Zn(Proline)2: a novel catalyst for the synthesis of dicoumarols
Zeba N.
Siddiqui
* and
Farheen
Farooq
Department of Chemistry, Aligarh Muslim University, Aligarh, 202002, India. E-mail: siddiqui_zeba@yahoo.co.in; Tel: +91 09412653054
First published on 20th May 2011
Abstract
A novel, greener approach was adopted for the synthesis of dicoumarols (3a–j) using Zn(Proline)2 as a mild, non-toxic, Lewis acid catalyst in water employing 4-hydroxycoumarin (1) and aromatic/heteroaromatic aldehydes (2a–j). The catalytic activity results suggest that the methodology adopted offers several advantages such as mild reaction conditions, low loading of catalyst, quantitative yields, short reaction time and operational simplicity.
Introduction
Dicoumarol (1a) (Scheme 1) is a naturally occurring anticoagulant drug that functions as a vitamin K antagonist. Chemically, it is designated as 3,3′-methylenebis[4-hydroxycoumarin] and obtained from metabolism of coumarin in the sweet clover (Melilotus alba and Melilotus officinalis) by bacteria Penicillium nigricans and Penicillium jensi.1,2 Dicoumarol and its synthetic derivative warfarin sodium (coumadin) have shown to decrease metastases in animal models.3 Moreover, warfarin sodium therapeutically as an anticoagulant has emerged as one of the most substantial classes of drugs for the treatment of a variety of cancers and has shown to improve tumor response rates.4–6 In clinical trials, such compounds have also demonstrated to have some activity against prostate cancer, malignant melanoma, and metastatic renal cell carcinoma.7–9 A recent study has revealed that the inhibition of NAD (P) H: quinone oxidoreductase (NQO1) with dicoumarol induces cell killing and oxidative stress in pancreatic cancer cells.10 Further, lanthanum(III) complexes of dicoumarol have been reported to show potent cytotoxic activity.11 Compounds with this ring system also possess various other pharmacological activities such as insecticidal, anthelmintic, hypnotic, antifungal, phytoalexin, HIV proteases inhibition, antimicrobial and antioxidant.12–19 During the last decades, several methods have been adopted for the synthesis of this important class of compounds and include use of DBU (1,8-diazabicyclo[5.4.0]undec-7-ene),20 (Et2AlCl3),21 refluxing ethanol or acetic acid,22 molecular iodine,23 POCl3 in dry DMF,24 manganous chloride,25 and thermal solvent free reaction conditions26etc. However, in spite of their potential utility some of the reported methods suffer from certain drawbacks such as long reaction time, expensive reagents, harsh conditions, low product yields and use of toxic catalysts that are harmful to environment. Therefore, to avoid these limitations there is still a need for the development of a new protocol for the synthesis of dicoumarol derivatives in terms of operational simplicity, reusability of the catalyst and economic viability.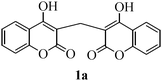 | ||
| Scheme 1 Structure of dicoumarol. | ||
The current environmental concerns encourage development of greener reaction conditions, where possible, and the tight legislation on the maintenance of green conditions in synthetic processes that insist on preventing the generation of waste products, avoiding the use of hazardous organic solvents, and minimizing the energy requirements.27 The use of aqueous reaction media has received considerable attention in the context of green chemistry. During recent years, water has attracted great interest as an inexpensive and environmentally benign solvent due to its specific properties.28 When organic compounds are suspended in water, their relative insolubility causes them to associate, diminishing the water–hydrocarbon interfacial area.29,30 In other words, the hydrophobic effect of water generates internal pressure and promotes the association of the reactants in the solvent cavity during the activation process and accelerates the reaction.31,32
Lewis-acid catalyzed organic reactions in water have attracted much attention in organic synthesis because they allow environmentally friendly processes under mild reaction conditions.33
Proline is the most prominent amino acid for the coordination of Zn, its secondary amino group and carboxylate function being ideally suited for Zn2+ in low coordination number, which makes Zn complex a moderately soft Lewis acid. Zn(Proline)2 is an efficient, stable, inexpensive, recyclable, water compatible, Lewis acid catalyst which is not dissociated under reaction conditions.34 This complex is soluble in water but insoluble in organic solvents, which allows simple and quantitative recovery of the catalyst.35 Zn(Proline)2 appears to be a particularly efficient catalyst for both enamine and enolate type catalyses.36 Among the various zinc-amino acid complexes, the Zn(Proline)2 catalyst shows higher activity.37 The use of Zn(Proline)2 as a powerful catalyst has been accepted for a wide range of organic transformations including aldol,38 direct nitroaldol,37 Hantzsch reaction,39 and Knovenagel condensation.40 It has also been used for the synthesis of 1,5-benzodiazepines,41 1,2-disubstituted benzimidazoles,34a quinoxaline,35 pyrano[2,3-d]pyrimidine,42 and pyrazoles.43 As a part of our ongoing research to develop efficient and environmentally benign synthetic methods in organic chemistry44,45 we report herein an efficient, one pot, extremely mild, green, and simple method for the preparation of dicoumarol derivatives in the presence of Zn(Proline)2 as water-tolerant, green Lewis acid catalyst in environmentally benign solvent, “water” as the reaction medium.
Results and discussion
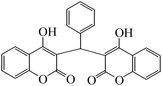
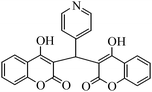
4-Hydroxycoumarin (1) ring at position 3 is highly reactive due to the presence of an electron donating OH group and an electron withdrawing carbonyl group. The presence of a carbon–carbon double bond and a lone pair of electrons from oxygen make the coumarin ring very convenient at position 3 to react with the carbonyl group of aldehydes (2a–j) to give dicoumarol derivatives (3a–j) (Scheme 2). To the best of our knowledge, Zn(Proline)2 has not been used as a catalyst for the synthesis of dicoumarols. This prompted us to test the catalytic activity of the viable catalyst for the synthesis of dicoumarol derivatives.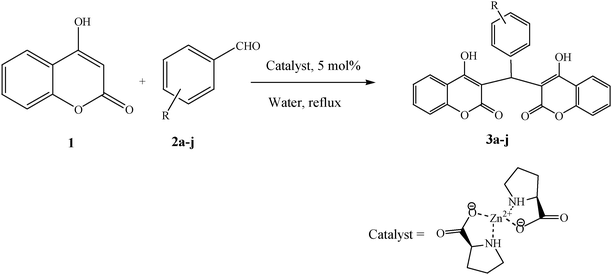 | ||
| Scheme 2 Synthesis of dicoumarol derivatives using the Zn(Proline)2 catalyst in water. | ||
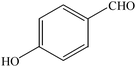
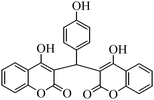
The aim of the present study was to develop an efficient protocol for Zn(Proline)2 catalyzed reaction of 4-hydroxycoumarin (1) with a variety of aromatic/heteroaromatic aldehydes (2a–j) to obtain the corresponding dicoumarol derivatives (3a–j) with excellent yield (91–96%) in a short span of time (5–9 min) (Table 1). Our initial investigations were concerned with the Zn(Proline)2 catalyzed reaction of 4-hydroxycoumarin and 4-hydroxybenzaldehyde as a model reaction. In order to optimize the reaction conditions including catalysts and solvents the reaction was conducted under various conditions and the results are listed in Tables 2 and 3.
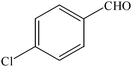
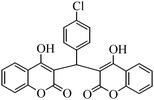
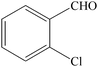
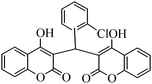
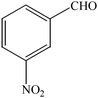
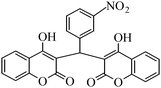
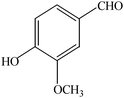
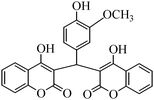
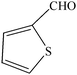
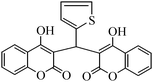
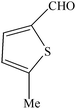
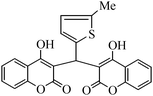
| Entry | Catalyst | Timea | Yieldb (%) |
|---|---|---|---|
| a All the reactions were monitored by TLC. b All yields refer to isolated products. | |||
| 1 | Zn(Proline)2 | 5 min | 96 |
| 2 | AlCl3 | 15 min | 89 |
| 3 | P2O5/silicagel | 20 min | 79 |
| 4 | NiCl2 | 15 min | 75 |
| 5 | 6MHCl | 15 min | 68 |
| 6 | CuCl2 | 35 min | 54 |
| 7 | L-Proline | 25 min | 40 |
| 8 | FeCl3 | 35 min | 25 |
| 9 | ZnCl2 | 48 h | 24 |
| 10 | PTS | No reaction | — |
In a comparative study a variety of catalysts were used in order to show the superiority of Zn(Proline)2 over other catalysts. In this context common Lewis acids of transition metals for the model reaction were used (Table 2). Our investigation revealed that the catalytic activity of various transition metal chlorides was found to be of the order Zn(Proline)2 > AlCl3 > P2O5/silica gel > NiCl2 > 6 M HCl > CuCl2 > L-Proline > FeCl3 > ZnCl2 > PTS. Initial examination indicated that Zn(Proline)2 was superior to other catalysts in terms of yield and reaction time. When the reaction was examined with AlCl3, P2O5/silica gel, 6 M HCl, NiCl2, CuCl2, the reaction was completed in relatively short time (15–35 min) and the product was also obtained in good to moderate yields, but they generated harmful waste, which poses environmental problems. Using L-Proline, ZnCl2 and FeCl3 the product (3b) was obtained in very low yields whereas no product formation was observed when the reaction was carried out in PTS (p-toluene sulfonic acid). From Table 1 it is, thus, clear that Zn(Proline)2 exhibited the highest catalytic activity with regard to the transformation of 4-hydroxycoumarin (1) to dicoumarol (3a–j). In the study of catalyst loading, the effect of the relative amounts of Zn(Proline)2 on the outcome of the model reaction was also studied. The best results were obtained with the use of 5 mol% of catalyst. When the reaction was performed by using less than 5 mol% (i.e. 2 mol% and 4 mol%) of the catalyst, the reactions were either incomplete or took longer time for completion.
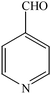
To compare the efficiency as well as capacity of the reaction under aqueous conditions, the model reaction was examined in the presence of Zn(Proline)2 in different solvents such as CH2Cl2, CH3CN, MeOH and CH3COOH (Table 3). The use of a relatively less polar aprotic solvent, CH2Cl2, furnished the product in very low yield after a long period of time (Table 3, entry 4). With the use of aprotic solvents of relatively high polarity, MeCN (Table 3, entry 5), either trace product formation was observed or no reaction was detected. In polar protic solvents MeOH and CH3COOH relatively high yield of the product was obtained under reflux conditions but it took a longer reaction time, within 35–45 min (Table 3, entries 2 and 3). However, when the reaction was performed in water, the product was obtained in excellent yield in 5 min (Table 3, entry 1). It was very fortunate that the best results were obtained for water, which is a desirable solvent from the green chemistry viewpoint. These excellent preliminary results encouraged us to further explore the applicability of the catalyst for the synthesis of other dicoumarol derivatives (Scheme 2 and Table 1). The structural assignment of all the compounds (3a–j) was done by elemental and spectroscopic data (IR, NMR and mass). The IR spectrum of the newly synthesized compound (3j) exhibited strong absorption bands at 1610 cm−1, 1689 cm−1 and 3137 cm−1 for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, coumarin carbonyl and OH groups respectively. The proton nuclear magnetic resonance spectroscopy (1H NMR) exhibited a sharp singlet at δ 6.42 for methine proton. Twelve aromatic protons (four protons of a pyridine moiety and eight protons of a coumarin unit) were discernible as multiplet in the range δ 6.90–8.64. Further confirmation for the structure (3j) was provided by mass spectroscopy which showed M+ at 413 as base peak. The proposed mechanism for the Zn(Proline)2 complex catalyzed synthesis of dicoumarol derivatives can be visualized as given in Scheme 3.
C, coumarin carbonyl and OH groups respectively. The proton nuclear magnetic resonance spectroscopy (1H NMR) exhibited a sharp singlet at δ 6.42 for methine proton. Twelve aromatic protons (four protons of a pyridine moiety and eight protons of a coumarin unit) were discernible as multiplet in the range δ 6.90–8.64. Further confirmation for the structure (3j) was provided by mass spectroscopy which showed M+ at 413 as base peak. The proposed mechanism for the Zn(Proline)2 complex catalyzed synthesis of dicoumarol derivatives can be visualized as given in Scheme 3.
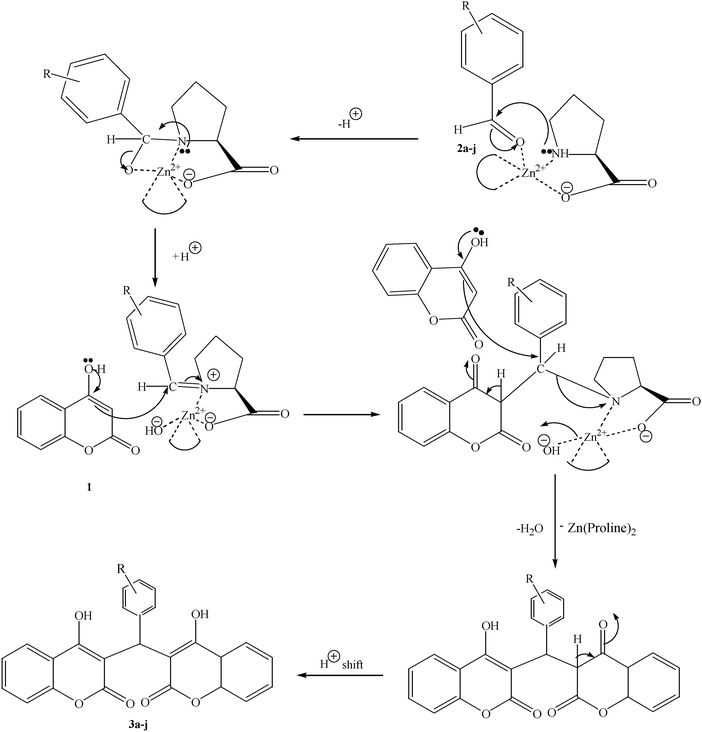 | ||
| Scheme 3 Plausible mechanism for the synthesis of dicoumarols (3a–j). | ||
Conclusions
In summary, we have successfully developed a simple and efficient methodology for the preparation of a wide variety of dicoumarols employing Zn(Proline)2 as the recyclable Lewis acid catalyst for the first time. The powerful catalytic activity of Zn(Proline)2 for these transformations can be sustained by the less reaction time as well as high product yields. Henceforth, this methodology works well and is environmentally benign and may prove beneficial to both academia and industry for the socio-economic change.Experimental
General
Melting points were taken in a Riechert Thermover instrument and are uncorrected. The IR spectra were recorded on a Perkin Elmer spectrometer in KBr, 1H NMR spectra on a Bruker DRX-300 spectrometer using tetra methyl silane (TMS) as an internal standard. Chemical shifts are reported in ppm downfield from TMS as internal standard and coupling constants J are given in Hz. Mass spectra were obtained on a DART-MS recorded on a JEOL-Accu TOF JMS-T100LC mass spectrometer having a DART source. The micro analytical data were collected on an Elementar vario EL III elemental analyzer. The Zn(Proline)2 catalyst was synthesized by the reported procedure.35 4-Hydroxycoumarin and aldehydes were of commercial grade and were used without further purification. The purity of all compounds was checked by TLC on glass plates coated with silica gel (E-Merck G254).Preparation of the Zn(Proline)2 catalyst
(L)Proline (20 mmol) was dissolved in absolute ethanol (50 mL) containing potassium hydroxide (20 mmol) and magnetically stirred for 15 min in a round-bottomed flask at room temperature. In order to maintain the metal to ligand ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 10 mmol of Zn(NO3)2·6H2O was dissolved in a small quantity of double distilled water and added in drops to the (L)Proline solution. The contents were vigorously stirred at room temperature for 6 h by using a magnetic stirrer. The Zn(Proline)2 complex was obtained as a white solid. It was collected by filtration and dried at 70 °C in vacuum for 6 h.
:2, 10 mmol of Zn(NO3)2·6H2O was dissolved in a small quantity of double distilled water and added in drops to the (L)Proline solution. The contents were vigorously stirred at room temperature for 6 h by using a magnetic stirrer. The Zn(Proline)2 complex was obtained as a white solid. It was collected by filtration and dried at 70 °C in vacuum for 6 h.
General procedure for the synthesis of dicoumarols (3a–j)
A mixture of 4-hydroxycoumarin (2 mmol), aldehyde (1 mmol), Zn(Proline)2 (5 mol%) and water (10 mL) was taken in a round-bottomed flask and refluxed on a heating mantle at 110 °C for specified time (Table 1). The progress of the reaction was monitored by thin-layer chromatography (TLC). Upon completion of the reaction, the mixture was cooled to room temperature. The crude product was extracted with dichloromethane, dried over anhydrous Na2SO4, and concentrated to furnish (3a–j). The catalyst was recovered by simple separation of aqueous and organic phases. The catalyst present in the aqueous layer was recovered by precipitating the aqueous layer by addition of acetone and used for the subsequent cycle.Recycling studies of the catalyst
Recycling studies were carried out in order to evaluate the catalytic activity of Zn(Proline)2. Thus, in a model reaction 4-hydroxycoumarin (2 mmol), 4-hydroxy benzaldehyde (1 mmol) and Zn(Proline)2 (5 mol%) in water (10 mL) were thoroughly mixed and refluxed for an appropriate time. The progress of the reaction was monitored by TLC (Table 4). After completion of the reaction the crude product was extracted with dichloromethane and the catalyst was recovered by separation of aqueous and organic phases. The catalyst was recovered by precipitating the aqueous layer by simple addition of acetone. Then, the catalyst was dried at 80 °C for 2 h and used for the next cycle, adopting the same procedure for all the recycling studies. The results (Table 4) revealed that the catalyst exhibited good catalytic activity up to three cycles without appreciable loss of its catalytic activity and no marginal difference in the yield of the product. The scope of the reaction was further investigated with different aldehydes with viable catalysts, and the results obtained were satisfactory.| Catalyst recycle | Timeb/min | Yieldc (%) |
|---|---|---|
| a Reaction of 4-hydroxycoumarin with 4-hydroxybenzaldehyde in the presence of 5 mol% of Zn(Proline)2. b Reaction progress monitored by TLC. c Isolated yield. | ||
| I | 5 | 96 |
| II | 5 | 95 |
| III | 5 | 95 |
| IV | 6 | 92 |
Spectroscopic data
Acknowledgements
Financial assistance in the form of major research project [F. No. 37-15/2009 (SR)] from University Grants commission, New Delhi, India, is gratefully acknowledged. The authors would also like to thank SAIF, CDRI, Lucknow, for spectral data.References
- H. Madari, D. Panda, L. Wilson and R. Jacobs, Cancer Res., 2003, 63, 1214–1220 CAS.
- G. J. Keating and R. O'Kennedy, John Wiley & Sons, West Sussex, United Kingdom, 1997, pp. 23–64.
- G. F. Smith, B. L. Neubauer, J. L. Sundboom, K. L. Best, R. L. Goode, L. R. Tanzer, R. L. Merriman, J. D. Frank and R. G. Herrmann, Thromb. Res., 1988, 50, 163–174 Search PubMed.
- S. Schulman and P. Lindmarker, N. Engl. J. Med., 2000, 342, 1953–1958 Search PubMed.
- D. D'Souza, L. Daly and R. D. Thornes, Ir. Med. J., 1978, 11, 605–608 Search PubMed.
- A. P. Chahinian, K. J. Propert, J. H. Ware, B. Zimmer, M. Perry, V. Hirsh, A. Skarin and S. Kopel, J. Clin. Oncol., 1989, 7, 993–1002 Search PubMed.
- R. D. Thornes, L. Daly, G. Lynch, B. Breslin, H. Browne, H. Y. Browne, T. Corrigan, P. Daly, G. Edwards and E. Gaffney, J. Cancer Res. Clin. Oncol., 1994, 120, S32–S34 Search PubMed.
- M. E. Marshall, K. Bulter and A. Fried, Mol. Biother., 1991, 3, 170–178 Search PubMed.
- J. L. Mohler, L. G. Gomella, E. D. Crawford, L. M. Glode, C. D. Zippe, W. R. Fair and M. E. Marshall, Prostate, 1992, 20, 123–131 Search PubMed.
- A. Lewis, M. Ough, L. Li, M. M. Hinkhouse, J. M. Ritchie, D. R. Spitz and J. J. Cullen, Clin. Cancer Res., 2004, 10, 4550–4558 Search PubMed.
- I. Kostava, I. Manolov, I. Nicolova, S. Konstantonov and M. Karaivanova, Eur. J. Med. Chem., 2001, 36, 339–347 Search PubMed.
- B. Musicki, A. M. Periers, P. Laurin, D. Ferroud, Y. Benedetti, S. Lachaud, F. Chatreaux, J. L. Haesslein, A. IItis and C. Pierre, Bioorg. Med. Chem. Lett., 2000, 10, 1695–1699 CrossRef CAS.
- M. E. Marshall, J. L. Mohler, K. Edmonds, B. Williams, K. Bulter, M. Ryles, L. Weiss, D. Urban, A. Beuschen, M. Markiewicz and G. Cloud, J. Cancer Res. Clin. Oncol., 1994, 120, S39–42.
- A. Maucher and E. J. von Angerer, J. Cancer Res. Clin. Oncol., 1994, 120, 502–504 Search PubMed.
- Z. H. Chohan, A. U. Shaikh, A. Rauf and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2006, 21, 741–748 Search PubMed.
- J. C. Jung, J. H. Lee, S. Oh, J. G. Lee and O. S. Park, Bioorg. Med. Chem. Lett., 2004, 14, 5527–5531 Search PubMed.
- H. Zhao, N. Neamati, H. Hong, A. Mazumder, S. Wang, S. Sunder, G. W. A. Milne, Y. Pommier and T. R. Burke, J. Med. Chem., 1997, 40, 242–249 Search PubMed.
- P. C. M. Mao, J. F. Mouscadet, L. Leh, C. Auclair and L. Y. Hsu, Chem. Pharm. Bull., 2002, 50, 1634–1637 CrossRef CAS.
- K. M. Khan, S. Iqbal, M. A. Lodhi, G. M. Maharvi, Z. Ullah, M. I. Choudhary, A. U. Rahman and S. Perveen, Bioorg. Med. Chem., 2004, 12, 1963–1968 Search PubMed.
- H. Hagiwara, N. Fujimoto, T. Suzuki and M. Ando, Heterocycles, 2000, 53, 549–552 Search PubMed.
- H. Hagiwara, S. Miya, T. Suzuki, M. Ando, I. Yamamoto and M. Kato, Heterocycles, 1999, 51, 497–500 Search PubMed.
- N. Hamdi, P. M. Carmen and P. Valerga, Eur. J. Med. Chem., 2008, 43, 2541–2548 Search PubMed.
- M. Kidwai, V. Bansal, P. Mothsra, P. Saxena, S. Rishi, R. K. Somvanshi, S. Dey and T. P. Singh, J. Mol. Catal. A: Chem., 2007, 268, 76–81.
- M. H. A. Elgamal, N. M. M. Shalaby, M. A. Shaban, H. Duddeck, B. Mikhova, A. Simon and G. Tóth, Monatsh. Chem., 1997, 128, 701–712 Search PubMed.
- J. N. Sangshetti, N. D. Kokare and D. B. Shinde, Green Chem. Lett. Rev., 2009, 2, 233–235 Search PubMed.
- H. R. Shaterian and M. Honarmand, Chin. J. Chem., 2009, 27, 1795–1800 Search PubMed.
- P. T. Anastas and J. Warner, Green Chemistry Theory and Practice, Oxford Univ. Press, Oxford, 1998 Search PubMed.
- (a) J. H. Clark, Green Chem., 1999, 1, 1–8 RSC; (b) H. Wang, S. X. Deng, Z. R. Shen, J. G. Wang, D. T. Ding and T. H. Chen, Green Chem., 2009, 11, 1499–1502 RSC; (c) S. Shirakawa and S. Kobayashi, Org. Lett., 2006, 8, 4939–4942 CrossRef CAS; (d) W. Zhuang and K. A. Jorgensen, Chem. Commun., 2002, 1336–1337 RSC; (e) A. J. Boersma, B. L. Feringa and G. Roelfes, Angew. Chem., Int. Ed., 2009, 48, 3346–3348 CrossRef CAS; (f) K. Manabe, N. Aoyama and S. Kobayashi, Adv. Synth. Catal., 2001, 343, 174–176 CrossRef CAS; (g) S. Kobayashi and K. Manabe, Acc. Chem. Res., 2002, 35, 209–217 CrossRef CAS; (h) N. Azizi, F. Arynasab and M. R. Saidi, Org. Biomol. Chem., 2006, 4, 4275–4277 RSC.
- S. Venkatraman and C.-J. Li, Tetrahedron Lett., 2001, 42, 781–784 CrossRef CAS.
- A. Lubineau and J. Auge, Springer, Berlin, 1999, vol. 206, pp. 1–39.
- M. C. Pirrung and K. D. Sarma, J. Am. Chem. Soc., 2004, 126, 444–445 CrossRef CAS.
- R. Breslow, Acc. Chem. Res., 1991, 24, 159–164 CrossRef CAS.
- (a) C. J. Li and T. H. Chang, Organic Reactions in Aqueous Media, Wiley, New York, 1997 Search PubMed; (b) S. Kobayashi, S. Nagayama and T. Busujima, J. Am. Chem. Soc., 1998, 120, 8287–8288 CrossRef CAS; (c) S. Kobayashi, Synlett, 1994, 689–701 CrossRef CAS; (d) S. Kobayashi, Lanthanides: Chemistry and Use in Organic Synthesis, Springer, Berlin, 1999 Search PubMed; (e) A. Yoshida, X. Hao, O. Yamazaki and J. Nishikido, Molecules, 2006, 11, 627–640 Search PubMed.
- (a) V. Ravi, E. Ramu, K. Vijay and A. S. Rao, Chem. Pharm. Bull., 2007, 55, 1254–1257 CrossRef CAS; (b) R. F. Lopez, J. Kofoed, M. Machuqueiro and T. Darbre, Eur. J. Org. Chem., 2005, 5268–5276 CrossRef CAS.
- M. M. Heravi, M. H. Tehrani, K. Bakhtiari and H. A. Oskooie, Catal. Commun., 2007, 8, 1341–1344 Search PubMed.
- J. Kofoed, T. Darbre and J. L. Reymond, Chem. Commun., 2006, 1482–1484 RSC.
- K. R. Reddy, C. V. Rajasekhar and G. G. Krishna, Synth. Commun., 2007, 37, 1971–1976 CrossRef CAS.
- J. Kofoed, J. L. Reymond and T. Darbre, Org. Biomol. Chem., 2005, 3, 1850–1855 RSC.
- (a) V. R. Sivamurugan, S. Kumar, M. Palanichamy and V. Murugesan, J. Heterocycl. Chem., 2005, 42, 969–974 CrossRef CAS; (b) V. Sivamurugan, A. Vinu, M. Palanichamy and V. Murugesan, Heteroat. Chem., 2006, 17, 267–271 CrossRef CAS.
- Z. N. Siddiqui, T. N. M. Musthafa, S. Praveen and F. Farooq, Med. Chem. Res., 2010 DOI:10.1007/s00044-010-9376-4.
- V. Sivamurugan, K. Deepa, M. Palanichamy and V. Murugesan, Synth. Commun., 2004, 34, 3833–3846 Search PubMed.
- M. M. Heravia, A. Ghodsa, K. Bakhtiaria and F. Derikvanda, Synth. Commun., 2010, 40, 1927–1931 Search PubMed.
- M. Kidwai, A. Jain and R. Poddar, J. Organomet. Chem., 2011, 696, 1939–1944 Search PubMed.
- T. N. M. Musthafa, Z. N. Siddiqui, F. M. Husain and I. Ahmad, Med. Chem. Res., 2010 DOI:10.1007/s00044-010-9386-2.
- Z. N. Siddiqui, F. Farooq and T. N. M. Musthafa, Green Chem. Lett. Rev., 2011, 4, 63–68 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |