOPG surface. Due to weak interactions with a well-ordered HOPG surface, silver particles are sintered into large agglomerates at the reaction temperatures (>150 °C). Formation of defects viaargon sputtering of the HOPG surface stabilizes the deposited silver particles against sintering.A procedure for preparation of stable silver nanoparticles on a HOPG surface was developed. This procedure includes a stage of surface defect formation (both vacancy defects on the surface and interlayer defects in the upper HOPG layers) followed by Ag deposition and a stage of the defect annealing (mainly interlayer defects) at T = 300 °C in UHV. The latter stage provides final stabilization of the system so that the silver particles prepared are seen in STM and SEM images after a long-term storage in the atmosphere (up to one month).
The prepared samples are suitable for in situ investigation in oxidation reactions, which proceed with the measurable rate at T < 250 °C (the upper limit of temperature stability). The low intensity of HOPG signals in O1s spectra also allows for the application of XPS in the studies of oxygen species adsorbed on the surface of metal particles supported on HOPG.
Acknowledgements
The project has been supported by RFBR (grant # 10-03-01138-a) and by Presidium of RAS (grant # 27.51). The authors are grateful to Gisela Weinberg from FHI for her help in SEM investigation.References
- Supported metals in catalysis, ed. J. A. Anderson and M. F. Garcia, Imperial College Press, London, 2005 Search PubMed.
- M. V. Twigg, Appl. Catal., B, 2007, 70, 2 CrossRef CAS.
- Catalytic naphtha reforming. Second edition revised and expanded, ed. G. J. Antos and A. M. Aitani, Marcel Dekker Inc., New York, Basel, 2004 Search PubMed.
- Fuel Cell Handbook, ed. J. H. Hirschenhofer, D. B. Stauffer and R. R. Engleman, Morgantown, West Virginia, 1998 Search PubMed.
- D. C. Lim, I. Lopez-Salido and Y. D. Kim, Surf. Sci., 2005, 598, 96 CrossRef CAS.
- H. Zhang, Q. Fu, Y. Yao, Z. Zhang, T. Ma, D. Tan and X. Bao, Langmuir, 2008, 24, 10874 CrossRef CAS.
- L. A. Langley, D. E. Villanueva and D. H. Fairbrother, Chem. Mater., 2006, 18, 169 CrossRef CAS.
- V. I. Bukhtiyarov, A. F. Carley, L. A. Dollard and M. W. Roberts, Surf. Sci., 1997, 381, L605 CrossRef CAS.
- D. C. Lim, I. Lopez-Salido, R. Dietsche, M. Bubek and Y. D. Kim, Surf. Sci., 2006, 600, 507 CrossRef CAS.
- R. A. van Santen and H. C. P. E. Kuipers, Adv. Catal., 1987, 35, 265 CAS.
- V. I. Bukhtiyarov, A. I. Nizovskii, H. Bluhm, M. Hävecker, E. Kleimenov, A. Knop-Gericke and R. Schlögl, J. Catal., 2006, 238, 260 CrossRef CAS.
- C. T. Campbell, J. Catal., 1985, 94, 436 CrossRef CAS.
- R. B. Grant and R. M. Lambert, J. Catal., 1984, 92, 364.
- S. N. Goncharova, E. A. Paukshtis and B. S. Bal'zhnimaev, Appl. Catal., A, 1995, 126, 67 CrossRef CAS.
- V. I. Bukhtiyarov, M. Hävecker, V. V. Kaichev, A. Knop-Gericke, R. W. Mayer and R. Schlögl, Phys. Rev. B, 2003, 67, 235422 CrossRef.
- V. I. Bukhtiyarov, A. I. Boronin, I. P. Prosvirin and V. I. Savchenko, J. Catal., 1994, 150, 268 CrossRef CAS.
- R. B. Grant and R. M. Lambert, J. Catal., 1984, 92, 364–375.
- Y. Lei, F. Mehmood, S. Lee, J. Greeley, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, R. J. Meyer, P. C. Redfern, D. Teschner, R. Schlögl, M. J. Pellin, L. A. Curtiss and S. Vajda, Science, 2010, 328, 224 CrossRef CAS.
- I. Lopez-Salido, D. C. Lim and Y. D. Kim, Surf. Sci., 2005, 588, 6 CrossRef CAS.
- A. Stable, K. Eichhorst-Gerner, J. P. Rabe and A. R. Gonzalez-Elipe, Langmuir, 1998, 14, 7324 CrossRef CAS.
- R. E. Palmer, S. Pratontep and H.-G. Boyen, Nat. Mater., 2003, 2, 443 CrossRef CAS.
- H. Hövel, Th. Becker, A. Bettac, B. Reihl, M. Tdchudy and E. J. Williams, J. Appl. Phys., 1997, 81, 154 CrossRef.
- L. L Wang, X. C. Ma, Y. Qi, P. Jiang, J. F. Jia, Q. K. Xue, J. Jiao and X. H. Bao, Ultramicroscopy, 2005, 105, 1 CrossRef.
- G. Zhang, S. Sun, M. Bostetter, S. Poulin and E. Sacher, J. Colloid Interface Sci., 2010, 350, 16 CrossRef CAS.
- I. Horcas, R. Fernandez, J. M. Gomez-Rodriguez, J. Colchero, J. Gomez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS.
- R. W. Joyner, M. W. Roberts and K. Yates, Surf. Sci., 1979, 87, 501 CrossRef CAS.
- M. Büttner and P. Oelhafe, Surf. Sci., 2006, 600, 1170 CrossRef.
- C. D. Wagner, Faraday Discuss. Chem. Soc., 1975, 60, 291 RSC.
- C. D. Wagner, L. H. Gale and R. H. Raymond, Anal. Chem., 1979, 51, 466 CrossRef CAS.
- http://www.srim.org/ .
- D.-Q. Yang and E. Sacher, Surf. Sci., 2002, 504, 125 CAS.
- D.-Q. Yang and E. Sacher, Surf. Sci., 2002, 516, 43 CrossRef CAS.
- A. P. Dementjev, K. I. Maslakov and A. V. Naumkin, Appl. Surf. Sci., 2005, 245, 128 CrossRef CAS.
- J.-C. Charlier, X. Gonze and J.-P. Michenaud, Phys. Rev. B, 1991, 43, 4579 CrossRef CAS.
- D. D. L. Chung, J. Mater. Sci., 2002, 37, 1475 CrossRef CAS.
- W. Choi, C. Kim and H. Kang, Surf. Sci., 1993, 281, 323 CrossRef CAS.
- D. Marton, K. J. Boyd, T. Lytle and J. W. Rabalais, Phys. Rev. B, 1993, 48, 6757 CrossRef CAS.
- J. R. Hahn and H. Kang, Phys. Rev. B, 1999, 60, 6007 CrossRef CAS.
- I. N. Kholmanov, J. Edgeworth, E. Cavaliere, L. Gavioli, C. Magnuson and R. S. Ruoff, Adv. Mater., 2011, 23, 1675–1678 CrossRef CAS.
- O. A. Baschenko, V. I. Bukhtiyarov and A. I. Boronin, Surf. Sci., 1992, 271, 493 CrossRef.
| This journal is © The Royal Society of Chemistry 2011 |