Active site modifications in pentaerythritol tetranitrate reductase can lead to improved product enantiopurity, decreased by-product formation and altered stereochemical outcome in reactions with α,β-unsaturated nitroolefins
Anna
Fryszkowska
a,
Helen
Toogood
b,
Michiyo
Sakuma
b,
Gill M.
Stephens
c,
John M.
Gardiner
a and
Nigel S.
Scrutton
*b
aSchool of Chemistry, University of Manchester, 131 Princess Street, Manchester M1 7DN, United Kingdom
bFaculty of Life Sciences, University of Manchester, 131 Princess Street, Manchester M1 7DN, United Kingdom. E-mail: nigel.scrutton@manchester.ac.uk; Fax: +44 0161 3068918; Tel: +44 0161 3065152
cDepartment of Chemical and Environmental Engineering, University of Nottingham, University Park, Nottingham NG7 2RD, United Kingdom
First published on 4th April 2011
Abstract
This work describes a site-directed mutagenesis study of pentaerythritol tetranitrate reductase (PETN reductase) to probe the role of key active site residues in influencing both product enantiopurity and the ratio of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vs. nitro-group reduction with 2-phenyl-1-nitropropene. Comparative biotransformations of wild type and single/double mutants of PETN reductase with 2-phenyl-1-nitropropene showed that one enzyme scaffold was capable of generating both enantiomeric products with improved enantiopurities by a manipulation of the reaction conditions and/or the presence of a one or two key mutations. These changes located at key active site residues were sufficient to moderately improve product enantiopurity, cause a switch in the major product enantiomer formed and/or promote or eliminate side-product formation. The mutation of substrate-binding residue Y351 to alanine and phenylalanine improved the biocatalytic potential of PETN reductase by the elimination of a competing side reaction. The crystal structures of three mutants at residue Y351 (PDB codes: 3P81, 3P84 and 3P8J) show that only subtle changes in the active site environment may be necessary to generate significantly improved biocatalysts.
C vs. nitro-group reduction with 2-phenyl-1-nitropropene. Comparative biotransformations of wild type and single/double mutants of PETN reductase with 2-phenyl-1-nitropropene showed that one enzyme scaffold was capable of generating both enantiomeric products with improved enantiopurities by a manipulation of the reaction conditions and/or the presence of a one or two key mutations. These changes located at key active site residues were sufficient to moderately improve product enantiopurity, cause a switch in the major product enantiomer formed and/or promote or eliminate side-product formation. The mutation of substrate-binding residue Y351 to alanine and phenylalanine improved the biocatalytic potential of PETN reductase by the elimination of a competing side reaction. The crystal structures of three mutants at residue Y351 (PDB codes: 3P81, 3P84 and 3P8J) show that only subtle changes in the active site environment may be necessary to generate significantly improved biocatalysts.
Introduction
Pentaerythritol tetranitrate reductase (PETN reductase) from Enterobacter cloacae PB2 is a member of the Old Yellow Enzyme (OYE; EC 1.6.99.1) family of enzymes.1–4 This FMN-containing oxidoreductase catalyses the NAD(P)H-dependent reduction of a variety of α,β-unsaturated aldehydes, ketones, nitroalkenes, carboxylic acids and derivatives (Scheme 1).5 A recent review of the OYE family highlighted the biocatalytic potential of these enzymes in the generation of a number of industrially useful compounds, such as substituted nitroalkanes.5 These synthons are versatile and inexpensive and can be converted readily to the corresponding amines, aldehydes, carboxylic acids, oximes, hydroxylamines, or denitrated compounds.6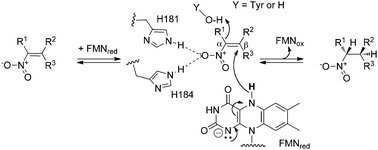 | ||
| Scheme 1 General mechanism and stereochemistry of nitroalkene reduction by PETN reductase. R1, R2 = CH3, or H. | ||
The potential usefulness of PETN reductase in the asymmetric reduction of a variety of (Z)- and (E)-α,β-unsaturated nitroolefins, such as 1-nitro-2-phenyl-propene 1a, has been demonstrated previously.4,7–9 Reactions of PETN reductase with (Z)-nitroolefins were rapid and yielded products with high enantiopurity. However biotransformations with (E)-nitroolefins showed a reduction in reaction rate and both product yield and enantiopurity.4,7 Previous models of (Z)- and (E)-nitroolefin binding to PETN reductase suggested that the highly conserved active site residue Y351 may prevent optimal binding of (E)-nitroolefins due to a clash between the two aromatic rings.4 Site-directed mutagenesis of Y351 to smaller residues may reduce or eliminate this clash, which in turn may improve the binding of (E)-1a and could possibly lead to an increase in the reaction rate and/or product enantiopurity. This was seen in previous studies of the reduction of a related substrate 2-methyl cinnamaldehyde by PETN reductase mutant Y351F, which showed a minor increase in product enantiopurity with no observed loss in product yield compared to wild type reactions.8 However, residue Y351 has been shown previously to be key in substrate binding due to its ability to hydrogen bond with some substrates and inhibitors in PETN reductase and other members of the classical OYE subclass.1,5,9–12
Recently we also described the screening of single site-saturated libraries of PETN reductase mutants that generated novel biocatalysts with altered or improved product selectivity.8 Previous iterative site-saturated mutagenesis studies of another OYE YqjM by others have shown that more dramatic improvements in biocatalytic functionality can often be achieved after at least 2 rounds of iterative saturation mutagenesis due to the cooperative effects of mutations.10 Therefore we generated a number of PETN reductase single and double mutants targeted at specific residues (T26, W102, H181, H184 and Y351) shown previously to be important in product enantioselectivity and side-product generation.8,9 Here we describe comparative biocatalytic reactions of the wild type and single/double mutants of PETN reductase with a variety of (E)-nitroolefins and other substrates and discuss the role of these residues in influencing product yield and/or enantioselectivity. We also determined the three-dimensional crystal structures of PETN reductase mutants Y351A, Y351F and Y351S to see what effect the residue changes have made to the protein structure both locally and globally.
Experimental
General
All reagents were of analytical grade. All biotransformations were set up within an anaerobic glove box (Belle Technology Ltd) under a nitrogen atmosphere (<5 ppm oxygen). The concentration of PETN reductase and substrates were determined by the extinction coefficient method using values described previously.4,7,13 All medium components were obtained from Formedium. Full gene sequences of all mutants were confirmed by DNA sequencing (Eurofins MWG Operon).Site-directed mutagenesis
PETN reductase His8-tagged mutants T26S, W102Y, H181N and H184N were prepared as described previously.8 Additional single and double mutants of PETN reductase were generated by site-directed mutagenesis using the Stratagene QuickChangeTM whole plasmid synthesis protocol. PCR reactions were performed using both the non-tagged3 and PETN reductase-His8 modified pONR1 template (pBluescript SK+)8 and the following oligonucleotides (T26S: GGCCCCACTTAGCCGTCTGCGCA, TGCGCAGAC GGCTAAGTGGGGCC; W102Y: TGCGGTTCAGCTGTATCA CACCGGTCGTATC, GATACGACCGGTGTGATACAGCTG AACCGCA; Y186F: GCTTCACTCTGCGCACGGTTTTCTGC TGCATCAGTT, AACTGATGCAGCAGAAAACCGTGCGC AGAGTGAAGC; Y351A: TGAAAGCTTCGCTGGCGGCG GCG, CGCCGCCGCCAGCGAAGCTTTCA; Y351F: TGAA AGCTTCTTTGGCGGCGGCG, CGCCGCCGCCAAAGAAGC TTTCA; Y351K: TGAAAGCTTCAAGGGCGGCGGCG, CGCC GCCGCCCTTGAAGCTTTCA and Y351S: TGAAAGCTTC TCTGGCGGCGGCG, CGCCGCCGCCAGAGAAGCTTTCA). Following template removal by selective restriction digest (DpnI), PCR products (50 ng) were transformed into the Escherichia coli strain JM109 (Promega) according to the manufacturer's protocol. Each mutant was grown on LB agar containing ampicillin (100 μg mL−1) for 24 h at 37 °C. Colonies (2–5) of each mutant were grown and fully sequenced, as above, to confirm the presence of the required mutations. The following mutants were obtained in using this process: Y186F, Y351A, Y351F, Y351K, Y351S and double mutants T26S/W102Y, T26S/H181N, T26S/H184N and T26S/Y351S.Enzyme production and purification
Non-His8-tagged PETN reductase mutants Y351A, Y351F and Y351S were prepared as described previously for wild type enzyme.1 PETN reductase-His8 wild type and mutant enzymes were prepared as described previously.9 Purity was assessed by SDS-PAGE (>90%) and the concentration of active (flavinated) enzyme was determined using the extinction coefficient method.4,7 All spectra were determined aerobically on a Cary UV-50 Bio UV–visible scanning spectrophotometer using a 1 mL quartz cuvette (Hellma) with a 1 cm path length.Chemistry
Chemicals and solvents obtained from commercial sources were of analytical grade. Substrates (5R)-carvone 7a, citral 8a and ketoisophorone 4a, and products (R/S)-8b and (R/S)-5b were obtained from commercial suppliers (Aldrich, Alfa Aesar and Avocado). (+)-Dihydrocarvone 7b was obtained from a commercial supplier as a mixture of 2 isomers (2R,5R)-7b and (2R,5S)-7b in a 77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 ratio (>99% ee). Nitroalkenes (E)-1a to (E)-4a and their respective nitroalkane products were synthesised as described previously.4,7,13
:20 ratio (>99% ee). Nitroalkenes (E)-1a to (E)-4a and their respective nitroalkane products were synthesised as described previously.4,7,13
Analytical procedures
HPLC analysis was performed using an instrument equipped with a UV detector. The reaction progress was monitored by TLC on standard silica gel plates. The determination of the yield, % conversion and % ee for all compounds were performed as described previously.4,7,9,13Biotransformations with PETN reductase
PETN reductase wild type and mutants were deoxygenated by passage through a BioRad 10DG column equilibrated in anaerobic reaction buffer. The concentration of NAD(P)H and substrates within the reactions were determined by the extinction coefficient method using values determined previously.7 Standard reactions (1.0 mL) were performed in buffer (50 mM KH2PO4/K2HPO4 pH 7.0) containing alkene (5 mM; added as a DMF solution with a 2% (v/v) final concentration), PETN reductase (2 μM), glucose-6-phosphate dehydrogenase (GDH; 8U), glucose-6-phosphate (20 mM) and NADP+ (6 μM). The reactions were shaken at 30 °C for 24–48 h at 130 rpm followed by reaction termination by extraction with ethyl acetate (0.9 mL) containing an internal standard. The extracts were dried using MgSO4 and analyzed by GC or HPLC to determine the % yield, % conversion, and enantiomeric excess as described previously.4,7,13Biphasic reactions (12 mL) with wild type and mutant PETN reductase were performed in buffer (50 mM KH2PO4/K2HPO4 pH 7.0; 7.2 mL), alkene in iso-octane (1.67 mM; 4.8 mL) with t-butylbenzene as an internal standard (25 μL), PETN reductase (1.4 μM), GDH (15 U), glucose-6-phosphate (3.12 mM) and NADP+ (1.4 μM). Products were extracted and analysed as above. The reactions were agitated at 130 rpm for 7 days at 30 °C. Additional reactions with wild type enzyme were carried out as above except the NADP+/GDH recycling system was replaced by NADPH (3.12 mM).
Crystallogenesis and data collection
Crystals of oxidised PETN reductase non-tagged mutants Y351A, Y351F and Y351S were grown using the sitting-drop method in buffer (100 mM sodium cacodylate pH 6.2 containing 100 mM sodium acetate, 16–18% isopropanol and 18–22% polyethylene glycol 3000) for 3 days at 20 °C. All crystals were flash frozen in liquid nitrogen in the absence of additional cryoprotectant. Full X-ray diffraction data sets of PETN reductase mutants Y351A (1.1 Å), Y351F (1.2 Å) and Y351S (1.0 Å) were collected from single crystals at the European Synchrotron Radiation Facility (Grenoble, France) on Station ID 14.4 (wavelength 0.97 Å; 100 K) using an ADSC CCD detector.Structure determination and refinement
All data sets were processed using the D*Trek software,14 and solved via molecular replacement using the coordinates for the acetate-bound PETN reductase structure (PDB code: 1H50).1 Model rebuilding and water addition was performed automatically using REFMAC combined with ARP/wARP.15 Positional and anisotropic B-factor refinement was performed using REFMAC516 (hydrogens included in the refinement), with alternate rounds of manual rebuilding of the model in COOT.17 The final models were refined to 1.10 Å, 1.20 Å and 1.00 Å resolution for PETN reductase mutants Y351A, Y351F and Y351S, respectively. The atomic coordinates and structure factors (pdb codes 3P84, 3P81 and 3P8J, respectively) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).Results and discussion
Mutant selection
We generated four site-directed mutants of PETN reductase (Y351A, Y351F, Y351K and Y351S) to see if altering the nature of residue Y351 could improve biocatalysis with nitroolefin substrates. These changes were designed to add new functionality to this residue, remove the terminal hydroxy-group, add flexibility to potentially increase the substrate-binding capacity of this residue, and maintain the hydrogen-bonding capacity with a shorter residue length. Given our prior success in generating single-site mutants with altered and/or improved biocatalytic potential,8 we also generated a series of single and double mutants to investigate possible cooperative or synergistic effects in enhancing the biocatalytic potential of the enzyme. The additional mutations (T26S, W102Y, H181N, H184N and Y186F) were chosen due to their involvement in, or influence on substrate binding and/or product enantiopurity in reactions with substrates such as (E)-1a.8 Previous iterative site-saturation studies with the OYE YqjM10 targeted residues equivalent to T26 and W102 in PETN reductase, but not H181, H184, Y186 or Y351 (the latter absent in YqjM). A number of single and double mutants at these residues and other specific sites were successful in improving the catalytic functioning and/or enantioselectivity towards a variety of substrates.Mutagenesis of the key substrate-binding residue H181 was previously shown to switch the biocatalytic activity towards oxime formation as opposed to reduction of the CC bond of (E)-1a to form the nitroalkane 1b.9 Similar mutations at a second key substrate-binding residue H184 had more moderate effects on the levels of oxime formation, with a higher retention of alkene reduction and significantly higher product enantiopurities. Two contrasting mechanisms of PETN reductase-catalysed oxime formation from (E)-α,β-unsaturated nitroolefins have been proposed recently. One mechanism proposes that nitro reduction occurs after alkene reduction,18 while a more recent mechanism asserts that nitro reduction occurs first.9 Further studies are required to determine the exact mechanism of oxime formation.
The mutant T26S showed a switch in product enantiopreference with (E)-1a, while mutant W102Y showed a dramatic increase in both the levels of oxime formation and alkane product ee.8 Another key active site residue Y186 was chosen due to its role as the proton donor in the majority of OYEs. However its role in PETN reductase is unclear due to the retention of significant catalytic ability of the Y186A mutant.12
All double mutants contained the mutation T26S, as previous studies had shown this mutation causes a switch in the major product enantiomer formed in reactions with (E)-1a.8 The additional mutations incorporated into the T26S mutant (W102Y, H181N, H184N and Y351S) were selected based on changes in the product yields and/or enantiopurity of the respective single mutants compared to wild type enzyme (Table 1). All single and double mutants were purified to >90% purity and exhibited similar FMN UV/Vis absorbance spectra as wild type enzyme (results not shown).
| Substrate | Product | Enzyme | Conv.b (%) | Yieldb (%) | ee (%) | Yield oximeb (%) | ||
|---|---|---|---|---|---|---|---|---|
| a Conditions: Reactions (1 mL) were performed in buffer (50 mM KH2PO4/K2HPO4 pH 7.0), alkene (5 mM; added as a DMF solution with 2% (v/v) final concentration), PETNR (2 μM), glucose-6-phosphate dehydrogenase (8 U), glucose-6-phosphate (20 mM) and NADP+ (6 μM). Reactions were agitated at 30 °C for 48 h at 130 rpm; b By GC using DB-Wax column; c By HPLC (Chiralcel OD column); d Significant oxime by-product detected, but not quantified; ND = not determined. | ||||||||
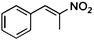
| (E)-1a |
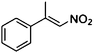
|
(S)-1b |
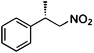
|
Wild type | >99 | 72 | 48 (S) | 18 |
| T26S | >99 | 89 | 37 (R) | 8 | ||||
| W102Y | 94 | 12 | 92 (S) | 40 | ||||
| H181N | 90 | 15 | 90 (S) | 40 | ||||
| H184N | 96 | 83 | 87 (S) | 12 | ||||
| Y186F | 96 | 83 | 69 (S) | 13 | ||||
| Y351A | 42 | 37 | 70 (S) | 0 | ||||
| Y351F | 88 | 80 | 68 (S) | 0 | ||||
| Y351K | 50 | 21 | 72 (S) | 5 | ||||
| Y351S | 60 | 19 | 75 (S) | 4 | ||||
| T26S/W102Y | 55 | 16 | 25 (S) | 16 | ||||
| T26S/H181N | 69 | 12 | 70 (S) | 7 | ||||
| T26S/H184N | 96 | 86 | 94 (S) | 7 | ||||
| T26S/Y351S | 46 | 23 | 25 (R) | 3 | ||||
| (E)-2a |
|
(R)-2b |
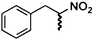
|
Wild type | >99 | 93 | 0 | ND |
| T26S | >99 | 92 | 0 | ND | ||||
| W102Y | >99 | 96 | 0 | ND | ||||
| H181N | 91 | 80d | 0 | ND | ||||
| H184N | >99 | 92 | 0 | ND | ||||
| Y351A | 86 | 74 | 14 | ND | ||||
| Y351F | 98 | 94 | 15 | ND | ||||
| Y351K | 90 | 86 | 14 | ND | ||||
| Y351S | >99 | 95 | 17 | ND | ||||
| (E)-3a |
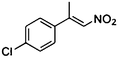
|
(S)-3b |
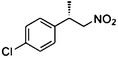
|
Wild type | 78 | 63d | 67 | ND |
| T26S | 65 | 55d | 82 | ND | ||||
| H181N | 70 | 19d | 98 | ND | ||||
| H184N | 78 | 64d | 87 | ND | ||||
| W102Y | 80 | 24d | 92 | ND | ||||
| Y186F | 74 | 59d | 63 | ND | ||||
| Y351A | 42 | 33 | 78 | ND | ||||
| Y351F | 62 | 50 | 83 | ND | ||||
| Y351K | 46 | 33 | 71 | ND | ||||
| Y351S | 48 | 37 | 89 | ND | ||||
| T26S/H184N | 74 | 65 | 92 | ND | ||||
| T26S/Y351S | 33 | 19 | 94 | ND | ||||
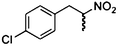
| (E)-4a |
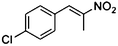
|
(R)-4b |
|
Wild type | 92 | 88 | 20 | ND |
| T26S | 84 | 84 | 13 | ND | ||||
| H181N | 36 | 30 | 8 | ND | ||||
| H184N | 80 | 68 | 25 | ND | ||||
| W102Y | 84 | 83 | 15 | ND | ||||
| Y351A | 30 | 26 | 21 | ND | ||||
| Y351F | 53 | 52 | 36 | ND | ||||
| Y351K | 39 | 32 | 29 | ND | ||||
| Y351S | 32 | 27 | 22 | ND |
Biotransformations with 1-aryl-2-nitropropenes and 2-aryl-1-nitropropenes
We performed anaerobic biotransformations of wild type and mutant PETN reductase enzymes against nitroolefin substrates (E)-1a to (E)-4a using a NADP+/glucose-6-phosphate dehydrogenase (G6PDH) recycling system as the source of reducing equivalents (Table 1). Biotransformations of wild type enzyme with (E)-1a typically go to completion under our reaction conditions to produce the alkane product (S)-1b with a moderate ee (48%) and 10–20% of the 2-phenylpropanal oxime by-product 1c.9,18–20 Other minor by-products of (E)-1a reduction have been previously identified as 2-phenylpropanal and 2-phenylpropan-1-ol, respectively.4,7,8 The latter product is most likely caused by the reduction of the aldehyde by-product by a contaminating E. coli keto-reductase.10Mutations of the highly conserved Y351 residue to alanine, lysine, serine and phenylalanine caused both a significant decrease in product yield and an increase in product (S)-1b enantiopurity (68–75%). Interestingly, mutants Y351A and Y351F did not produce detectable levels of oxime 1c, while mutants Y351K and Y351S showed a decrease in oxime 1c formation with lower (S)-1b yields. Therefore, mutations at residue Y351 appear to have had the opposite effect as those at positions H181 or H184, which lead to an increased level of by-product formation.9 This may be due to a reduction in substrate binding in a conformation compatible with nitro-reduction. As mutant Y351F showed similar levels of product (S)-1b yield as wild type enzyme, this mutant shows great potential producing cleaner and more specific biotransformations with (E)-α,β-unsaturated nitroolefins and other bulky substrates where direct binding of Y351 OH atom to the substrate is not thought to occur. Biotransformations with the related p-chloro-substituted 2-phenyl-1-nitropropene (E)-3a showed only minor differences in the product(s) between wild type PETN reductase and mutants (Table 1). The majority of the mutants had similar conversions and yields as wild type enzyme, often with a significant increase in product ee (78–98% vs. 67%). However with 1-aryl-2-nitropropene substrates (E)-2a and (E)-4a (Table 1), the majority of the mutants had decreased conversions and product yield. The poor product enantioselectivity obtained with substrate (E)-2a and (E)-4a are consistent with previous studies with PETN reductase and other OYEs,4,13,20,21 and are a result of product racemisation under the reaction conditions.7 Therefore the mutants screened did not have a major impact on influencing substrate-binding conformations with these substrates in a manner which would significantly increase the ee.
These results combined with previous work show that single point mutations of PETNR can generate biocatalysts yielding products with improved enantiopurity (Y351X), opposite enantiomeric product formed (T26S)8 and even a switch in the major reaction catalysed (H181X).9 Therefore we generated double mutants to see if any of these altered enzyme properties would act synergistically to improve the biotransformations further. To do so, comparative work between the single and the double mutants was necessary (Table 1). As seen previously,8 reactions of mutant T26S with (E)-1a showed similar conversions and product yields as wild type enzyme, with the major product switched to (R)-1b. Biotransformations of H181N and H184N with (E)-1a were in agreement with previous results,9 with mutant H181N showing a dramatic reduction in product yield due to an increase in the amount of the oxime 1c formation. Both mutants produced (S)-1b with high ee's (87–90%), with H184N showing similar yields and oxime levels as the wild type enzyme. The mutation of a putative proton donor Y186 to phenylalanine showed only minor effects in biotransformations with (E)-1a. Both a small increase in product yield and enantiopurity were seen, with similar yields of oxime detected. Clearly this residue does not play an essential role as a proton donor, and has only a minor influence in orienting substrate binding with this substrate.
Mutant W102Y was found to behave in a similar manner to H181N due to the formation of 40% of the oxime by-product 1c, and a small yield of alkane (S)-1b with high enantiopurity (92%; Table 1). Interestingly, previous studies with a similar mutant W102F produced the opposite enantiomeric product in reactions with the substrate α-methyl-cinnamaldehyde compared to wild type enzyme.8 In addition, kinetic and structural studies of PETN reductase W102Y and W102F mutants showed tighter binding kinetics with picric acid and an alteration in the mode of substrate binding.22 The equivalent residue in the OYE YqjM is alanine (A102), and a substitution to tyrosine produced a mutant that reduced 3-methyl-2-cyclohexenone to form the alkane product with a slightly higher yield and ee than wild type enzyme.10 Clearly the nature of the residue at position 102 in OYEs plays an important role in influencing the substrate-binding conformation(s), with the mutation to tyrosine apparently increasing the tendency of the substrate to bind in a conformation compatible with nitro-group reduction in PETN reductase.
An analysis of the biotransformations of the double mutants showed surprisingly little cooperative effects of the two respective mutations (Table 1). Only mutant T26S/Y351S showed retention of the switch of enantiopreference to form the (R)-1b product, with an overall lower ee of 25%. This mutant also maintained the lower levels of oxime formation seen in both single mutants, however there was a significant decrease in both conversion and product yields. Double mutants containing H181N or H184N retained their effects on product yield and increased enantiopurity, however the low yields of oxime detected in the T26S/H181N reactions suggested significant levels of an unidentified side-product(s) were present. The double mutant T26S/W102Y showed retention of wild type-like (S)-1b formation and oxime levels, but with lower product ee and yields. Therefore a combination of mutations, each imparting specific biocatalytic changes in isolation, does not guarantee any or all of the changes will be maintained in the multiple mutated enzyme.
Biotransformations of (E)-3a with other single mutants showed significant levels of oxime by-product formation. The effects of the double mutants with p-chloro-substituted 2-phenyl-1-nitropropene substrate (E)-3a were mostly additive; with mutant T26S/H184N showing the best overall results of an increase in product ee although with a slight decrease in yield. Mutant T26S/Y351S generated the (S)-3b product with high enantiopurity, although there was a dramatic reduction in product yield. Less dramatic effects were seen in the reactions of the mutants with 1-phenyl-2-nitropropene substrates (E)-2a and (E)-4a (Table 1). Some mutants gave similar product yields and racemic product 2b as wild type (T26S, W102Y and H184N) while a small amount of the equivalent oxime by-product was detected in reactions with mutant H181N. These results show that the effects of individual mutations is often substrate-specific, and it is unlikely that only one or two improved mutants would be sufficient to improve the reactivity of PETN reductase against all industrially-useful substrates.
Biphasic reactions with 1-aryl-2-nitropropenes and 2-aryl-1-nitropropenes
Previous studies with PETN reductase have shown that variations in reaction conditions may have a dramatic impact on product yields and enantiopurities.4,7 We performed comparative biotransformations of wild type PETN reductase and mutants T26S and H184N against nitroolefins (E)-1a to (E)-3a under biphasic conditions using a NADP+/glucose-6-phosphate dehydrogenase recycling system as the cofactor (Table 2). Additional wild type reactions were performed using NADPH to show the effect of the source of reducing equivalents on the reaction. The most dramatic differences in product ee were seen in reactions with substrate (E)-1a. Wild type enzyme performed poorly, showing a lower ee (7%) than under aqueous conditions. However both T26S and H184N mutants showed a dramatic increase in product ee compared to wild type enzyme, although only T26S showed an improvement compared to reactions performed under aqueous conditions (67% vs. 37%). The best results of (E)-1a reduction to form the (S)- or (R)-product are obtained by T26S/H184N and T26S mutants, performed under aqueous and biphasic conditions, respectively. Surprisingly, wild type and mutant reactions with substrates (E)-3a and (E)-4a gave lower product enantiopurities than the equivalent aqueous reactions. Therefore reaction optimisation studies with each enzyme-substrate pair is needed to ensure the best product enantiopurities are obtained.7| Substrate | Product | Enzyme | Cofactor | Conv.b (%) | Yieldb (%) | ee (%) | ||
|---|---|---|---|---|---|---|---|---|
| a Conditions: Reactions (12 mL) were performed in buffer (50 mM KH2PO4/K2HPO4 pH 7.0; 7.2 mL), alkene in iso-octane (1.67 mM; 4.8 mL) with t-butylbenzene as an internal standard (25 μL), PETNR (1.4 μM), glucose-6-phosphate dehydrogenase (15 U), glucose-6-phosphate (3.12 mM) and NADP+ (1.4 μM). Reactions were agitated at 30 °C for 7 days at 130 rpm. Additional reactions with wild type enzyme were carried out as above except the NADP+/glucose-6-phosphate dehydrogenase recycling system was replaced by NADPH (3.12 mM); b By GC using DB-Wax column; c By HPLC (Chiralcel OD column); GDH=NADP+/Glucose-6-phosphate dehydrogenase recycling system. | ||||||||
| (E)-1a |
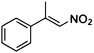
|
(S)-1b |
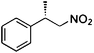
|
Wild type | NADPH | 56 | 30 | 13 (S) |
| GDH | 100 | 52 | 7 (S) | |||||
| T26S | GDH | 100 | 84 | 67 (R) | ||||
| H184N | GDH | 82 | 59 | 78 (S) | ||||
| (E)-2a |
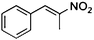
|
(R)-2b |
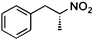
|
Wild type | NADPH | 98 | 98 | 34 (S) |
| GDH | 100 | 93 | 15 (S) | |||||
| T26S | GDH | 100 | 95 | 14 (S) | ||||
| H184N | GDH | 98 | 92 | 10 (S) | ||||
| (E)-3a |
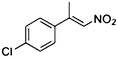
|
(S)-3b |
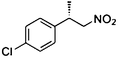
|
Wild type | NADPH | 77 | 48 | 73 (S) |
| GDH | 64 | 40 | 64 (S) | |||||
| T26S | GDH | 60 | 38 | 60 (S) | ||||
| H184N | GDH | 60 | 46 | 83 (S) |
Model of the binding mode of (Z)- vs. (E)-β-alkyl-β-arylnitro-alkenes by PETN reductase
The reduction of nitroolefins (Z)- and (E)-isomers of β-alkyl-β-arylnitroalkenes by wild type PETN reductase,4 Baker's yeast21,23 and crude extracts of Clostridium sporogenes13 is an enantioconvergent process, as the same enantiomeric product is produced from both isomers. In the absence of a co-crystal structure of (Z)- or (E)-1a-bound PETN reductase, high-resolution structural information combined with kinetics and/or enantiopreference of the reaction with wild type and T26S mutant enables us to discern important determinants of substrate binding. Prior kinetic studies of wild type enzyme showed the reduction of (Z)-1a is rapid and quantitative, yielding almost enantiopure products.4 This contrasts with the reduction of (E)-1a that is considerably slower (>20-fold) with poorer product enantiopurities. These results suggest substrate binding of (Z)-1a is more optimal compared to (E)-1a, and the lower ee's with the latter substrate are suggestive of multiple substrate binding conformations (Scheme 2).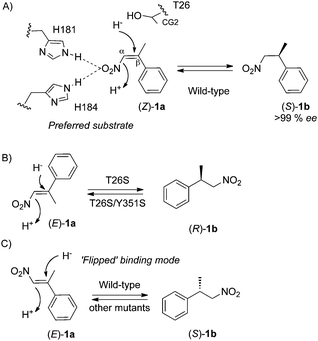 | ||
| Scheme 2 Proposed binding mode(s) of the nitroolefin substrates (Z)- and (E)-1a by PETN reductase. | ||
A model of the preferred substrate (Z)-1a bound to wild type PETN reductase shows both the nitro-group and the phenyl ring pointing towards the ribityl chain of FMN (Scheme 2A). In this position, the nitro-group interacts with His181 and/or His184, which positions the β-methyl group close to the CG2 atom of residue T26. In the case of (E)-1a, two possible conformations are proposed, where either the nitro-group or the phenyl ring are pointing downwards (Scheme 2B and C). Prior kinetic and structural studies with wild type PETN reductase suggested the presence of the side chain CG2 atom of T26 may clash with some substrates and inhibitors, and the substitution to S26 may eliminate this.1,7,12,24 This could explain why (S)-1b is the preferred enantiomeric product as the binding mode to produce (R)-1b is likely to cause a clash between the substrate phenyl ring and T26 CG2 atom (Scheme 2B). In contrast, the preferred enantiomeric product with T26S mutant is (R)-1b, suggesting that the absence of a substrate phenyl to T26 CG2 clash allows the substrate to bind in its preferred mode with the nitro-group pointing towards the ribityl chain of FMN. This effect was overridden by double mutants T26S/W102Y, T26S/H181N and T26S/H184N as the effects of both mutations combined to cause the retention of (S)-1b as the preferred product. Interestingly, the equivalent residue in YqjM is a conserved cysteine (C26; thermophilic-like subclass of OYE), and a substitution to aspartate and glycine produced a mutant that reduced 3-methyl-2-cyclohexenone to form the opposite enantiomeric product compared to wild type enzyme. This suggests residue 26 is a key substrate-orienting residue in OYEs.10 Therefore site-directed mutagenesis to either eliminate or add a potential clash with selected substrate(s) might be a simple approach to alter the enantiopreference and/or product enantiopurity of the reaction.
Biotransformations of mutants with other activated alkene substrates
Activity towards four additional substrates, namely ketoisophorone 5a, maleimide 6a, (R)-carvone 7a and citral 8a were studied with a number of the single and double mutants (Table 3). Biotransformations with substrates 5a and 6a showed the majority of the mutants retained wild type-like activity. An exception was the double mutant T26S/Y351S that showed a dramatic decrease in conversion and product yield, but retention of high ee with substrates 5a–6a and 8a. Given this mutant showed similar results with substrate (E)-1a and (E)-3a–4a, this suggests an overall worsening of substrate binding in this mutant, making it potentially less useful in industrial biotransformations.| Substrate | Product | Enzyme | Time (h) | Conv.b (%) | Yieldb (%) | ee (%) | ||
|---|---|---|---|---|---|---|---|---|
| a Conditions: Reactions (1 mL) were performed in buffer (50 mM KH2PO4/K2HPO4 pH 7.0), alkene (5 mM; added as a DMF solution with 2% (v/v) final concentration), PETNR (2 μM), glucose-6-phosphate dehydrogenase (8 U), glucose-6-phosphate (20 mM) and NADP+ (6 μM). Reactions were agitated at 30 °C at 130 rpm for 24 to 48 h; b By GC using DB-Wax column; c By GC (DB-Wax column) or HPLC (Chiralcel OD column); d Significant unidentified by-product detected. | ||||||||
| 5a |
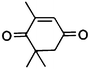
|
(R)-5b |
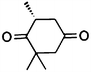
|
Wild type | 48 | >99 | 80 | 95 |
| T26S | 48 | >99 | 89 | 95 | ||||
| H184N | 48 | >99 | 83 | 96 | ||||
| Y186F | 48 | >99 | 91 | 94 | ||||
| T26S/H184N | 48 | >99 | 89 | 83 | ||||
| T26S/Y351S | 48 | 19 | 14 | 94 | ||||
| 6a |
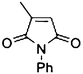
|
(R)-6b |
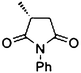
|
Wild type | 48 | >99 | >99 | >99 |
| T26S | 48 | >99 | >99 | >99 | ||||
| H184N | 48 | >99 | >99 | >99 | ||||
| Y186F | 48 | >99 | >99 | 73 | ||||
| T26S/H184N | 48 | >99 | >99 | >99 | ||||
| T26S/Y351S | 48 | 45 | 44 | >99 | ||||
| (5R)-7a |
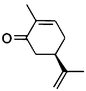
|
(2R,5R)-7b |
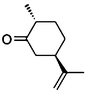
|
Wild type | 48 | >99 | 76 | 94 de |
| T26S | 48 | >99 | 95 | 94 de | ||||
| W102Y | 48 | >99 | 73 | 92 de | ||||
| H181N | 48 | 24 | 13 | 75 de | ||||
| H184N | 48 | >99 | 94 | 96 de | ||||
| Y186F | 48 | >99 | 82 | 10 de | ||||
| Y351K | 48 | 38 | 28 | 78 de | ||||
| Y351S | 48 | 40 | 27 | 94 de | ||||
| T16S/W102Y | 48 | 43 | 28 | 93 de | ||||
| T26S/H181N | 48 | 10 | 7 | 76 de | ||||
| T26S/H184N | 48 | 99 | 76 | 93 de | ||||
| T26S/Y351S | 48 | 99 | 74 | 93 de | ||||
| 8a |
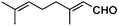
|
(S)-8b |
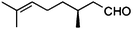
|
Wild type | 48 | >99 | 33d | 93 |
| T26S | 24 | >99 | 33d | >95 | ||||
| H184N | 24 | >99 | 33d | >95 | ||||
| Y186F | 48 | >99 | 33d | 78 | ||||
| T26S/H184N | 48 | 91 | 26d | 97 | ||||
| T26S/Y351S | 48 | 46 | 11d | 93 |
Interestingly, mutant Y186F showed a significant decrease in product enantiopurity with substrates 6a–8a, with near racemic products obtained with (5R)-7a (Table 3). This shows residue Y186 plays an important role in determining the product enantiopurity in addition to influencing the overall kinetics of the reaction seen in previous studies.12 Due to the close proximity of residue Y186 to the substrate-binding site, it is possible that mutations could influence the substrate-binding conformation(s). Protonation of at least some substrates by PETN reductase was found to be water-mediated rather than via residue Y186.12 However this does not exclude the possibility that Y186 may be a proton donor in some cases, as seen by the position of the Y186 OH atom in the active site of wild type PETN reductase structures. Therefore we cannot exclude the possibility that the decrease in product ee of mutant Y186F reactions with substrates 6a–8a is not partially influenced by the absence of a proton-donating capacity.
Reactions of the mutants with (5R)-carvone 7a typically showed poorer results than wild type enzyme, except for H184N which gave a higher product yield. The majority of the mutants showed a reduction in conversion, product yield and ee, although double mutants T26S/H184N and T26S/Y351S retained wild type-like activity. No improvement in product yield and enantiopurity was obtained with mutant biotransformations with substrate 8a. All reactions produced significant quantities of side-products, and both double mutants gave a lower product yield. Clearly the mutants tested had no significant impact on improving the reaction outcome with substrate 8a, highlighting the importance of screening multiple libraries of mutants and optimising reactions conditions to improve biocatalytic reactions.
Crystal structures of three Y351X mutants
The crystal structures of PETN reductase mutants T26S, H181N, H184N, Y186F and picric acid-bound W102Y were determined previously.8,9,12,22 These structures all showed only minor changes in the active site architecture compared to wild type enzyme. However, no structures were known for PETN reductase mutated at residue Y351, a key substrate-binding residue which influences product yield, ee and the ability form the side product oxime 1c. In the p-hydroxybenzaldehyde inhibitor-bound structure of OYE1, the aldehyde carbonyl group of the ligand was shown to interact with the equivalent residue Y375,11 while in LeOPR1 (Y358) this residue is within van der Waals contact distance of the alkyl chain of substrate 12-oxophytodienoic acid (OPDA).25 We determined atomic resolution crystal structures of mutants Y351A, Y351F and Y351S to 1.1, 1.2 and 1.0 Å resolution, respectively, to see what impact the mutations have on the overall active site architecture. The data collection and refinement statistics can be found in Table 4.| Parametersa | PETNR Y351A | PETNR Y351F | PETNR Y351S |
|---|---|---|---|
| a Highest resolution shell is shown in parentheses; Rmerge = ∑hkl ∑i |Ii(hkl) − [I(hkl)]|/∑hkl ∑iIi(hkl), where Ii(hkl) is the intensity of the ith observation of unique reflection hkl; Redundancy = total number of reflections/total unique reflections; Rwork = ∑‖Fobs| − |Fcalc‖/∑|Fobs|, where Fobs and Fcalc are observed and model structure factors, respectively; Rfree was calculated by using a randomly selected set (5%) of reflections. | |||
| Space group | P212121 | P212121 | P212121 |
| Cell dimensions a, b, c/Å | 57.43, 70.33, 88.88 | 56.68, 68.84, 88.91 | 57.49, 70.31, 88.97 |
| Resolution (Å) | 30.0–1.1 (1.14–1.10) | 37.4–1.2 (1.23–1.19) | 30.0–1.0 (1.04–1.00) |
| R merge (%) | 4.3 (6.5) | 7.7 (24.8) | 5.1 (19.2) |
| I/σI | 21.9 (15.3) | 10.6 (4.4) | 14.9 (3.9) |
| Completeness (%) | 92.4 (97.7) | 99.3 (99.7) | 93.6 (85.4) |
| Redundancy | 4.0 (3.8) | 3.8 (3.7) | 3.6 (1.8) |
| Number of reflections | 534692 | 426148 | 657952 |
| Unique reflections | 135417 | 111179 | 181871 |
| R work/Rfree | 12.4/14.0 (10.1/11.7) | 12.9/14.7 (17.4/20.0) | 13.4/14.8 (25.4/28.2) |
| RMS deviations | |||
| Bond angles (°) | 1.325 | 1.381 | 1.406 |
| Bond lengths (Å) | 0.009 | 0.010 | 0.009 |
| Ramachandran plot | |||
| Allowed region (%) | 95.9 | 95.4 | 95.6 |
| Additionally allowed region (%) | 3.8 | 4.4 | 4.1 |
All structures were found to be virtually identical to the wild type PETN reductase structure (PDB code: 1H50),1 including the presence of an acetate ion interacting with residues His181 and His184 (Fig. 1A–C). The FMN of each structure was modelled with a minor ‘butterfly-bend’ in the isoalloxazine ring of the FMN prosthetic group, as seen in recent PETN reductase mutant structures.8,9 The omit maps |F0| − |Fc| of each structure show a clear absence of density for a complete tyrosine side chain. Instead, the density of residue 351 was consistent with presence of a mutation to alanine, phenylalanine and serine side chains (Fig. 1A–C, respectively). In addition, each structure showed a significant shift in the backbone positions of two surface loops near the substrate-binding site (T239–D244; T273–K279; Fig. 1) including a shift in the orientation and position of the backbone and side chain atoms of surface residue D360 and active site residue Q241 (results not shown). The crystal structures of mutants Y351A and Y351S showed additional changes in position of residues A127–T131 and I141–T145 (Fig. 1C). Unlike the recent PETN reductase inhibitor-bound and mutant crystal structures,8,9 the orientation of the FMN ribityl chain was consistent with previously determined wild type structures.1
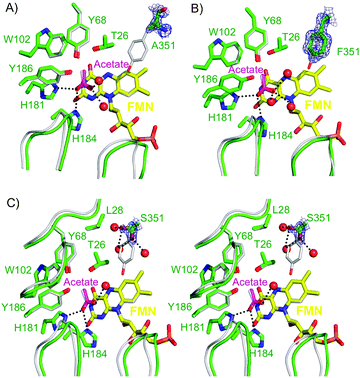 | ||
| Fig. 1 X-ray crystal structures of the active site of PETN reductase mutants (A) Y351A and (B) Y351F superimposed with wild type enzyme. (C) Stereo X-ray crystal structure of the active site of PETNR mutant Y351S superimposed with wild type enzyme. Two positions of atoms Cβ and OG of Y351S have been modelled. All mutant enzyme residues and FMN are shown as atom coloured sticks with green and yellow carbons, respectively. Wild type residues (PDB code: 1H50)1 are shown as atom coloured lines with grey carbons, while waters and interactions are shown as red spheres and black dotted lines, respectively. Loops of mutant enzyme and wild type residues A127-T131; I141-T145 (top left), T239-D244 (left) and T273-K279 (right) are shown as green cartoons and grey ribbons, respectively. The omit |Fo| − |Fc| map of the mutated residue is contoured at 1 σ (green mesh). All figures were generated in Pymol.17 | ||
Residue Y351 in wild type PETN reductase is positioned above the si-face of the FMN, with the OH group positioned about 5 Å from the acetate O atom.1 The side chain atom OH coordinates with 1–2 water molecules in the absence of a bound substrate or inhibitor. Crystal structures of the trinitrotoluene- and picric acid-bound PETN reductase showed the OH atom of residue Y351 forms a hydrogen bond with the O41 atom of one of the nitro-groups of each substrate.22,26 A similar interaction was seen with the O1 atom of the nitro-group of the inhibitor (E)-1-(4′-hydroxyphenyl)-2-nitroethene in the recent PETN reductase co-crystal structure.9 Interestingly, the side chain of Y351 is altered in position in one of the progesterone-bound PETN reductase structures (1H60), causing a significant clash between residue atoms OH and CZ with the FMN isoallozaxine atoms C7 and C7M.1 Therefore, this residue may be involved in positioning or anchoring some substrates and inhibitors in the correct orientation for alkene and/or nitro-group reduction to occur.
In all three structures, there was a loss of wild type-like interactions between residue 351 atom OH and water molecules. However there was maintenance of wild type-like interactions between the backbone residues N and O and water molecules (results not shown). Mutant Y351F differs from wild type enzyme by the absence of the atom OH, and the resultant phenylalanine residue backbone position was shifted compared to wild type enzyme (Fig. 1B). The mutation from Y351 to both alanine and phenylalanine generated hydrophobic residues without the presence of a terminal polar group. Interestingly, these mutants were unable to produce the oxime by-product (E)-1c, suggesting the substrate is bound in a position incompatible with nitro-group reduction. Therefore the nature of the residue at position 351 is important in influencing the catalytic promiscuity of the enzyme by allowing or preventing substrate (E)-1a to undergo nitro-group reduction. A similar role was proposed for key substrate-binding residues H181, and to a lesser extent H184.9
The crystal structure of PETN reductase mutant Y351S revealed two conformations for the side chain atoms CB and OG (Fig. 1C). In both conformations, the OG atom is interacting with 2 water molecules, while in one position there is a minor clash between S351 OG and L28 CD1 atoms. This mutant generated significantly lower product yields with the nitroolefins (E)-1a and (E)-3a -(E)-4a, and (5R)-carvone 7a (Table 1 and 3). As neither position of the S351 side chain is in a position to interact with the known position of bound substrates and inhibitors, this suggests that the decrease in product yield may be due to a reduction of substrate-binding in an active conformation due to the effective elimination of a ligand-anchoring residue.
Conclusions
Modification of the substrate-binding pocket of OYEs by site-directed mutagenesis can have a dramatic impact on the biocatalytic outcomes with some substrates. The enantiopurity and yields of the desired product may also be influenced by reaction condition parameters such as organic solvent content, reaction time and pH.7 Our studies have shown that as little as 1–2 mutations in PETN reductase can improve product enantiopurity, and also result in a switch in the major enantiomer formed. Similar results have been seen with site-saturation mutagenesis studies of OYE1 and YqjM.10,24 In addition, single specific mutations can directly impact on the catalytic promiscuity of the reaction by either promoting or reducing the amount of side-product formation.9 Two conserved active site residues Y351 and Y186 were mutated, and some cases, product yields, enantiopurity and side-product formation were affected by the mutations highlighting the importance of these residues in the orientation of substrate binding. This in turn impacted on the promiscuity of the enzyme by affecting the ratio of alkene vs. nitro-group reduction with some substrates.These results have shown that it is not always necessary to screen enzymes from a number of different organisms to find a pair of biocatalysts capable of generating two different enantiomeric products. Another approach of screening libraries of site-saturated mutants against a variety of substrates under different reaction conditions may be sufficient to find a biocatalyst suitable for each biotransformation. In addition, modification of an enzyme in favour of increasing or decreasing side-product formation does not necessarily require extensive mutagenesis. The incorporation of two mutations each showing individual changes in catalytic functionality did not necessarily result in an additive change in enzyme performance. The biocatalytic ability of the resultant enzyme was dependent on a combination of the changes in the active site of the enzyme, and the effect may be dependent on the most dominant influence.
The study of a variety of mutants can also aid in the prediction of the substrate-binding conformation(s) and provide explanations for the preferred enantiomeric product obtained. This was seen in the switch in product enantiopreference for wild type vs. T26S reduction of (E)-1a and aided in the understanding of the role of the T26 CG2 atom in substrate binding orientation. A comparison of the crystal structures of 8 of the mutants studied here showed that in all cases the effects of the individual mutations were minor, suggesting only subtle changes in the active site environment may be needed to generate significantly improved biocatalysts.
Acknowledgements
We thank Victoria Hare for assistance in the generation and crystallisation of the Y351A/F/S mutants. This work was funded by the UK Biotechnology and Biological Sciences Research Council (BBSRC). NSS is a BBSRC Professorial Research Fellow and a Royal Society Wolfson Merit Award holder. GS is a BBSRC Research Development Fellow.References
- T. M. Barna, H. Khan, N. C. Bruce, I. Barsukov, N. S. Scrutton and P. C. Moody, J. Mol. Biol., 2001, 310, 433–447 CrossRef CAS.
- P. R. Binks, C. E. French, S. Nicklin and N. C. Bruce, Appl. Environ. Microbiol., 1996, 62, 1214–1219 CAS; N. J. Mueller, C. Stueckler, B. Hauer, N. Baudendistel, H. Housden, N. C. Bruce and K. Faber, Adv. Synth. Catal., 2010, 352, 387–394 CrossRef CAS.
- C. E. French, S. Nicklin and N. C. Bruce, J. Bacteriol., 1996, 178, 6623–6627 CAS.
- H. S. Toogood, A. Fryszkowska, V. Hare, K. Fisher, A. Roujeinikova, D. Leys, J. M. Gardiner, G. M. Stephens and N. S. Scrutton, Adv. Synth. Catal., 2008, 350, 2789–2803 CrossRef CAS.
- H. S. Toogood, J. M. Gardiner and N. S. Scrutton, ChemCatChem, 2010, 2, 892–914 Search PubMed.
- N. Ono, The nitro group in organic synthesis, Wiley-VCH, New York-Chichester, 2001 Search PubMed.
- A. Fryszkowska, H. S. Toogood, M. Sakuma, J. M. Gardiner, G. M. Stephens and N. S. Scrutton, Adv. Synth. Catal., 2009, 351, 2976–2990 CrossRef CAS.
- M. E. Hulley, H. S. Toogood, A. Fryszkowska, D. Mansell, G. M. Stephens, J. M. Gardiner and N. S. Scrutton, ChemBioChem, 2010, 11, 2433–2447 Search PubMed.
- H. S. Toogood, A. Fryszkowska, M. Sakuma, G. M. Stephens, J. M. Gardiner and N. S. Scrutton, ChemBioChem, 2011, 12(5), 738–749 Search PubMed.
- D. J. Bougioukou, S. Kille, A. Taglieber and M. T. Reetz, Adv. Synth. Catal., 2009, 351, 3287–3305 CrossRef CAS.
- K. M. Fox and P. A. Karplus, Structure, 1994, 2, 1089–1105 CrossRef CAS.
- H. Khan, T. Barna, N. C. Bruce, A. W. Munro, D. Leys and N. S. Scrutton, FEBS J., 2005, 272, 4660–4671 Search PubMed.
- A. Fryszkowska, K. Fisher, J. M. Gardiner and G. M. Stephens, J. Org. Chem., 2008, 73, 4295–4298 CrossRef CAS.
- J. W. Pflugrath, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 1718–1725 CrossRef.
- A. Perrakis, R. Morris and V. S. Lamzin, Nat. Struct. Biol., 1999, 6, 458–463 CrossRef CAS.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef.
- W. L. DeLano, The pymol user's manual, DeLano Scientific, Palo Alto, CA, USA, 2002 Search PubMed.
- K. Durchschein, B. Ferreira-da Silva, S. Wallner, P. Macheroux, W. Kroutil, S. M. Glueck and K. Faber, Green Chem., 2010, 12, 616–619 RSC.
- B. V. Adalbjornsson, H. S. Toogood, A. Fryszkowska, C. R. Pudney, T. A. Jowitt, D. Leys and N. S. Scrutton, ChemBioChem, 2010, 11, 197–207 CrossRef.
- R. Stuermer, B. Hauer, M. Hall and K. Faber, Curr. Opin. Chem. Biol., 2007, 11, 203–213 CrossRef CAS.
- Y. Kawai, Y. Inaba and N. Tokitoh, Tetrahedron: Asymmetry, 2001, 12, 309–318 CrossRef CAS.
- H. Khan, T. Barna, R. J. Harris, N. C. Bruce, I. Barsukov, A. W. Munro, P. C. Moody and N. S. Scrutton, J. Biol. Chem., 2004, 279, 30563–30572 CrossRef CAS.
- H. Ohta, N. Kobayashi and K. Ozaki, J. Org. Chem., 1989, 54, 1802–1804 CrossRef CAS.
- S. K. Padhi, D. J. Bougioukou and J. D. Stewart, J. Am. Chem. Soc., 2009, 131, 3271–3280 CrossRef CAS.
- C. Breithaupt, J. Strassner, U. Breitinger, R. Huber, P. Macheroux, A. Schaller and T. Clausen, Structure, 2001, 9, 419–429 Search PubMed.
- H. Khan, R. J. Harris, T. Barna, D. H. Craig, N. C. Bruce, A. W. Munro, P. C. Moody and N. S. Scrutton, J. Biol. Chem., 2002, 277, 21906–21912 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |