Interaction of ethene and ethyne with bare and hydrogenated Ir4 clusters. A density functional study†
Galina P.
Petrova
a,
Georgi N.
Vayssilov
*a and
Notker
Rösch
*b
aFaculty of Chemistry, University of Sofia, 1126 Sofia, Bulgaria. E-mail: gnv@chem.uni-sofia.bg
bDepartment Chemie & Catalysis Research Center, Technische Universität München, 85747 Garching, Germany. E-mail: roesch@mytum.de
First published on 24th June 2011
Abstract
The paper reports a computational study of species that can be formed during ethene hydrogenation on iridium clusters. The simulated concentrations of the complexes (C2Hm)Ir4Hn (m = 2–5, 0 ≤ n ≤ 14 − m) based on calculated Gibbs free energies suggest at low temperature and high hydrogen pressure π-bonded ethene to be the dominant species at the Ir4 cluster covered by hydrides. At higher temperature and lower H2 pressure, this model predicts ethylidyne and, subsequently, di-σ-coordinated ethyne with a minor or zero amount of hydride ligands on the metal cluster. Ethyl, vinyl, and vinylidene species were calculated to be less stable over the range of the hydrogen coverage studied. Ethane desorption from the most stable complex was calculated to be thermodynamically favorable for systems in which at least three hydride ligands will remain on the metal cluster after desorption. Adsorption of one of these organic ligands and/or hydrogen results in an oxidation of the metal moiety; this effect is more pronounced in complexes with ethylidyne, vinyl, and vinylidene. The calculated vibrational spectra of ethylidyne on Ir4Hn clusters agree well with available experimental data for this species on iridium surfaces and supported metal particles. The spectra of the various organic species in the region of C–H stretching modes (3300–2700 cm−1) were calculated to overlap, in particular in the presence of hydride ligands on the metal moiety.
1 Introduction
Transition metals are traditionally applied as industrial catalysts for hydrocarbon transformation reactions such as hydrogenation, dehydrogenation, methane conversion, cyclization processes, etc. Various experimental studies of hydrogenation processes occurring on surfaces or supported metal clusters provide information about the changes in the structure of the metal moiety as a result of adsorption and catalyzed processes based on a detailed analysis (XPS, EXAFS) of the adsorption complexes. Infra-red (IR) spectroscopy may be applied for identifying the coordination mode of the organic adsorbates and, in some cases, may reveal information on intermediates of (de)hydrogenation processes.Ethene adsorption on transition metal surfaces and various transformations of the organic adsorbate on the surface have been studied both experimentally1–6 and by theoretical modeling.7–9 To benefit from the high catalytic activity of the transition metal, to decrease the amount of metal used, and eventually to control the selectivity of the reaction, attempts have been made to prepare and test the catalytic activity of small metal particles anchored on various types of supports.10–13 Therefore, the interaction of ethene and ethyne with transition metal clusters, both in the gas phase or supported on metal–oxide surfaces, has been the subject of many experimental15–23 and theoretical24–26 investigations. These systems and the resulting processes also are relatively simple and amenable to computational modeling.
In previous theoretical studies we modeled the adsorption of hydrogen on zeolite-supported tetrahedral metal clusters27–29 as well as on charged Ir4 species in the gas phase.30 The obtained results showed that up to 6 H2 molecules can be adsorbed dissociatively on such iridium clusters, either free or zeolite-supported. According to the thermodynamic model derived from these calculations31 and the comparison with experimental metal–metal distances,27 the iridium clusters are covered with hydrogen both after preparation and during alkene hydrogenation. To clarify the influence of the hydrogen coverage on the adsorption properties of the cluster and thus, on catalytic reactions, we report here the results from computational modeling of various species that can be formed under reaction conditions for ethene hydrogenation, adsorbed on bare and hydrogenated Ir4Hn clusters with n = 0, 3, 6, 9. Though ignoring the potential support effect, this approach allowed us to model a large variety of complexes: ethene and ethyne coordinated in π- or di-σ-fashion to Ir4Hn clusters, as well as several intermediate species suggested to exist on metal catalysts by experiments1–4,10–21 or computational modeling:5,7,8ethyl (H3CH2C–), ethylidyne (H3CC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) ), vinyl (H2CHC–), and vinylidene (H2CC
), vinyl (H2CHC–), and vinylidene (H2CC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ). The obtained series of structures were analyzed regarding their stability, geometry, and electronic structure to compare them with available pertinent experimental data from EXAFS and XPS. The vibrational spectra of the adsorption complexes were also simulated because IR and high-resolution electron energy loss spectroscopy (HREELS) are often applied in experiments to identify organic adsorbates on metal surfaces. The tetragonal iridium clusters were selected for this study because experimental information from IR and EXAFS spectroscopies is available from the works of Gates and co-workers for ethene and propene hydrogenation over such supported clusters.14,15
). The obtained series of structures were analyzed regarding their stability, geometry, and electronic structure to compare them with available pertinent experimental data from EXAFS and XPS. The vibrational spectra of the adsorption complexes were also simulated because IR and high-resolution electron energy loss spectroscopy (HREELS) are often applied in experiments to identify organic adsorbates on metal surfaces. The tetragonal iridium clusters were selected for this study because experimental information from IR and EXAFS spectroscopies is available from the works of Gates and co-workers for ethene and propene hydrogenation over such supported clusters.14,15
2 Results and discussion
We first describe the structures of the modeled adsorption complexes of various organic moieties C2Hm on bare and hydrogenated Ir4Hn clusters. Then we discuss the relative stability of the complexes of the same chemical composition to clarify which organic adsorbate is the most stable at a certain amount of hydrogen. Subsequently, we analyze how the electron density distributions of the adsorption complexes varies and we compare them to those of Ir4Hn clusters. To assist in a potential experimental discrimination of the adsorbed organic species we report their calculated vibrational frequencies and compare them to pertinent experimental data. Direct information on the complexes modeled (ethene on Ir4Hn) is limited to structural features (from EXAFS) and experimental vibrational spectra in the v(C–H) region.14,15 Therefore, we compare the model results also to experimental or computational studies of related systems, e.g., ethene-derived species on clusters of Pt or Pd or on single crystal surfaces of iridium.2.1 Structure of the complexes
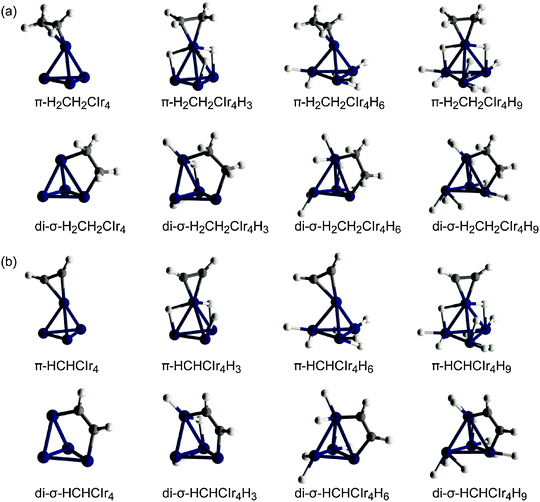
We modeled eight types of organic adsorbates on the metal cluster (Scheme 1; Fig. 1 and 2). The initial positions of the hydride ligands in the complexes were adopted from the optimized structures of the Ir4Hn clusters reported earlier.27,30 Of course, more isomers are possible than those structures examined in the present work. Yet, the trends derived from the models allow one to estimate how hydride ligands affect the preferred types and coordination modes of the organic moieties in the adsorption complexes.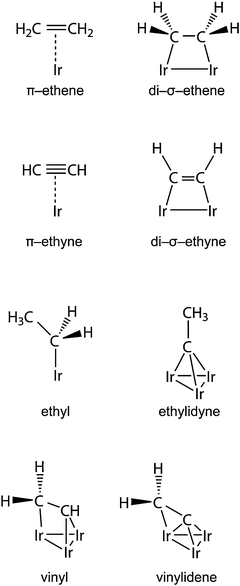 | ||
| Scheme 1 | ||
 | ||
| Fig. 1 Sketches of the most stable structures of the adsorption complexes of π- and di-σ-coordinated (a) ethene and (b) ethyne on bare and hydrogenated Ir4Hn clusters, n = 0, 3, 6, 9. | ||
 | ||
| Fig. 2 Sketches of the optimized structures obtained by adsorption of (a) ethyl, (b) ethylidyne, (c) vinyl, and (d) vinylidene on Ir4Hn clusters, n = 0, 3, 6, 9. | ||
The increasing number of hydrogen ligands has only a minor influence on the local structures of the π-coordinated C2H4 at the clusters (Fig. 1a); crucial interatomic distances are quite constant: Ir–C = 213 ± 2 pm, C–C = 144 ± 2 pm. The C–C distance is intermediate between the lengths of single and double bonds calculated for ethane, 153 pm, and ethene, 134 pm, in the gas phase. The isomer with 3 H ligands coordinated at distorted bridge Ir–Ir positions of π-H2CH2CIr4H3 is energetically preferred. With increasing hydrogen loading of the cluster, structures with all H-ligands atop Ir atoms are more stable (Fig. 1, Fig. S1a in the ESI†). Di-σ coordination of ethene at various hydrogenated metal moieties results in longer C–C bonds, up to 152 ± 1 pm; this corresponds to a single C–C bond and sp3 hybridization of the C atoms, in agreement with the tetrahedral coordination of the C atoms. In complexes with lower hydrogen coverage of the metal moiety, the Ir–C distances, 202–205 pm, are calculated to be shorter than in π-complexes of ethene (Table 1).
| Structure | ΔEa | E ads b | ΔEDAc | ΔGa | ΔGadsb | ΔGDAc | 〈Ir–Ir〉d | ΔRe | Ir–Cd | C–C | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C2H4 | C2H2 | C2H4 | C2H2 | |||||||||
| a Relative energy ΔE (relative Gibbs free energy ΔG) of the complexes. b Energy Eads (Gibbs free energy ΔGads) of adsorption of ethene/ethyne on the cluster Ir4Hn. c Energy, ΔEDA (Gibbs free energy, ΔGDA) of H2 dissociative adsorption (per H atom). d Average Ir–Ir distance in the metal particle. According to experimental data (ref. 14) for tetrahedral iridium particles adsorbed on γ-Al2O3, the coordination number of Ir is estimated at 3.3 with respect to other Ir atoms, 〈Ir–Ir〉 = 265–268 pm, and at 0.3–0.5 with respect to C atoms, Ir–C = 186–193 pm. e Difference between the longest and shortest intermetallic distance in the cluster. f The structure is obtained during the optimization of the complex H3CH2CIr4 as one of the hydrogen atoms of the organic ligand was found to migrate spontaneously to the metal particle. | ||||||||||||
| Ethene | ||||||||||||
| π-H2CH2CIr4 | −213 | −213 | −169 | −169 | 248 | 7 | 211, 211 | 144 | ||||
| π-H2CH2CIr4H f | −276 | −208 | −63 | −235 | −166 | −67 | 251 | 33 | 212, 213 | 144 | ||
| π-H2CH2CIr4H3 | −408 | −212 | −65 | −371 | −170 | −67 | 256 | 26 | 211, 211 | 145 | ||
| π-H2CH2CIr4H6 | −672 | −182 | −76 | −609 | −129 | −73 | 257 | 22 | 214, 215 | 142 | ||
| π-H2CH2CIr4H9 | −834 | −197 | −69 | −762 | −146 | −66 | 266 | 8 | 211, 212 | 144 | ||
| di-σ-H2CH2CIr4 | −209 | −209 | −165 | −165 | 249 | 19 | 204, 205 | 151 | ||||
| di-σ-H2CH2CIr4H3 | −424 | −227 | −71 | −385 | −184 | −73 | 253 | 71 | 203, 204 | 153 | ||
| di-σ-H2CH2CIr4H6 | −669 | −179 | −77 | −608 | −128 | −74 | 261 | 36 | 202, 205 | 152 | ||
| di-σ-H2CH2CIr4H9 | −814 | −178 | −67 | −734 | −117 | −63 | 265 | 13 | 210, 210 | 151 | ||
| Ethyne | ||||||||||||
| π-HCHCIr4 | −76 | −289 | −26 | −250 | 248 | 16 | 197, 198 | 132 | ||||
| π-HCHCIr4H3 | −256 | −273 | −60 | −205 | −229 | −60 | 254 | 17 | 197, 197 | 132 | ||
| π-HCHCIr4H6 | −528 | −252 | −75 | −456 | −201 | −72 | 257 | 24 | 199, 200 | 131 | ||
| π-HCHCIr4H9 | −660 | −237 | −65 | −581 | −189 | −62 | 267 | 7 | 202, 202 | 131 | ||
| di-σ-HCHCIr4 | −114 | −327 | −63 | −287 | 246 | 83 | 192, 193 | 139 | ||||
| di-σ-HCHCIr4H2 | −245 | −310 | −66 | −200 | −281 | −68 | 246 | 68 | 192, 193 | 139 | ||
| di-σ-HCHCIr4H3 | −307 | −324 | −64 | −263 | −286 | −67 | 253 | 74 | 193, 195 | 137 | ||
| di-σ-HCHCIr4H6 | −526 | −250 | −69 | −461 | −205 | −66 | 262 | 39 | 196, 196 | 137 | ||
| di-σ-HCHCIr4H9 | −666 | −243 | −61 | −579 | −186 | −57 | 266 | 11 | 200, 202 | 136 | ||
The optimized structures of π- and di-σ-coordinated ethyne (Fig. 1b) were obtained via formal dehydrogenation of the corresponding ethene complexes. The structure of the Ir4Hn moiety in these two series of complexes resembles the corresponding structures with π- and di-σ-coordinated ethene. The higher C–C bond order in the HCHCIr4Hn complexes is reflected in a shorter C–C distance, by more than 10 pm compared to the corresponding complexes of ethene. The C–C distance of π-coordinated ethyne, 131–132 pm, is 2–3 pm shorter than the distance of free ethene, 134 pm. The C–C bond of di-σ-coordinated ethyne, 136–139 pm, and bond angles close to 120° suggest sp2 hybridization of the C atoms and a bond order close to 2. The Ir–C distances in both series of ethyne complexes (197–202 pm for π- and 192–202 pm for di-σ) are 10–15 pm shorter than the corresponding distances of adsorption complexes of ethene. The Ir–C distances of the di-σ structures of HCHCIr4Hn (n = 0–6), 192–196 pm (Table 1), are close to the experimentally observed Ir–C distances, 186–193 pm.14 The Ir–C distances of di-σ-coordinated ethyne increase with increasing hydride coverage, by up to 10 pm, similarly to the structures of di-σ-bonded ethene.
Fig. 2a shows the structures of adsorption complexes of the ethyl species. All attempts to obtain a structure of an H3CH2CIr4 complex on bare Ir4 failed because during optimization one of the hydrogen centers of the CH3 group of the ethyl radical migrated to the metal moiety resulting in a stable π-coordinated complex of ethene on the cluster Ir4H; that complex follows the trends discussed for the series of π-coordinated ethene structures. Stable adsorption of ethyl species was found on the hydrogenated iridium clusters Ir4Hn for n = 3, 6, and 9. In these complexes the ethyl ligand is coordinated via an Ir–C σ-bond to the metal tetrahedron. The Ir–C distances, 205 ± 2 pm, in the model complexes are somewhat longer than the experimental values, 186–193 pm.14 As expected, the C–C distance of H3CH2CIr4Hn, 153–154 pm, is close to the C–C bond of free ethane.
To account for the peculiar effect of the hydrogen loading on the stability of the complexes containing an ethylidyne (H3CC) ligand (see also section 2.2), we modeled structures with 0–4, 6, and 9 H ligands. For complexes with up to 6 hydrides, the organic ligand coordinates to a facet of the metal tetrahedron forming three Ir–C σ-bonds of 198 ± 2 pm (averaged over all ethylidyne complexes; Fig. 2b). However, when nine hydride ligands saturate the bonding capacity of the metal moiety, one Ir–C bond elongates to 228 pm at the expense of two shorter Ir–C distances, 193 and 195 pm. Thus, the complex H3CCIr4H9 exhibits two Ir–C bonds only, with ethylidyne bridging an Ir–Ir bond. The C–C bond in all ethylidyne complexes is slightly shorter than a single C–C bond.
A vinyl ligand also coordinates to a facet of the metal tetrahedron; the CH fragment attaches to an Ir–Ir bond and the CH2 fragment forms only one Ir–C bond (Fig. 2c). The Ir–C bonds of adsorbed vinyl, 204 ± 4 pm, are longer than those of adsorbed ethylidyne. The local structure of the organic ligand corresponds to sp3 hybridization of both C centers, as corroborated by the C–C distance, 151 ± 4 pm. Further “dehydrogenation” of the organic adsorbate results in adsorption complexes where the vinylidene moiety forms similar local structures as the vinyl adsorbates. The vinylidene fragment is coordinated at a facet of the cluster where an H-free C center connects to three metal atoms with Ir–C distances of 188–215 pm while the CH2 fragment binds to a single Ir atom at a slightly longer distance, 214–222 pm (Fig. 2d). The C–C bond of adsorbed vinylidene, calculated at 143–146 pm, is between a single and a double C–C bond; the CH2 group exhibits a local structure compatible with C sp2 hybridization, but slightly distorted due to the coordination to the metal particle.
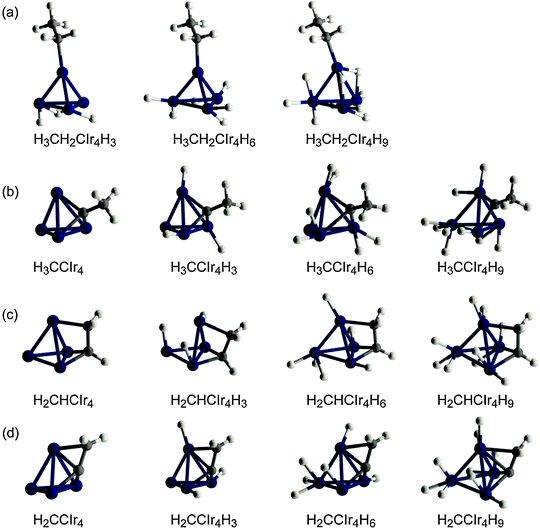
The Ir4 moiety of most adsorption complexes preserves a tetrahedral shape upon relaxation despite the presence of the organic and the hydride ligands. Partial exceptions are four complexes with low H loading (n = 0, 3) in which either the di-σ-coordination of ethene or ethyne, or the bridging coordination of hydrogen ligands in a vinyl complex, results in very long Ir–Ir distances, above 300 pm (Fig. 1 and 2c). Previously, we had calculated metal–metal distances of free and zeolite-supported Ir4Hn to increase from 247 pm in a bare cluster (n = 0) with hydrogen loading in almost linear fashion to 273 pm for n = 12, i.e., about 6.5 pm per 3 H ligands.27,28,30 Also for the π and di-σ complexes of ethene or ethyne, we calculated average distances 〈Ir–Ir〉 that increase almost linearly with the number of hydrogen ligands on the metal moiety (Fig. 3a). Compared to our previous results for Ir4Hn,27 an organic ligand induces a slightly steeper increase of 〈Ir–Ir〉 with the hydrogen loading. Also, EXAFS experiments14 show such an increase for alumina-supported Ir4 clusters during the catalytic hydrogenation of ethene. However, this observed elongation of 〈Ir–Ir〉 could be due to either the presence of an organic adsorbate or H2 dissociative adsorption because the reaction gas mixture contains H2 and C2H4.
 | ||
| Fig. 3 Average 〈Ir–Ir〉 distances in the complexes (C2Hm)Ir4Hn as a function of the number n of H atoms coordinated to the metal particle: (a) π- and di-σ-coordinated ethene (blue and red triangles, respectively); π- and di-σ-coordinated ethyne (blue and red circles, respectively); (b) ethyl (magenta diamonds); ethylidyne (green squares); vinyl (dark blue stars); vinylidene (cyan crosses). The black line in both panels corresponds to Ir4Hn species in the gas phase (〈Ir–Ir〉 = 247.8 + 1.71 n; ref. 27). | ||
Fig. 3b shows how the type of the other four organic ligands—ethyl, ethylidyne, vinyl or vinylidene—affects the change of the 〈Ir–Ir〉 distances for different hydrogen loadings. Adsorption complexes of ethyl feature a similar trend of 〈Ir–Ir〉 with hydrogen loading as determined for Ir4Hn clusters without organic ligand. However, the trends in the complexes of ethylidyne, vinyl, or vinylidene differ notably. These organic ligands cause an elongation of 〈Ir–Ir〉 to ∼258 pm even in the absence of hydrogen ligands; hence this distance is 10 pm longer than in the bare metal cluster. Increasing the hydrogen loading above n = 3 results in longer 〈Ir–Ir〉 distances where the increment per H ligand is similar to that of complexes of ethene or ethyne (Fig. 3).
2.2 Stability issues
Tables 1 and 2 provide key energy characteristics of the systems studied. All complexes of the various model series were found to be stable with respect to the reference: the bare iridium cluster, ethene and the appropriate number of H2 in the gas phase. To estimate the energetic preference among the complexes (C2Hm)Ir4Hn (m = 2–5, n = 0, 3, 6, 9, or 12) with different organic adsorbates as a function of the amount of hydrogen in the system, we compared the stability of the complexes with the same chemical composition, i.e., as function of the total number k = n + m of hydrogen atoms in the system, including both hydride ligands of the metal particle and H substituents of the adsorbed organic moiety (Fig. 4a). Such a comparison allows us to determine the thermodynamically most stable form of a given chemical composition and to highlight the direction of the thermodynamic driving force for chemical transformations at a defined amount of hydrogen in the system. In this scheme, ΔG values of the hydrogenated complexes Ir4Hn without organic adsorbate can be ascribed to a non-interacting system of Ir4Hn and C2H4. All organic adsorbates on Ir4Hn were calculated to be more stable than this non-interacting reference system, by up to 310 kJ mol−1. For low amounts of hydrogen, k ≤ 4, di-σ-coordinated ethyne was found to be the most stable adsorbed species, followed by ethylidyne for 5 ≤ k ≤ 8, and π-bonded ethene for a large amount of hydrogen, k ≥ 9.| Structure | ΔEa | ΔEDAb | ΔGa | ΔGDAb | 〈Ir–Ir〉c | ΔRd | Ir–Cc | C–C |
|---|---|---|---|---|---|---|---|---|
| a Relative energy ΔE (relative Gibbs free energy ΔG) of the complexes. b Energy, ΔEDA (Gibbs free energy, ΔGDA) of H2 dissociative adsorption (per H atom). c Average Ir–Ir distance in the metal particle. According to experimental data (ref. 14) for tetrahedral iridium particles adsorbed on γ-Al2O3, the coordination number of Ir is estimated at 3.3 with respect to other Ir atoms, 〈Ir–Ir〉 = 265–268 pm, and to 0.3–0.5 with respect to C atoms, Ir–C = 186–193 pm. d Difference between the longest and shortest intermetalic distance in the cluster. | ||||||||
| Ethyl | ||||||||
| H3CH2CIr4H3 | −409 | −376 | 251 | 22 | 203 | 154 | ||
| H3CH2CIr4H6 | −690 | −628 | 257 | 36 | 204 | 153 | ||
| H3CH2CIr4H9 | −821 | −755 | 264 | 8 | 207 | 154 | ||
| Ethylidyne | ||||||||
| H3CCIr4 | −124 | −83 | 257 | 27 | 197, 198, 198 | 152 | ||
| H3CCIr4H | −232 | −108 | −189 | −106 | 258 | 30 | 196, 197, 199 | 152 |
| H3CCIr4H2 | −341 | −109 | −303 | −110 | 258 | 34 | 196, 197, 198 | 152 |
| H3CCIr4H3 | −455 | −111 | −416 | −111 | 257 | 32 | 199, 197, 198 | 151 |
| H3CCIr4H4 | −499 | −94 | −462 | −95 | 258 | 32 | 196, 199, 200 | 151 |
| H3CCIr4H6 | −556 | −72 | −493 | −68 | 261 | 30 | 193, 195, 228 | 150 |
| H3CCIr4H9 | −713 | −65 | −629 | −61 | 267 | 43 | 190, 196 | 149 |
| Vinyl | ||||||||
| H2CHCIr4 | −53 | −6 | 256 | 38 | 202, 204, 208 | 151 | ||
| H2CHCIr4H3 | −211 | −53 | −163 | −52 | 262 | 48 | 204, 205, 204 | 152 |
| H2CHCIr4H6 | −525 | −79 | −459 | −75 | 260 | 11 | 200, 204, 208 | 151 |
| H2CHCIr4H9 | −678 | −69 | −590 | −65 | 268 | 16 | 201, 205, 204 | 150 |
| Vinylidene | ||||||||
| H2CCIr4 | −8 | 39 | 256 | 29 | 196, 196, 203, 217 | 146 | ||
| H2CCIr4H3 | −250 | −81 | −197 | −79 | 256 | 27 | 188, 200, 215, 222 | 144 |
| H2CCIr4H6 | −502 | −82 | −440 | −80 | 266 | 32 | 192, 200, 203, 222 | 143 |
| H2CCIr4H9 | −610 | −67 | −527 | −63 | 272 | 31 | 195, 205, 213, 214 | 146 |
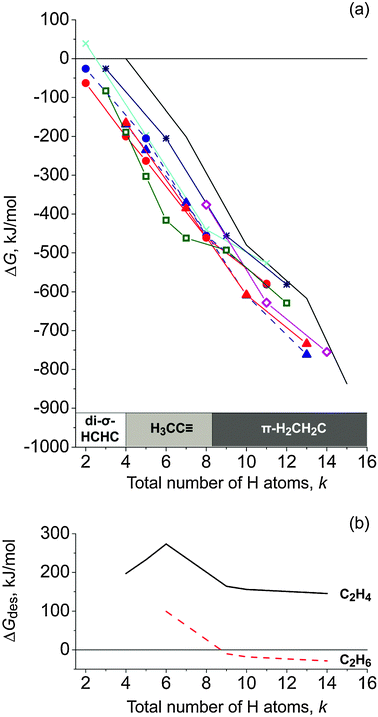 | ||
| Fig. 4 (a) Relative Gibbs free energy (298 K) of the modeled adsorption complexes (C2Hm)Ir4Hn and Ir4Hn (m = 2–5; n = 0, 3, 6, 9, 12) as a function of the total number of H atoms in the system (k = n + m): ethene—triangles, ethyne—circles, ethyl—diamonds (magenta), ethylidyne—squares (green), vinyl—stars (dark blue), vinylidene—crosses (cyan). Blue (dashed line) color is applied for π-coordinated ethene or ethyne; red—for di-σ-bonded species. The relative Gibbs free energies of Ir4Hn clusters are presented by a solid black line (ref. 27). (b) Gibbs free energy for the desorption of ethene (solid black line) or ethane (dashed red line) from the most stable complex (C2Hm)Ir4Hn for a given k. | ||
Ethyl, vinyl, and vinylidene complexes were calculated to be the least stable species over the whole range of hydrogen coverage of the iridium cluster.
The relative stability of the complexes in each of the series (except for ethylidyne), varies linearly with k, ΔG = A + B × k; the corresponding RMS values are 0.96–0.99 (Table S1 of the ESI†). From coefficient B one can estimate the average Gibbs free energy, ΔGDA, of dissociative adsorption of hydrogen on the metal cluster in the presence of an organic ligand. The resulting average values of ΔGDA per H atom for the complexes with π- or di-σ-bonded ethene or ethyne are −68, −64, −64 and −58 kJ mol−1, respectively. For the complexes of ethylidyne, H3CCIr4Hn, the relative Gibbs free energy depends on the amount of hydrogen in the complex in a more complicated fashion. Here exist two subsets, n ≤ 3 and n > 3, characterized by straight lines of different slopes (Fig. 4a). In the former region, corresponding to the adsorption of the first 3 H ligands at the cluster H3CCIr4, the slope is −111 kJ mol−1, but is only −34 kJ mol−1 in the second region. This strong change in free energy trend can be related to the attachment of the first three hydride ligands in terminal positions to each of the Ir centers bound to the ethylidyne. When these positions are occupied, the next H ligands are bound much weaker to the distant Ir atom or as a second ligand to one of the iridium atoms coordinating the organic ligand.
From the relative Gibbs free energies of the species modeled we calculated their concentrations (Fig. 5) as function of temperature and hydrogen pressure P(H2); see the ESI for details.† At low temperature, 298 K, only the most stable species at highest hydrogen loading is formed, π-H2CH2CIr4H10. Accordingly, with increasing temperature (up to 473 K; Fig. 5a), ethylidyne species H3CCIr4H3 appears at low P(H2), while π-bonded ethene remains dominant at higher P(H2). For temperatures in the range of 573–673 K (Fig. 5b) di-σ-HCHCIr4 becomes dominant at low P(H2), at increasing the hydrogen pressure this is followed by ethylidyne and π-bonded ethene. Decreasing the total pressure in the system has a similar effect on the type of the dominant species as the increase of the temperature. Thus, among the various types of systems (C2Hm)Ir4Hn considered here, only three types of species are dominant within wide ranges of temperature and hydrogen pressure. Some minor amounts of adsorbed ethene or ethylidyne species (with various amounts of hydrogen on the metal cluster) can be seen in the high temperature plots (T ≥ 473 K) with total concentrations at most ∼5% at 473 K and 15% at 673 K.
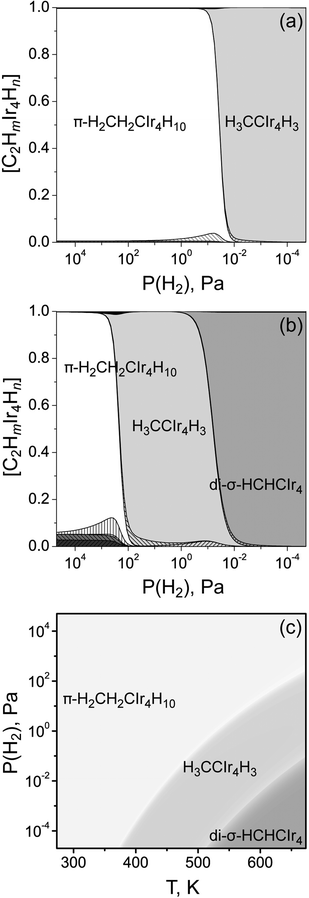 | ||
| Fig. 5 Calculated concentrations of different adsorbed species at (a) T = 473 K and (b) T = 673 K. The P(H2)/T diagram (c) shows the dominant adsorbed species. For all panels, Ptotal = 1.013 × 105 Pa and P(C2H4) = 1.00 × 104 Pa. The minority species in panels (a) and (b) are π-H2CH2CIr4H9, H3CCIr4H4, π-HCHCIr4H12, di-σ-H2CH2CIr4H10, π-H2CH2CIr4H9, H3CCIr4H4, H3CCIr4H3, di-σ-HCHCIr4H. | ||
Following these observations we constructed a P(H2)/T diagram (Fig. 5c) showing the type of the dominant species. π-H2CH2CIr4H10 are the only species for a large part of the diagram. At higher temperatures and lower values of the hydrogen pressure this model predicts the species H3CCIr4H3 and, subsequently, di-σ-HCHCIr4 to form. The appearance of these species (in particular at higher temperatures) follows the order described above for the dependence of ΔG[(C2Hm)Ir4Hn] on the total number k of hydrogen atoms in the system. In experiment, ethene hydrogenation on supported size-selected clusters typically is carried out at a total pressure ∼1 × 105 Pa, P(H2) and P(C2H4) in the region 5 × 103 to 4 × 104 Pa, and T = 273–294 K.14,15 Other experimental studies were conducted at higher temperatures, up to 410 K or 473 K.13,21 Under these conditions our simulations (Fig. 5c) suggest that the dominant species will be π-bonded ethene with a high hydrogen coverage of the metal cluster.
Our previous modeling of hydrogenated M4clusters on zeolite support27,28 suggested that the optimum amount of adsorbed hydrogen on these clusters is lower than that of clusters of the same size in the gas phase because some coordination sites on the metal framework are blocked by the support. Therefore, changes in the type of the most stable organic species on supported Ir4Hn clusters is expected to occur at lower amounts of adsorbed hydrogen, i.e., at lower hydrogen pressure. Thus, π-bonded ethene can be expected as the dominant species at even lower values of P(H2) than those shown in Fig. 5c.
The calculated values of ΔEDA (Tables 1 and 2), between −61 kJ mol−1 and −77 kJ mol−1, show that ethene or ethyne as ligand of the metal particle slightly reduce the binding energy of hydrogen on the iridium cluster, compared to bare Ir4 species for which the average energy of dissociative adsorption of hydrogen from the gas phase was calculated at −74 kJ mol−1.30 In contrast, the complexes with ethylidyne show notable increase of the ΔEDA value for the three initial H ligands, i.e., this organic ligand substantially modifies the energy for dissociative adsorption of hydrogen on the iridium moiety, as described above. As the type of the most stable organic species on the cluster varies with hydrogen loading, the calculated Gibbs free energy for the desorption of ethene or ethane from the complex (C2Hm)Ir4Hn depends non-monotonously on k = n + m (Fig. 4b). While the desorption of ethene is calculated to be always endergonic (by at least 150 kJ mol−1), the desorption of ethane becomes thermodynamically favorable for complexes containing at least 9 hydrogen atoms, i.e., in which at least three hydride ligands will remain on the metal cluster after desorption.
The above discussion addresses a large variety of rather different chemical processes, namely adsorption of ethene or ethyne on bare (Ir4) or hydride metal clusters (Ir4Hn), changes of the adsorption mode (π- or di-σ-coordinated), transformations between different adsorbed species, e.g., dehydrogenation of ethene in H2CH2CIr4Hn to ethyne and 2 hydride ligands, coordinated to the metal particle. As can be seen from the provided data, the adsorption of ethene and ethyne on the bare and hydrogenated iridium clusters is exergonic and the Gibbs free energy for the adsorption varies from −117 kJ mol−1 to −184 kJ mol−1 for ethene and from −186 kJ mol−1 to −287 kJ mol−1 for ethyne. For ethene adsorption on the metal particle, we were not able to identify a clear dependence of the adsorption energy on the number of adsorbed H ligands on the metal clusters. This is not so for the adsorption energy of ethyne: it becomes less negative with increasing hydrogen loading of the cluster, by 5–6 kJ mol−1 per H. At low hydrogen loading, we calculated the most negative Gibbs free energy of ethyne adsorption in the di-σ-mode, below −250 kJ mol−1, but at an H/Ir ratio of 1.5 the adsorption becomes notably less favorable as ΔGads rises sharply by more than 70 kJ mol−1. Concomitantly, the Ir–C distances elongate, by up to 7 pm.
The dehydrogenation of ethene in the complexes H2CH2CIr4Hn to adsorbed ethyne, with the formation of H2 in the gas phase, was calculated to be endergonic for both types of coordination and the energy required for the processes depends on the amount of hydrogen on the metal cluster.
Valero et al.24 using a density functional method (PW91 functional) calculated the adsorption energy of ethene in di-σ- and π-modes to a Pd4 cluster as −86 and −118 kJ mol−1, respectively. Those calculations suggested that π-H2CH2CPd4 is more stable than di-σ-H2CH2CPd4, by 32 kJ mol−1 in the gas phase and by 10 kJ mol−1 on dehydrated alumina. For Pd4 on a hydrated alumina surface, di-σ-coordinated ethene was identified as the preferred species. The adsorption energy of ethene on the surface Pd(111),7 obtained with periodic DFT (PW91) slab models, was similar for di-σ coordination, −71 to −90 kJ mol−1 (in absolute terms, the adsorption energy decreases with the coverage), but notably lower for π-coordination, −57 to −74 kJ mol−1, due to the lack of low coordinated corner metal atoms. The binding to the Pt(111) surface8 was calculated stronger, −105 to −121 kJ mol−1 (di-σ coordination) and −60 to −84 kJ mol−1 (π-coordination). The binding of ethene to the bare Ir4, estimated in the present study, is notably stronger, with calculated adsorption energies of −209 kJ mol−1 (di-σ) and −213 kJ mol−1 (π), likely due to coordinative undersaturation of the metal atoms in the cluster. In a DFT model study with the same functional, ethyne was calculated to adsorb on Pd(111)7 at a three-fold site with both carbon centers close to sp3 hybridization with an adsorption energy of −203 kJ mol−1. This type of coordination was also obtained for di-σ bound ethyne on a Pt10 cluster25 with a similar adsorption energy, −209 kJ mol−1 (using the B3LYP method). We calculated the adsorption energy of ethyne on Ir4 in the same coordination mode, on a facet of the Ir4 cluster with each of the carbon atoms coordinating two Ir centers, also to be close to these values, −224 kJ mol−1. However, this structure is 86 kJ mol−1 less stable than the most stable di-σ complex of ethyne, where the molecule is bound to an Ir–Ir bond and both C atoms are in sp2 hybridization (Fig. 1b, Scheme 1, and Table 1), with an adsorption energy calculated at −310 kJ/mol. The low stability of ethyl, vinyl, and vinylidene adsorbates, compared to adsorbed ethene or ethylidyne species, parallels their lower stability on open metal surfaces, e.g., see the reaction energy profiles in ref. 7.
2.3 Electron density distribution
First we will consider the adsorption complexes of the organic species under discussion on the bare metal cluster, without hydride ligands. As expected, adsorption of an organic ligand results in an oxidation of the metal moiety which is confirmed by both the overall stabilization of electronic levels of the metal centers and their larger atomic charges. The stabilization of the Ir 4f levels, averaged over all metal centers, increases roughly in the order π-C2H4 < π-C2H2 < di-σ-C2H2 ≈ di-σ-C2H4 ≈ H3CC < H2CHC < H2CC, as the Ir atoms bound to the organic moiety are affected more strongly. The energy shifts of all complexes, except the π-coordinated ethene and ethyne, calculated at 0.51–0.77 eV, are close to the surface core level shift, 0.55 eV, observed for C2H4 adsorbed on Ir(111)1 and to the shift of ∼0.6 eV of the Pt 4f levels of surface atoms, measured for propene adsorbed on Pt(111).32
Adsorption of ethene on the bare metal cluster also results in a stabilization of the C 1s levels (estimated with respect to ethene in the gas phase). For π-coordinated ethene, the C 1s levels are stabilized least, only 0.17 eV. In complexes of ethyne and di-σ-ethene, this stabilization is 0.51–0.88 eV, whereas the stabilization is much more significant, 1.81 eV and 2.11 eV, in complexes of ethylidyne or vinylidene, respectively, where the ligand binds via three C–Ir bonds.
As a whole, the average energy shift of the Ir 4f levels in the set of modeled complexes reflects the changes in the electronic structure as characterized by the potential-derived charges (see Fig. 6). The combined effect of the organic adsorbate and the hydrogen on the metal species is very similar to that observed in the case of clusters Ir4Hn,30 as the maximum stabilization of the Ir 4f levels reaches 1.75 eV, calculated for the complex H2CCIr4H9 (Fig. 6d).
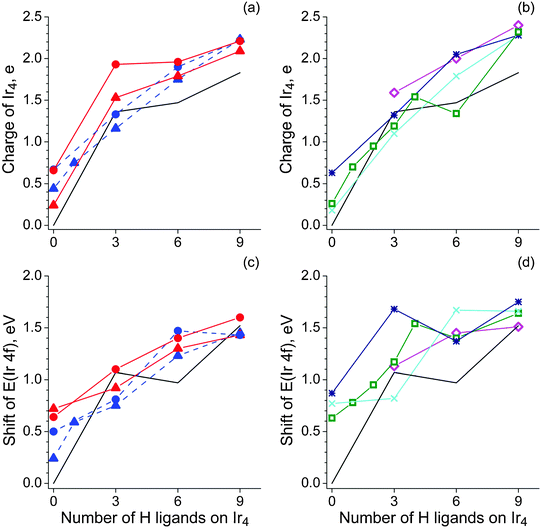 | ||
| Fig. 6 Potential-derived charges of the Ir4 moiety in the (C2Hm)Ir4Hn complexes (a, b) and average shifts of the Ir 4f core levels (c, d) with respect to the corresponding bare clusters as a function of the total number n of hydrogen atoms adsorbed on the metal moiety: ethene—triangles, ethyne—circles, ethyl—diamonds (magenta), ethylidyne—squares (green), vinyl—stars (dark blue), vinylidene—crosses (cyan). Blue (dashed line) color is applied for π-coordinated ethene/ethyne; red—for di-σ-bonded. In all panels, the corresponding values for the clusters Ir4Hn are presented as a solid black line (ref. 30). | ||
2.4 Infrared spectra of the complexes
As already mentioned in the Introduction, the formation of different organic adsorbates on open metal surfaces or supported metal clusters can be detected by IR spectroscopy or HREELS which may help in distinguishing the type of bonding. Therefore, we also simulated vibrational spectra of all modeled complexes and analyzed them in the light of the available experimental data for ethene adsorbed on iridium or during the catalytic hydrogenation of ethene on iridium. The calculated vibrational frequencies of the C–H and C–C modes of all modeled adsorption complexes are provided in Tables S4 and S5 of the ESI.† Comparison between the theoretically obtained (not corrected) and available experimental data for ethene and ethane in the gas phase (Table S3 in the ESI†) shows that the C–C vibrational modes are simulated with good accuracy, the difference between computed and experimental values is ∼10 cm−1. The calculated harmonic frequencies of the C–H vibrational modes deviate more from experiment, up to 50 cm−1, likely due to missing anharmonic contributions. The calculated IR frequencies of various complexes (see the ESI†) do not show a clear dependence on the number of the hydrogen ligands on the cluster. Fig. 7 presents intervals in which some specific IR bands of each type of adsorbate C2Hm in the complexes (C2Hm)Ir4Hn can be expected as the number n of H ligands varies on the metal moiety. The frequency intervals are defined within each series n = 0, 3, 6, 9. Fig. 7 also shows the experimental data,2,3,13,15,17 available for some of the organic species—ethene, ethyl, and ethylidyne—detected or suggested on supported metal clusters or open iridium surfaces.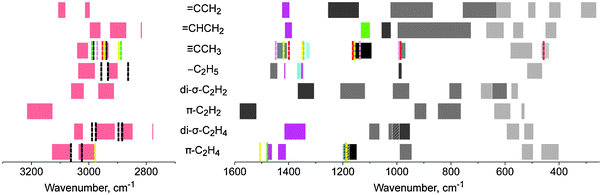 | ||
| Fig. 7 Theoretically obtained spectra of the organic ligand in the complexes (C2Hm)Ir4Hn (m = 2–5; n = 0, 3, 6, 9): C–H, C–C and Ir–C vibrational modes in red, dark grey, and light grey, respectively; CH3 symmetric deformation—cyan; CH2 scissoring deformation—magenta; C–H deformation mode in >CH(CH2) group—green; other CH deformations—grey. Experimental data for some species are presented as dashed lines: ethene and ethyl on alumina-supported Ir4 species (ref. 14; 288 K, 4 × 104 Pa H2, 2.7–3.9 × 104 Pa C2H4)—black; ethylidyne on Ir(111) surface (ref. 2; 180 K, 4–5 × 10−5 Pa C2H4)—red; ethene and ethylidyne on Irn/Al2O3 (ref. 17; 298 K)—yellow; ethene and ethylidyne on Irn/Al2O3 (ref. 18; 90 K; n ∼ 350; sample saturated with ethene)—green; ethylidyne on Ir(210) surface (ref. 3; 90 K, 3 Langmuir C2H2)—pink. | ||
At least two types of modeled species have overlapping IR bands almost anywhere in the pertinent frequency ranges, i.e., in the two intervals 3200–2850 cm−1 and 1500–400 cm−1. Only π-coordinated ethyne can be clearly distinguished, by both its C–H and C–C stretching modes which are higher than those of the other species. The overlap of the bands of different species to some extent is due to the presence of hydride ligands which shift the bands unpredictably, to variable degrees and in opposite directions. Therefore, the type of the organic species on hydrogenated clusters can hardly be determined unambiguously following only one type of vibrations.
The calculated vibrational frequencies can be compared with experimental data for ethene adsorbed on surfaces Ir(111) and Ir(210) or on large iridium particles, or for ethene and H2 adsorption on Ir4 clusters. π-Coordinated ethene on alumina-supported iridium particles (18 Å in diameter) after ethene exposure was experimentally detected via the IR bands at 1188, 1504, and 2978 cm−1 assigned to C–C stretching, CH2 scissoring, and C–H stretching modes (yellow lines in Fig. 7).17 The experimental value for the C–C stretching mode matches well the theoretical frequencies, 1148–1198 cm−1, while the deviation of the scissoring mode is ∼25 cm−1. Experimental data in the C–H stretching region during ethene hydrogenation are available for Ir4 clusters (as those modeled here) supported on alumina (black lines in Fig. 7); they have been assigned to π- and di-σ-coordinated ethene and ethyl.14 According to the P(H2)/T diagram of Fig. 5c, under experimental conditions, one expects the metal cluster to have a large number of adsorbed hydride ligands and π-coordinated ethene to be the dominant species. The C–H bands, assigned experimentally to ethyl species, also fit in the range of frequencies that are representative for di-σ-coordinated ethene and ethyne, or ethylidyne, and a clear assignment is possible only after inspecting other regions of the IR spectra. Similarly to the experimental data, our calculations suggest that the C–H stretching modes of di-σ-coordinated ethene should be observed at lower values, by ca. 40–60 cm−1, compared to the π-coordinated species.
Ethylidyne is the most stable surface species formed after ethene adsorption on various single-crystal surfaces of iridium. Our calculated vibrational frequencies for the H3CC species agree well with the experimental results of Marinova et al.2 (red lines in Fig. 7) for ethene adsorption on Ir(111) and those of Chen et al.3 (pink lines in Fig. 7) for ethyne adsorption on Ir(210). The experimental frequencies of all types of bands, Ir–C, C–C, and C–H stretching and CH3 deformation modes, essentially fall in the interval calculated with our models; some deviations occur for C–H stretching vibrations. Ethylidyne was also identified on alumina-supported iridium particles17 with specific bands at 1156 cm−1 (C–C stretching), 1350 and 1418 cm−1 (CH3 symmetric and asymmetric deformations, respectively), 2896 and 2947 cm−1 (C–H stretching). From Fig. 7 one notes that for ethylidyne species the bands assigned to C–C stretching and CH3 deformation modes fall in the corresponding computationally defined regions. The gap between the two experimentally observed C–H stretching modes, 51 cm−1, matches the gap between the two intervals in the C–H region of the theoretical spectra, 61 cm−1. However, by absolute value, the calculated C–H frequencies overestimate the experimental one by ∼50 cm−1, which may be attributed both to the influence of the anharmonicity of C–H vibrations and accuracy of our computational approach for simulating the vibrational frequencies. The observed good correspondence between the experimental data for ethylidyne on relatively large supported particles and open metal surfaces, on the one hand, and our calculated IR spectrum of the model complexes H3CCIr4Hn, on the other hand, can be rationalized by the fact that in the model complexes the H3CC moiety is coordinated to a facet of the metal cluster and, thus, resembles the coordination mode on metal surfaces on which ethylidyne also coordinates at a three-fold site.
3 Models and computational details
Electronic structure calculations were carried out with the linear combination of Gaussian-type orbitals fitting–functions density functional method (LCGTO-FF-DF)33,34 as implemented in the program PARAGAUSS.35,36 We employed the gradient-corrected exchange–correlation functional suggested by Becke (exchange) and Perdew (correlation) (BP).37 A scalar relativistic variant of the LCGTO-FF-DF method was applied as the relativistic effects were described by explicitly treating all electrons with a Douglas–Kroll–Hess approach of second order.34,38,39 The Kohn–Sham (KS) orbitals were represented by Gaussian-type basis sets, contracted in generalized form: (6s1p) → [4s1p] for H,40 (9s5p1d) → [5s4p1d] for C40 and (21s17p12d7f) → [9s8p6d4f] for Ir.41,42 The auxiliary basis set, used in the LCGTO-FF-DF method to represent the Hartree part of the electron–electron interaction, was derived from the orbital basis set in a standard fashion.33 On the iridium atoms we supplemented these auxiliary basis sets by five p- and five d-type polarization exponents, constructed as geometric series with a factor 2.5, starting with 0.1 au and 0.2 au for p- and d-exponents, respectively. Only the p-type series was added at hydrogen centers.According to the EXAFS data of Gates et al.14,15iridium forms catalytically active tetrahedral clusters anchored on faujasite-type zeolites. Therefore, in the current study, we modeled adsorption of organic species only on Ir4 moieties of tetrahedral shape although theoretical modeling of Ir4 clusters in the gas phase43 showed that the high-spin square-planar structure of Ir4 is 49 kJ mol−1 more stable than the singlet tetrahedral isomer. Exploring tetrahedral models also allowed a direct comparison of some simulated characteristics (interatomic distances, IR features of the organic ligands) with available experimental data. As the current study focuses on organic adsorbates that can be formed during ethene hydrogenation on the metal cluster we examined species C2Hm (m = 2–5) on bare and hydrogenated iridium clusters: π-H2CH2CIr4Hn, di-σ-H2CH2CIr4Hn, π-HCHCIr4Hn, di-σ-HCHCIr4Hn, H3CH2CIr4Hn, H3CCIr4Hn, H2CHCIr4Hn, H2CCIr4Hn. In analogy to our previous studies of hydrogen adsorption on M4clusters,27,28,30 only complexes with 0, 3, 6, or 9 hydrogen ligands were considered. Such simplified models of hydrogenated clusters feature initial configurations of the ligands similar to those obtained for analogous zeolite-supported moieties.27,28 We also modeled complexes with n = 1, 2, and 4 to delineate the rather complex conditions of how the stability of the H3CCIr4Hn species depends on the number n of the hydrogen ligands (section 2.2). Structures were optimized without any symmetry constraints. For the most stable structures we calculated harmonic vibrational frequencies of all degrees of freedom and showed these structures to be local minima by ensuring the absence of imaginary vibrational frequencies. However, other isomeric structures of the species studied are likely to exist. For some of the complexes with adsorbed ethene and ethyne we checked for 2–4 configurations of the hydride ligands on the Ir4 moiety (Fig. S1 of the ESI†); in the following, we report only the most stable isomers.
To analyze the electron density distributions of the complexes, we determined atomic charges by fitting the electrostatic potential (potential derived charges, PDCs).44 As an additional characteristic, we estimated average shifts of core level binding energies for the Ir 4f shells as changes of KS energies relative to the corresponding orbital energies of bare tetrahedral Ir4. In the same spirit we determined variations of binding energies of C 1s core levels of organic adsorbates. Positive values of the calculated shifts represent a stabilization of the core levels with respect to the reference. Although experimental core level energies cannot directly be compared with energies of KS orbitals, changes of KS energies with respect to a reference allow one to estimate core level shifts and have proven useful when analyzing experimental data, e.g., from XPS.45
We evaluated the stability of the species by the energies, ΔE, and Gibbs free energies, ΔG, of the (C2Hm)Ir4Hn complexes relative to the corresponding quantities of bare Ir4 as well as ethene molecule and the appropriate number of hydrogen molecules in the gas phase according to the formal reactions.
| Ir4 + C2H4 + (m + n − 4)/2 H2 → (C2Hm)Ir4Hn, | (1) |
| ΔE[(C2Hm)Ir4Hn] = E[(C2Hm)Ir4Hn] − E[Ir4] − E[C2H4] − (n + m − 4)/2 E[H2], | (2) |
The energy Eads and the Gibbs free energy ΔGads of adsorption of ethene (or ethyne) at the bare (Ir4) and hydrogenated clusters (Ir4Hn) were calculated with respect to the corresponding Ir4Hn cluster and ethene (or ethyne) in the gas phase:
| Eads[(C2Hm)Ir4Hn] = E[(C2Hm)Ir4Hn] − E[Ir4Hn] − E[C2Hm], for m = 4 or 2 | (3) |
4 Summary
The structure and stability of adsorption complexes of bare and hydrogenated iridium tetramers with various organic species, which can be formed under the reaction conditions for ethene hydrogenation, were computationally studied at the density functional level. We considered π- and di-σ-coordination of ethene or ethyne to Ir4Hn clusters as well as analogous complexes of various reaction intermediates—ethyl (H3CH2C–), ethylidyne (H3CC), vinyl (H2CHC), and vinylidene (H2CC).
Calculated relative Gibbs free energies of the complexes (C2Hm)Ir4Hn with respect to ethene in the gas phase allowed us to estimate the concentrations of various species depending on the reaction conditions. At low hydrogen coverage (high temperature and low hydrogen pressure) di-σ-coordinated ethyne is the dominant species on the cluster. At lower temperatures and higher values of the pressure P(H2) ethylidyne and π-bonded ethene are predicted to be the dominant species. Similarly to theoretical data7,8 from modeling of organic species on metal surfaces, complexes of ethyl, vinyl and vinylidene were determined to be less stable than adsorbed ethene or ethylidyne, in the whole range of hydrogen coverage of the iridium cluster. The Gibbs free energy of ethene adsorption on Ir4Hn was calculated from −117 kJ mol−1 to −184 kJ mol−1, depending on the coordination mode of the organic ligand and the hydrogen loading of the cluster. The Gibbs free energy of ethyne adsorption was found to be even higher (by absolute value) at low hydrogen coverage, −287 kJ mol−1 for the preferred di-σ-bonding. However, with increasing hydrogen loading of the cluster this value is reduced by more than 100 kJ mol−1. The desorption of ethane from the most stable complex was calculated to be thermodynamically favorable for complexes in which at least three hydride ligands will remain on the metal cluster after desorption.
Coordination of ethene or ethyne in π-fashion does not affect the structure of the metal moiety, whereas di-σ-bonding of these molecules or binding of an intermediate (ethylidyne, vinyl) results in an elongation (and cleavage) of some of the intermetallic bonds. Analogously to our previous investigations of hydrogen adsorption on metal clusters,27,28 increasing hydrogen loading increases the average Ir–Ir distances. The computed Ir–C distances in most cases vary in the range 188–205 pm which corresponds to the distances observed by EXAFS. Only for π-coordinated ethene Ir–C are distances above 211–215 pm.
From an analysis of the charges and 4f shifts of Ir centers, both the adsorption of the organic ligand and hydrogen was concluded to result in an oxidation of the metal atoms. The C 1s core levels of the organic species are also stabilized upon adsorption. In general, all electronic effects are more pronounced in the complexes with intermediate species (ethylidyne, vinyl, vinylidene) than in the complexes of the molecular adsorbates ethene or ethyne.
The theoretically obtained vibrational spectra of the series of model complexes with different hydrogen loading, to some extent, can be compared to IR and HREELS data. However, the spectra in the C–H region (2700–3300 cm−1) are rather complex and overlap for different adsorbates and adsorption modes. Therefore, this spectral range alone will not allow an unambiguous assignment of the type of the adsorbate, in particular in the presence of hydride ligands on the metal moiety.
Acknowledgements
This work was supported by the Bulgarian National Science Fund (Contract D002-184/08 and National Center for Advanced Materials UNION), Deutsche Forschungsgemeinschaft, and Fonds der Chemischen Industrie (Germany). We gratefully acknowledge generous computing resources at Leibniz Rechenzentrum München.References
- S. Lizzit and A. Baraldi, Catal. Today, 2010, 154, 68–74 CrossRef CAS.
- Ts. S. Marinova and K. L. Kostov, Surf. Sci. Lett., 1987, 181, A106–A107 CrossRef CAS; Ts. S. Marinova and D. V. Chakarov, Surf. Sci., 1987, 192, 275–282 CrossRef; Ts. S. Marinova and K. L. Kostov, Surf. Sci., 1987, 181, 573–585 CrossRef.
- W. Chen, I. Ermanoski, Q. Wu, T. E. Madey, H. H. Hwu and J. G. Chen, J. Phys. Chem. B, 2003, 107, 5231–5242 CrossRef CAS.
- A. Priebe, A. Pucci and A. Otto, J. Phys. Chem. B, 2006, 110, 1673–1679 CrossRef CAS.
- G. N. Vayssilov, Adv. Colloid Interface Sci., 1993, 47, 25–57 CrossRef CAS.
- D. Stacchiola, F. Calaza, T. Zheng and W. T. Tysoe, J. Mol. Catal. A: Chem., 2005, 228, 35–45 CrossRef CAS.
- L. V. Moskaleva, Z.-X. Chen, H. A. Aleksandrov, A. B. Mohammed, Q. Sun and N. Rösch, J. Phys. Chem. C, 2009, 113, 2512–2520 CAS; Z.-X. Chen, H. A. Aleksandrov, D. Basaran and N. Rösch, J. Phys. Chem. C, 2010, 114, 17683–17692 Search PubMed.
- Z.-J. Zhao, L. V. Moskaleva, H. A. Aleksandrov, D. Basaran and N. Rösch, J. Phys. Chem. C, 2010, 114, 12190–12201 CAS.
- J. Andersin, N. Lopez and K. Honkala, J. Phys. Chem. C, 2009, 113, 8278–8286 Search PubMed; J. Andersin and K. Honkala, Surf. Sci., 2010, 604, 762–769 CrossRef CAS.
- Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger and J. Weitkamp, Wiley-VCH, Weinheim, 1997 Search PubMed.
- Z. Ma and F. Zaera, Surf. Sci. Rep., 2006, 61, 229–281 CrossRef CAS.
- I. Lee, R. Morales, M. A. Albiter and F. Zaera, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15241–15246 CrossRef CAS.
- G. A. Somorjai, A. M. Contreras, M. Montano and R. M. Rioux, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10577–10583 CrossRef CAS; C.-K. Tsung, J. N. Kuhn, W. Huang, C. Aliaga, L.-I. Hung, G. A. Somorjai and P. Yang, J. Am. Chem. Soc., 2009, 131, 5816–5822 CrossRef.
- A. M. Argo, J. F. Odzak and B. C. Gates, J. Am. Chem. Soc., 2003, 125, 7107–7115 CrossRef CAS.
- A. M. Argo and B. C. Gates, J. Phys. Chem. B, 2003, 107, 5519–5528 CrossRef CAS; O. S. Alexeev, F. Li, M. D. Amiridis and B. C. Gates, J. Phys. Chem. B, 2005, 109, 2338–2349 CrossRef; A. M. Argo, J. F. Odzak, J. F. Goellner, F. S. Lai, F.-S. Xiao and B. C. Gates, J. Phys. Chem. B, 2006, 110, 1775–1786 CrossRef; A. Kulkarni, R. J. Lobo-Lapidus and B. C. Gates, Chem. Commun., 2010, 46, 5997–6015 RSC.
- T. P. Beebe, Jr. and J. T. Yates, Jr., J. Phys. Chem., 1987, 91, 254–257 CrossRef.
- S. B. Mohsin, M. Trenary and H. J. Robota, J. Phys. Chem., 1991, 95, 6657–6661 CrossRef CAS.
- M. Frank, M. Bäumer, R. Kuhnemuth and H.-J. Freund, J. Vac. Sci. Technol., A, 2001, 19, 1497–1501 CAS.
- G. M. Koretsky and M. B. Knickelbein, J. Chem. Phys., 1997, 107, 10555–10566 CrossRef CAS.
- C. De La Cruz and N. Sheppard, J. Chem. Soc., Faraday Trans., 1997, 93, 3569–3576 RSC.
- M. K. Ko and H. Frei, J. Phys. Chem. B, 2004, 108, 1805–1808 CrossRef CAS; W. Wasylenko and H. Frei, J. Phys. Chem. B, 2005, 109, 16873–16878 CrossRef.
- M. N. Mikhailov, L. M. Kustov and V. B. Kazansky, Catal. Lett., 2008, 120, 8–13 CrossRef CAS.
- C. Adlhart and E. Uggerud, Chem.–Eur. J., 2007, 13, 6883–6890 CrossRef CAS.
- M. C. Valero, P. Raybaud and P. Sautet, J. Catal., 2007, 247, 339–355 CrossRef CAS; F. Delbecq, D. Loffreda and P. Sautet, J. Phys. Chem. Lett., 2010, 1, 323–326 CrossRef.
- R. M. Watwe, B. E. Spiewak, R. D. Cortright and J. A. Dumesic, J. Catal., 1998, 180, 184–193 CrossRef CAS.
- J. Shen, J. M. Hill, R. M. Watwe, B. E. Spiewak and J. A. Dumesic, J. Phys. Chem. B, 1999, 103, 3923–3934 CrossRef CAS.
- G. P. Petrova, G. N. Vayssilov and N. Rösch, J. Phys. Chem. C, 2007, 111, 14484–14492 CAS.
- P. St. Petkov, G. P. Petrova, G. N. Vayssilov and N. Rösch, J. Phys. Chem. C, 2010, 114, 8500–8506 CAS.
- G. P. Petrova, G. N. Vayssilov and N. Rösch, Chem. Phys. Lett., 2007, 444, 215–219 CrossRef CAS.
- G. P. Petrova, G. N. Vayssilov and N. Rösch, Phys. Chem. Chem. Phys., 2010, 12, 11015–11020 RSC.
- G. P. Petrova, G. N. Vayssilov and N. Rösch, J. Phys. Chem. C, 2008, 112, 18572–18577 CAS.
- E. Janin, S. Ringler, J. Weissenrieder, T. Akermark, U. O. Karlsson, M. Gothelid, D. Nordlund and H. Ogasawara, Surf. Sci., 2001, 482–485, 83–89 CrossRef CAS.
- B. I. Dunlap and N. Rösch, Adv. Quantum Chem., 1990, 21, 317–339 CrossRef CAS.
- N. Rösch, S. Krüger, M. Mayer and V. A. Nasluzov, in Recent Development and Applications of Modern Density Functional Theory. Theoretical and Computational Chemistry, ed. J. M. Seminario, Elsevier, Amsterdam, 1996, vol. 4, pp. 497–566 Search PubMed.
- T. Belling, T. Grauschopf, S. Krüger, M. Mayer, F. Nörtemann, M. Staufer, C. Zenger and N. Rösch, in High Performance Scientific and Engineering Computing, Lecture Notes in Computational Science and Engineering, ed. H. -J. Bungartz, F. Durst and C. Zenger, Springer, Heidelberg, 1999, vol. 8, pp. 439–453 Search PubMed.
- T. Belling, T. Grauschopf, S. Krüger, F. Nörtemann, M. Staufer, M. Mayer, V. A. Nasluzov, U. Birkenheuer, A. Hu, A. V. Matveev, A. M. Shor, M. S. K. Fuchs-Rohr, K. M. Neyman, D. I. Ganyushin, T. Kerdcharoen, A. Woiterski, A. B. Gordienko, S. Majumder and N. Rösch, PARAGAUSS version 3.0, Technische Universität München, München, 2004 Search PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS; J. P. Perdew, Phys. Rev. B, 1986, 33, 8822–8824 CrossRef; J. P. Perdew, Phys. Rev. B, 1986, 34, 7406 CrossRef.
- O. D. Häberlen and N. Rösch, Chem. Phys. Lett., 1992, 199, 491–496 CrossRef.
- N. Rösch, A. Matveev, V. A. Nasluzov, K. M. Neyman, L. Moskaleva and S. Krüger, in Relativistic Electronic Structure Theory. Part II: Applications, Theoretical and Computational Chemistry Series, ed. P. Schwerdtfeger, Elsevier, Amsterdam, 2004, vol. 14, pp. 656–722 Search PubMed.
- F. B. van Duijneveldt, IBM Res. Report RJ 945, 1971.
- O. Gropen, J. Comput. Chem., 1987, 8, 982–1003 CrossRef CAS.
- K. M. Neyman, C. Inntam, V. A. Nasluzov, R. Kosarev and N. Rösch, Appl. Phys. A: Mater. Sci. Process., 2004, 78, 823–828 CrossRef CAS; K. M. Neyman, C. Inntam, A. V. Matveev, V. A. Nasluzov and N. Rösch, J. Am. Chem. Soc., 2005, 127, 11652–11660 CrossRef.
- C. Bussai, S. Krüger, G. N. Vayssilov and N. Rösch, Phys. Chem. Chem. Phys., 2005, 7, 2656–2663 RSC.
- B. H. Besler, K. M. Merz and P. A. Kollman, J. Comput. Chem., 1990, 11, 431–439 CrossRef CAS.
- G. N. Vayssilov and N. Rösch, J. Catal., 1999, 186, 423–432 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Figure with relative stability of various optimized isomers of the complexes obtained by ethene/ethyne adsorption at Ir4Hn clusters; table with coefficients of the linear functions ΔG = A + B × (k = n + m) showing the dependence of the relative Gibbs free energy of the model complexes (C2Hm)Ir4Hn (m = 2–5; n = 0, 3, 6, 9) on the total number of H atoms in the system, k = n + m; description of the procedure for determining the concentration of the different species formed after H2 and C2H4 adsorption on Ir4 cluster; table with experimental vibrational frequencies of ethene or ethene-derived species adsorbed on supported iridium species or an iridium surface; table comparing experimental and theoretical spectra of ethene and ethyne in the gas phase and adsorbed on iridium clusters in π- or di-σ-mode; tables with C–H and C–C vibrational frequencies in the model adsorption complexes (C2Hm)Ir4Hn of ethene, ethyne, ethyl, ethylidyne, vinyl, and vinylidene; tables with characteristics of the electronic structure of the modeled adsorption complexes. See DOI: 10.1039/c1cy00114k |
| This journal is © The Royal Society of Chemistry 2011 |