Nature of vanadium species on vanadium silicalite-1 zeolite and their stability in hydroxylation reaction of benzene to phenol†
Bin
Guo
,
Liangfang
Zhu
,
Xiaoke
Hu
,
Qian
Zhang
,
Dongmei
Tong
,
Guiying
Li
and
Changwei
Hu
*
Key Laboratory of Green Chemistry and Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, P.R. China. E-mail: gchem@scu.edu.cn; chwehu@mail.sc.cninfo.net; Fax: +86 28 85411105; Tel: +86 28 85411105
First published on 30th June 2011
Abstract
The vanadium silicalite-1 (VS-1) zeolite catalysts with different vanadium contents were prepared by a modified hydrothermal synthesis method, and the one with the highest vanadium content (1.9 wt%) was treated by ammonium acetate (NH4OAc), H2O2 and/or acetic acid (HOAc). The samples were characterized by ICP, XRD, XPS, TPR, and DR UV-Vis to reveal the effect of the state of the vanadium species on the catalysts and to examine their leaching behavior under different conditions. The presence of H2O2 removed the vanadium oligomer loaded on the extra-framework of the VS-1. Highly dispersed distorted octahedral VV species located in the pore of zeolite were unstable in HOAc. The mononuclear octahedral VV, tetrahedral VV and VIV species in the framework showed good stability under the hydroxylation conditions. The VS-1 catalyst with 1.0 wt% of vanadium incorporation in the framework showed a stable hydroxylation activity yielding about 11% phenol (TOF = 63.0) with above 90% selectivity.
1. Introduction
Phenol is commercially produced mainly by the cumene process, which involves three-steps and suffers from a need for high pressure and high temperature. Some other disadvantages such as low one-pass yield of phenol (∼5%), low atomic efficiency (61.8%), and a large amount of acetone by-product also exist in this process. The most useful alternative method is the one-step synthesis of phenol by direct oxidation of benzene under mild conditions, which is a challenging and fantastic work from the economical and environmental points of view.Studies on the direct oxidation of benzene to phenol with various metal-based catalysts have been reported in literature.1–6 In our previous studies, vanadium species were found to be responsible for the catalytic hydroxylation of benzene with hydrogen peroxide in the liquid phase.7–9 For example, [VO2]+ was indicated to be the active phase in the homogeneous hydroxylation of benzene with sodium metavanadate or V-substituted heteropolyacids (HPAs), where the catalytic activity was proved to be affected by the chemical environment of [VO2]+.7–11 Industrially, heterogeneous catalysts have some advantages over homogeneous ones such as catalyst recovery and recycling. VIVOx and/or VVOx presenting in several vanadium-supported catalysts prepared by ion exchange or impregnation were reported to be active for the hydroxylation reaction.12–17 However, the use of supported vanadium catalysts in the hydroxylation reaction always suffered from the leaching of the active vanadium species, especially in acidic solution.18 Thus, the stabilization of the active vanadium sites was critical for obtaining active and actually heterogeneous catalysts. For this goal, several V-containing microporous and mesoporous zeolites have been reported as effective hydroxylating catalysts.12,17,19,20 The recognition of the nature of the vanadium species on the V-substituted zeolite was also investigated by combinational techniques and theoretical calculations.21–23
Genti et al. reported the formation of extra-framework vanadium with high vanadium content and the co-existence of different vanadium species on VS-1 prepared by a hydrothermal method.24 On the other hand, the V-beta and V-ZSM-5 zeolites with higher vanadium contents on the zeolitic framework prepared with post-synthesis showed significant vanadium leaching when used in the liquid phase reactions.12 Acetic acid was usual employed as the solvent in the hydroxylation of benzene with vanadium-based catalysts, however the acidity of acetic acid led to leaching of the V species. In most hydroxylation reactions in the liquid phase, H2O2 was employed as the oxidant. So, the recognition of different vanadium species on the vanadium substituted zeolites and the investigation of their stability during the hydroxylation remained attractive and important subjects for the development of new stable vanadium catalysts.
In this paper, VS-1 catalysts with different vanadium contents were prepared and applied in the selective oxidation of benzene to phenol with hydrogen peroxide in acetic acid solvent. To study the stability of different vanadium species existing on VS-1 during the hydroxylation conditions, the fresh catalyst was separately treated with the solvent, oxidant, and a combination of the solvent and oxidant. The leaching of vanadium species in the treatment and the spectroscopic characterization of the treated catalysts are discussed in detail.
2. Experimental
2.1 Preparation of VS-1
The VS-1 catalysts were prepared by direct hydrothermal synthesis method as described in literatures with subtle modification.25,26 In a typical preparation procedure, 0.05 mol tetrapropylammonium hydroxide (TPAOH) (25%), 10 mL water and 0.07 mol isopropanol were added slowly to a beaker containing 0.5 mol tetraethyl orthosilicate (TEOS). The presence of isopropanol could prevent the hydrolysis of silicate. The mixture was stirred for 30 min before adding designed amount of VOSO4 dissolved in 5 mL water in a dropwise manner. Then another 0.2 mol TPAOH (25%) was added, followed by addition of 16 mL of water. After being thoroughly stirred for 10 min at room temperature, the gel was heated to 343 K and stirred for another 3 h. During the heating process, an appropriate amount of water was added to compensate for the evaporation loss. The resulting gel was transferred to an autoclave and heated at 448 K for about 150 h. After crystallization, the product was separated from the mother liquor by vacuum filtration, washed with water, and dried at 393 K overnight. Then the powder was calcined at 823 K in dry air for 8 h, obtaining the VS-1 catalyst samples 1 to 5, as indicated in Table 1.| Cat. no. | Treatment condition | Vanadium content (wt%) | Vanadium content (mol%) | ||
|---|---|---|---|---|---|
| Nominal | Actual | Nominal | Actual | ||
| a Reaction condition: 0.1 g catalyst 5, 11.25 mmol benzene, 29 mmol H2O2, 30 mL solvent, 343 K, 2 h. | |||||
| 1 | Fresh | 0.5 | 0.2 | 0.3 | 0.1 |
| 2 | Fresh | 1.0 | 0.7 | 0.5 | 0.4 |
| 3 | Fresh | 1.5 | 1.3 | 0.8 | 0.5 |
| 4 | Fresh | 2.0 | 1.7 | 1.1 | 0.7 |
| 5 | Fresh | 2.5 | 1.9 | 1.5 | 1.0 |
| 5A | NH4OAc | — | 1.0 | — | 0.5 |
| 5B | HOAc | — | 1.7 | — | 0.9 |
| 5C | H2O2 | — | 1.3 | — | 0.7 |
| 5D | HOAc + H2O2 | — | 1.1 | — | 0.6 |
| 3A | NH4OAc | — | 1.0 | — | 0.5 |
| 4A | NH4OAc | — | 1.0 | — | 0.5 |
| 5-Useda | Hydroxylation | — | 1.1 | — | 0.5 |
2.2 Treatment of VS-1
The fresh VS-1 catalyst 5 was treated with designed solutions at 343 K for 24 h. The solutions used were aqueous ammonium acetate (NH4OAc), acetic acid, hydrogen peroxide (H2O2 30 wt%), or a mixture of acetic acid and hydrogen peroxide (HOAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) : H2O2 = 10:1, v/v). The resultant samples were named as 5A, 5B, 5C, and 5D. The VS-1 catalyst 3 and 4 were also treated with aqueous ammonium acetate, and the resultant samples were named as 3A and 4A. All the above samples were separated by filtration, and dried at 343 K in air before being characterized.
: H2O2 = 10:1, v/v). The resultant samples were named as 5A, 5B, 5C, and 5D. The VS-1 catalyst 3 and 4 were also treated with aqueous ammonium acetate, and the resultant samples were named as 3A and 4A. All the above samples were separated by filtration, and dried at 343 K in air before being characterized.
2.3 Characterization of the catalyst
Elemental analysis was carried out by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) (IRIS Advantage ER/S). XRD experiments were conducted on a LTD DX-1000 CSC diffraction instrument under Cu-Kα radiation (λ = 1.54056 Å). Data were collected in the 2θ range of 5–35° with a step of 0.0544°, using the continuous scanning mode. H2-TPR was conducted in a brass U-tube reactor. The consumption of H2 during the reduction from room temperature to 1073 K was measured using a thermal conductivity detector (TCD). XPS measurements were carried out with a XSAM 800 spectrometer using Al Kα radiation. The binding energy (BE) was calibrated with XPS signals of C1s at 284.8 eV, and a Linear background was subtracted from all spectra. Then peak fitting was performed using a 80/20 Lorentz-Gauss function. UV-Vis DR spectra were obtained with a TU-1901 spectrometer equipped with a reflectance attachment. BaSO4 was used as the reference material. Before characterization, the samples were evacuated at 298 K for 12 h. The spectra were deconvoluted into sub-bands by Peakfit v4.12 software.2.4 Activity test
The VS-1 samples were used as catalysts for the oxidation of benzene with hydrogen peroxide in acetic acid. The oxidation reactions were carried out in a two-necked 50 mL round bottom flask equipped with a reflux condenser and a stirrer in open air. In a typical procedure, 0.1 g VS-1 catalyst and 1 mL (11.25 mmol) benzene were added to 30 mL glacial acetic acid, then 3 mL (29.7 mmol) H2O2 (30 wt%) was added. The mixture was stirred at 343 K for 2 h under reflux. After the reaction, the resulting mixture was analyzed by HPLC (equipped with a C18 column) with a UV-visible detector. An internal standard material, o-cresol, was used to quantify the phenol and by-products formed. The conversion of benzene, selectivity to phenol and turn over frequency (TOF) were calculated as:| Yield of phenol (%) = Mole amount of phenol × (Initial mole amount of benzene)−1 × 100 |
| Selectivity to phenol (%) = Mole amount of phenol × (Mole amounts of phenol + hydroquinone + catechol + hydroquinone)−1 × 100 |
| TOF (h−1) = Mole amount of phenol × (Mole amount of vanadium sites)−1 × (Reaction time)−1 |
3. Results and discussion
3.1 Vanadium contents on VS-1
The actual vanadium contents on VS-1 catalysts were detected by elemental analysis, and the nominal and actual values are given in Table 1. It was obvious that the actual vanadium contents on the catalysts were lower than the nominal values, implying that a part of vanadium species did not incorporate into the solid samples and might be washed away during the synthesis process.Table 1 also displayed the vanadium contents on treated catalyst 5. The initial high vanadium content on sample 5 declined in various degrees, depending on the composition of the solutions. A vanadium content of about 1.0 wt% was stable on sample 5A after being treated by NH4OAc. As indicated in Table 1, the vanadium content in samples 3A and 4A obtained from the treatment of samples 3 and 4 by NH4OAc was about 1.0 wt%. It was reported that the extra-framework vanadium species loosely bound to the zeolite wall were easily washed out by aqueous solution of ammonium acetate.18 Thus, the content of framework vanadium species was about 1.0 wt%, which might be the threshold value for vanadium to enter into the framework of VS-1 prepared by the present method. According to literature, the leaching of active metal ions on the catalysts depended on several factors, such as oxidant, solvent, structure of the catalyst, and even the substrate.12,27 Then the leaching of vanadium on VS-1 during the treatment was investigated by ICP. The sample treated with HOAc (5B) lost only 0.2 wt% content of vanadium, indicating that most vanadium species in the sample were stable in acetic acid solution. The content of vanadium on sample 5C was 1.3 wt% (0.6 wt% leached), indicating that the leaching of vanadium was more serious in H2O2 than in acetic acid. The vanadium content on sample 5D was 1.1 wt% (0.8 wt% leached), which means the combination of oxidant and acidic solution washed out more vanadium species than each of them used alone. The comparison of the vanadium content over 5B, 5C and 5D showed that H2O2 played an important role on the leaching of vanadium, and the combination of acetic acid and H2O2 resulted in almost equivalent leaching as with NH4OAc.
3.2 XRD
All of the fresh VS-1 samples were white powders, the XRD patterns of which were characteristic of microporous and highly crystalline molecular sieves with MFI topology (Fig. 1a).23,25,26,28 The absence of typical diffraction peaks of vanadium species may be due to the lack of obvious formation of vanadium oxide aggregates, even in the sample with the highest vanadium content (1.9 wt%). However, the XRD patterns could not indicate whether the vanadium species were present actually within the framework, because they could also be present on the external surface as amorphous vanadium oxides.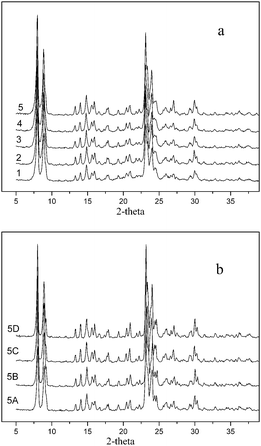 | ||
| Fig. 1 The XRD spectra of the fresh VS-1 catalysts 1–5 and treated catalyst 5. (a) Fresh VS-1 catalysts 1–5; (b) treated VS-1 sample 5. | ||
The XRD patterns of treated VS-1 were displayed in Fig. 1b. No detectable difference was observed in the XRD patterns of each treated samples. It implied that no crystalline change in the process of treatment, and the framework of zeolite was firm under the present acidic or oxidative conditions.
3.3 XPS
The fresh VS-1 catalysts were characterized by XPS and the results are shown in Table 2. It was generally accepted that the BE value of the tetravalent or pentavalent vanadium species located within the zeolite was discrepant with that of the VIV or VV oxides. Centi et al. reported that the BE of the framework vanadium in VS-1 zeolite prepared by a hydrothermal method was 516.8 eV, between the values of VIV and VV oxides.24 However, it was presumed that the vanadium species with different structures in the zeolite would exhibit different BE values. A small contribution from a reduced vanadium species would influence the overall BE value of the vanadium species. To distinguish the chemical environment of the vanadium species, regardless of their locations in VS-1, the V2p3/2 peaks were fitted to VV (517.5 eV) and VIV (516.4 eV). In addition, two kinds of oxygen species were detected with the O1s BE values of 530.5 eV (Oa) and 532.5 eV (Ob), respectively, being assigned to oxygen bonded with vanadium (V–O–V, V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, V–O–Si) and silicon (O–Si–O).29,30 Detailed XPS data of the O1s and V2p3/2 region, as well as the surface content of the fresh VS-1 catalysts were shown in Table 2. It was found that the surface vanadium content increased with the increase of average vanadium content, and the vanadium content of the surface was higher than that of the average for all samples. The enrichment of vanadium species on the surface implied that vanadium species formed outside the framework of zeolite, even on catalyst 1 with the lowest vanadium content (0.2 wt%). That is, both framework and extra-framework vanadium species formed in the hydrothermal synthesis process. The results were in accordance with the foregoing ICP results and earlier reports.24,31,32 Furthermore, the proportion of Oa/(total oxygen) remained almost stable with increasing of the surface vanadium content, implying that two vanadium atoms are linked by one oxygen atom, as in a V–O–V bond. That is, the polynuclear V species (oligomeric V species or vanadium oxides) formed on the surface of VS-1 with higher vanadium contents.
O, V–O–Si) and silicon (O–Si–O).29,30 Detailed XPS data of the O1s and V2p3/2 region, as well as the surface content of the fresh VS-1 catalysts were shown in Table 2. It was found that the surface vanadium content increased with the increase of average vanadium content, and the vanadium content of the surface was higher than that of the average for all samples. The enrichment of vanadium species on the surface implied that vanadium species formed outside the framework of zeolite, even on catalyst 1 with the lowest vanadium content (0.2 wt%). That is, both framework and extra-framework vanadium species formed in the hydrothermal synthesis process. The results were in accordance with the foregoing ICP results and earlier reports.24,31,32 Furthermore, the proportion of Oa/(total oxygen) remained almost stable with increasing of the surface vanadium content, implying that two vanadium atoms are linked by one oxygen atom, as in a V–O–V bond. That is, the polynuclear V species (oligomeric V species or vanadium oxides) formed on the surface of VS-1 with higher vanadium contents.
| Cat. no. | O1s | V2p3/2 | Surface content of vanadium (mol%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Oaa | Obb | VIV | VV | ||||||
| BE (eV) | Area (%) | BE (eV) | Area (%) | BE (eV) | Area (%) | BE (eV) | Area (%) | ||
|
a The oxygen bonded with vanadium (V–O–V, VO, V–O–Si).
b The oxygen bonded with silica (Si–O–Si).
c The catalyst 5 was separated after hydroxylation reaction and evacuated at 298 K for 12 h.
|
|||||||||
| 1 | 530.5 | 9.0 | 532.5 | 91.0 | 516.3 | 65.0 | 517.5 | 35.0 | 0.4 |
| 2 | 530.5 | 7.5 | 532.5 | 92.5 | 516.3 | 61.9 | 517.5 | 38.1 | 0.7 |
| 3 | 530.6 | 8.1 | 532.6 | 91.9 | 516.4 | 64.1 | 517.5 | 35.9 | 0.8 |
| 4 | 530.3 | 7.5 | 532.5 | 92.5 | 516.5 | 72.4 | 517.3 | 27.6 | 1.1 |
| 5 | 530.3 | 9.8 | 532.7 | 90.2 | 516.4 | 73.9 | 517.6 | 26.1 | 1.4 |
| 5A | 530.5 | 5.5 | 532.7 | 94.5 | 516.4 | 21.8 | 517.0 | 78.2 | 0.9 |
| 5-Usedc | 530.7 | 10.1 | 533.1 | 89.9 | 516.3 | 30.2 | 517.4 | 69.8 | 0.8 |
The data listed in Table 2 indicated that both the amounts of surface VIV and VV species increased by increasing total surface vanadium contents, while the VIV content increased more obviously. In addition, the content of Oa and total surface vanadium decreased obviously after being treated by NH4OAc (sample 5A). The surface VIV/VV molar ratio turned to be 21.8/78.2, much lower than the value (73.9/26.1) before being treated. That is to say, most of the VIV species were washed off during the treatment. Thus, it was deduced that at least two kinds of VIV species existed in the fresh catalyst 5. The increasing vanadium contents on VS-1 mainly resulted in the formation of the surface VIV oligomer, which could be removed by NH4OAc.24,26 Another form of VIV was probably located in the framework of VS-1, which could not be washed away easily.
3.4 TPR
To further recognize the vanadium species on VS-1, the catalysts were characterized by H2-TPR experiments and the results were shown in Fig. 2.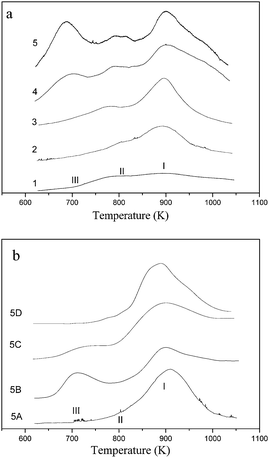 | ||
| Fig. 2 H2-TPR profiles of the fresh and treated VS-1 catalysts. (a) Fresh VS-1catalysts 1–5; (b) treated VS-1 sample 5. | ||
On catalyst 1, two weak reduction peaks I and II in the range 840–950 K and 750–830 K, respectively, were observed. These two reduction peaks shifted slightly to higher temperatures with increasing vanadium contents on catalysts 2 and 3. Furthermore, the area of the higher-temperature reduction peak I became larger while no peak area variation of the lower-temperature reduction peak II was observed. When the vanadium content was higher than 1.3 wt%, a third reduction peak centered at about 710 K (peak III) appeared on catalyst 4 and its peak area augmented notably on catalyst 5. As shown in Fig. 2b, the H2-TPR profile of treated VS-1 changed in different degrees. The TPR profile of sample 5A contained only one reduction peak at about 910 K (peak I). As discussed above, only framework vanadium species existed on the sample 5A. It was reported that a possible product for reduction of VV in the framework of VS-1 was VIV, and the deep reduction of framework VIV to VIII occurred at 1520 K.32,33 Therefore the appearance of peak I was ascribed to the reduction of VV to VIV within silica framework according to literature.18
The peaks II and III disappeared on the NH4OAc treated catalyst 5 (sample 5A). This indicated that the assignment of these two peaks to extra-framework was reasonable. The combination of the results of TPR and XPS made us assume that peak III originated from the reduction of the extra-framework oligomer, which was loaded on the surface of VS-1. The corresponding vanadium species to peak II could not be assigned to the oligomer or cluster because it was accompanied by framework vanadium species, even on the sample with the lowest vanadium content. According to the literature,34 the highly dispersed vanadium on the silica-based support could be reduced at about 800 K. So the peak II was tentatively attributed to the reduction of the VV species situated in the channel of the zeolite.
The peaks I and III were observed in the profile of sample 5B. Compared with the profile of fresh sample 5, peak II decreased significantly after acetic acid treatment. As shown by ICP results, a small part of the vanadium leached and this part of the vanadium was assigned to highly dispersed vanadium oxide on VS-1 which could be reduced at 750–830 K. Thus, the vanadium oligomer and the vanadium species on the framework were stable in acetic acid solution. After VS-1 was treated by H2O2, the areas of peak II and III decreased obviously (Fig. 2b, 5C). It indicated that the leaching of extra-framework vanadium species were serious in the presence of H2O2. It was interesting to find that peaks II and III disappeared after being treated by the mixture of H2O2 and HOAc (Fig. 2b, 5D). So it was deduced that the leaching of vanadium species from the extra-framework might be completed in the presence of the mixture solution.
Based on the above discussions, it was assumed that there were at least four kinds of vanadium species formed on the fresh catalysts 4 and 5, i.e. two located at the silica framework and two presented outside of the framework. Most of the framework vanadium existed as VV while most of the extra-framework vanadium as VIV. Highly dispersed vanadium species in the channel of zeolite could be washed out by acetic acid solution, and vanadium oligomers could be washed out by H2O2. Framework vanadium species were stable in the mixture of acetic acid and H2O2.
3.5 DR UV-Vis
DR UV-Vis spectroscopy was a useful technique for obtaining information about the coordination environment and oxidation states of the metal ions in the molecular sieves. The UV-Vis DR spectra of the fresh, treated and used VS-1 catalysts (5-used) are displayed in Fig. 3. To discuss them in detail, the spectra were fitted into various sub-bands, as plotted in Fig. 4.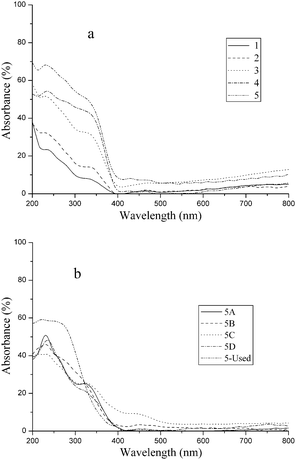 | ||
| Fig. 3 UV-Vis DR spectra of the fresh, treated and used VS-1 catalysts. (a) Fresh VS-1 catalysts 1–5; (b) treated VS-1 sample 5. | ||
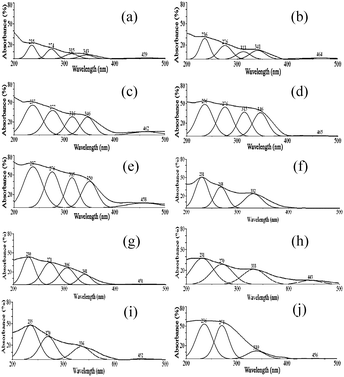 | ||
| Fig. 4 The peak-fitting of the UV-Vis DRs profiles for the VS-1 catalysts. (a) Catalyst 1; (b) catalyst 2; (c) catalyst 3; (d) catalyst 4; (e) catalyst 5; (f) 5A; (g) 5B, (h) 5C, (i) 5D, (j) 5-used. | ||
All of the catalysts exhibited strong charge-transfer (CT) bands below 500 nm. The spectra of the fresh VS-1 were dominated by four characteristic oxygen-to-metal CT bands. According to literature, the CT band centered at about 230–240 nm was assigned to the tetrahedral structure of (SiO)3VVO located in the silica framework.20,23 It indicated that vanadium species with tetrahedral structure existed in all VS-1 samples, irrespective of whether they are fresh or treated. The electronic absorption peak centered at 270–280 nm was inconspicuous in fresh VS-1, however, it became more significant in sample 5A and 5-used. The vanadium species corresponding to this peak could not be washed out by NH4OAc (Fig. 4h). It indicated that these vanadium species were located in the framework. The vanadium with a tetrahedral structure has a similar UV absorption peak at around 270–280 nm.24,33 So the band at 270–280 nm was ascribed to the (SiO)2(OH)VIVO species with tetrahedral structure that also located within the framework. The CT band at 320–350 nm was probably attributed to the isolated or oligomeric octahedral VV species on some vanadium-supported catalysts,19,35 however it was assigned to the framework vanadium with an octahedral structure by Tielens et al.31 As the intensity of the 320–350 nm band decreased in 5A, the electronic absorption peak at 320–350 nm was assigned as the overlapping of the CT bands of octahedral structures of VV both in framework and extra-framework. In addition, a CT band at 450 nm was observed on samples 1 to 5, and 5C, but it disappeared on 5A, 5B, and 5D (Fig. 4). This peak was tentatively assigned to highly dispersed vanadium species outside the framework. We could not estimate which octahedral VV species was located within the micropores of the zeolite, although the distorted one seems the most reasonable possibility. The d–d transitions above 600 nm were indicative of the presence of VIV on the catalysts, although the electronic spectra were relatively weak compared with the CT transition bands. The absorption peak at 310–320 nm was obviously observed on the catalysts with higher vanadium content (Fig. 4d and e), and was also observed in the treated sample 5B. However, this peak decreased sharply in 5A and 5D, so that it was hardly recognized. The change of the strength of the band at 310–320 nm with different treatments made it reasonable to identify a band at this wavelength. So the band at 310–320 nm was tentatively attributed to the extra-framework VIV oligomer with hexahedral or octahedral structure.
As shown in Fig. 4b, the absorption peaks at around 230, 270, 310, and 330 nm were observed on 5B. There was a weak absorption peak at 445 nm, and its strength decreased obviously comparing to fresh sample 5. It indicated that both the framework and extra-framework vanadium species were stable in acetic acid except the VV species with distorted octahedral structure outside the framework. According to the TPR profile of sample 5B, the vanadium species highly dispersed on the VS-1 disappeared after being treated with HOAc solution. The combination of TPR and DR UV-Vis confirmed that the distorted octahedral structure of VV species was loaded in the pore channel of zeolite.
For 5C (Fig. 4h), the absorption peaks at around 230, 270, 330, and 445 nm were found though the intensity of the 330 nm-peak decreased compared with fresh catalysts 4 and 5. Obviously, the framework vanadium species and the distorted octahedral structure of VV species outside the framework were stable in the presence of hydrogen peroxide, while a part of the octahedral structures of VV leached. The intensity of the absorption peak at around 330 nm was similar to 5A. It indicated that the octahedral structures of VV on the framework of VS-1 were stable in the presence of H2O2. The decrease of the 330 nm-peak was assigned to the leaching of the octahedral structures of VV on the extra-framework. The absence of a band at 310 nm revealed that the extra-framework VIV leached in the presence of H2O2.
After being treated by mixture of acetic acid and hydrogen peroxide (5D), the UV-Vis DR spectrum of sample 5D exhibited three peaks at around 230, 270, and 330 nm (Fig. 4i). It indicated that the vanadium species in framework with a tetrahedral structure were stable, no matter whether it contained VIV or VV. The absence of band at 310 nm revealed that the extra-framework vanadium oligomer leached in the presence of H2O2 and HOAc. The decrease in intensity of the 340 nm-peak indicated that a part of VV species with octahedral structure were unstable in H2O2 and HOAc. However, comparing the DR UV-Vis results of 5A and 5D, the intensity of 320–350 nm almost unchanged after being treated by the mixture of H2O2 and HOAc, so it was deduced that octahedral structures of VV on the extra-framework leached. The assignment of the different vanadium species are summarized in the supporting information.†
3.6 Activity and stability of the VS-1 catalysts
To examine the recyclability of various vanadium species on VS-1 in the hydroxylation of benzene, the catalysts 1 to 5 were consecutively used for five cycles, and the results were shown in Table 3.| Cat. no. | Runs | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|---|
| a Reaction conditions: 0.1 g catalyst, 11.25 mmol benzene, 29 mmol H2O2, 30 mL HOAc, 343 K, 2 h. | ||||||
| 1 | Yphenol (%) | 3.7 | 2.2 | 2.1 | 2.1 | 2.0 |
| Sphenol (%) | 94.3 | 95.1 | 96.0 | 96.3 | 95.9 | |
| TOF | 106.1 | 126.2 | 120.5 | 120.5 | 114.8 | |
| 2 | Yphenol (%) | 7.0 | 5.2 | 5.0 | 4.9 | 5.0 |
| Sphenol (%) | 93.3 | 93.2 | 93.9 | 94.8 | 95.1 | |
| TOF | 57.4 | 59.7 | 57.4 | 56.3 | 57.4 | |
| 3 | Yphenol (%) | 13.2 | 12.1 | 11.5 | 11.7 | 10.9 |
| Sphenol (%) | 91.2 | 90.4 | 91.0 | 90.6 | 92.4 | |
| TOF | 57.8 | 63.1 | 66.0 | 67.1 | 62.5 | |
| 4 | Yphenol (%) | 13.9 | 12.3 | 11.7 | 11.2 | 11.3 |
| Sphenol (%) | 84.8 | 90.1 | 91.1 | 91.0 | 90.8 | |
| TOF | 46.9 | 58.8 | 61.0 | 64.3 | 64.5 | |
| 5 | Yphenol (%) | 14.5 | 12.0 | 11.8 | 11.2 | 11.0 |
| Sphenol (%) | 76.1 | 91.4 | 91.8 | 90.0 | 91.6 | |
| TOF | 43.7 | 52.9 | 67.7 | 64.3 | 63.0 |
On all 1–5 catalysts, the yield of phenol decreased after the first use due to leaching of V species. On catalyst 1 and 2, the yield of phenol was almost unchanged after two consecutive runs with high selectivity above 93%. The vanadium content of used 1 and 2 catalysts was kept at 0.1% and 0.5% respectively after the second run. Based on the above discussion, the leaching part of vanadium would be the extra-framework vanadium species. These results indicated that the extra-framework vanadium species were not stable in the hydroxylation reaction. On the other hand, the extra-framework vanadium species on catalysts 1 and 2 mainly existed in a highly dispersed form. The slight variation of TOF between catalyst 1 or catalyst 2 in the consecutive runs showed that these highly dispersed vanadium species also showed compatible catalytic performance.
For catalyst 3, a 13.2% yield of phenol was obtained with about 91% selectivity to phenol on the fresh catalysts, and the TOF was 57.8. The yield of phenol decreased slightly (TOF above 60) and the selectivity to phenol kept above 90% in the four consecutive runs. The vanadium content decreased to 1.0% after the first run, and no variation was observed in the following runs. In the first run catalyst 4, 13.7% yield of phenol (TOF = 46.9) was obtained with 84.8% selectivity to phenol. In the second run, the yield decreased to 12.3%, however the selectivity increased to 90.1%. Similar results were obtained and no vanadium was detected in the resultant solution in the three consecutive runs. On catalyst 5, 14.5% yield of phenol was obtained with about 76.1% selectivity to phenol on the fresh catalysts, and the TOF was 43.7. After the first run, the phenol yield decreased to about 12.0% with about 90% selectivity to phenol (TOF = 52.9). The yield of phenol kept at about 11.0% with above 90% selectivity in the other three consecutive runs. The ICP results indicated that the vanadium content remained at about 1.0% after the first two runs. The ICP results listed in Table 1 indicated that the vanadium contents on fresh catalysts 3, 4 and 5 were higher than 1.0 wt%, and about 1.0 wt% vanadium content was stable on these catalysts after they were used in the hydroxylation reaction. It was indicated in Table 4 that these three catalysts showed a similar stable catalytic performance after being used 2 or 3 times, the yield of phenol was about 11% and the TOF was about 63 with above 90% selectivity to phenol. A series of control experiments were carried out to test the heterogeneous nature of the catalyst (the experiment is described in the supporting information†), the results showed that the framework vanadium species exhibited heterogeneous catalytic performance. These results indicated that the leached part of vanadium species had lower catalytic efficiency and accelerated deep oxidation of phenol, because the removal of them increased the selectivity to phenol. No variation of the vanadium content on the used catalyst was detected for the following successive runs, suggesting that the remained vanadium species was stable.
To explore the state variation of the vanadium species during the hydroxylation, the used catalyst 5 was characterized by XPS and DR UV-Vis, as fresh catalyst 5 contained all kinds of vanadium species as mentioned above. The results of XPS (Table 2) revealed the existence of VV and VIV species though the amount of VIV decreased, indicating the leaching of surface VIV species during the hydroxylation. Three CT bands at about 236, 271, and 339 nm were observed in the DR UV-Vis spectrum (Fig. 4j). It suggested that the framework vanadium species, both in tetrahedral and octahedral structures, were stable to acetic acid and hydrogen peroxide in the hydroxylation. However, the extra-framework vanadium species, leached out from the VS-1 zeolite, probably exhibited a homogeneous nature for the hydroxylation. In spite of other experimental factors, the nature of the vanadium species on the zeolite and the reaction conditions seemed to affect the leaching more significantly.
Furthermore, the catalytic performance of reused VS-1 catalysts 3, 4 and 5 showed a very similar TOF value (62.5–64.5). The high TOF was attributed to the framework vanadium species including VV and VIV species in the tetrahedral structure and VV in the octahedral structure. That is to say, these three kinds of vanadium species were highly efficient active centres in the hydroxylation reaction. So, incorporating the vanadium atom into the framework of zeolite could increase its catalytic performance.
The detected vanadium content on the NH4OAc treated and used catalysts 3, 4, and 5 in the present work was about 1.0 wt% (Table 1). This confirmed that the maximal incorporated content of vanadium into the silica framework was about 1.0 wt% for VS-1 prepared by a direct hydrothermal method. The excess use of vanadium resource mostly led to the formation of extra-framework vanadium species on the surface of VS-1, which bound loosely with the zeolite walls. In contrast with the post-synthesis method, the extra-framework vanadium species formed even when the controlled vanadium content was low. The reuse of the catalyst showed that the tetrahedral VIV and VV species and octahedral VV on the framework of VS-1 were stable, and the VS-1 was actually a heterogeneous catalyst. The highly dispersed distorted octahedral VV species, hexahedral or octahedral VIV oligomer and octahedral VV on the extra-framework were easily removed from the VS-1 surface by HOAc and/or H2O2. This part of vanadium species was unstable during the hydroxylation reaction, though it also had activity in the hydroxylation. In addition, it promoted the deep oxidation of phenol.
4. Conclusions
The threshold of vanadium entering into the silica framework was about 1.0 wt%, while extra-framework vanadium mainly formed on the catalyst with higher vanadium content. The vanadium species located in the silica framework existed as mononuclear octahedral VV, tetrahedral VV and VIV structures containing VO double bond, and were stable in acetic acid and/or hydrogen peroxide. During the present hydroxylation of benzene, these kinds of vanadium species showed good stability. The extra-framework vanadium was mainly present as hexahedral or octahedral VIV oligomers, together with two kinds of framework-like octahedral VV species possibly in the micropores of the zeolite, more or less distorted. Among these vanadium species, the oligomer vanadium species were unstable in oxidant and highly dispersed octahedral VV could dissolve in acetic acid. The catalyst with a vanadium content of 1.0 wt% in the framework gave a stable phenol yield of 11.0% (TOF = 63.0) with above 90% selectivity to phenol in the direct hydroxylation of benzene with H2O2.
Acknowledgements
The financial supports from the National Natural Science Foundation of China (No. 20872102, 20901053, and 21021001), PCSIRT (No. IRT0846), and characterization of the catalysts from Analytic and Testing Center of Sichuan University are greatly appreciated.References
- R. R. Fernandes, M. V. Kirillova, J. A. L. da Silva, J. da Silva and A. J. L. Pombeiro, Appl. Catal., A, 2009, 353, 107 CrossRef CAS.
- H. Abbo and S. Titinchi, Appl. Catal., A, 2009, 356, 167 CrossRef CAS.
- D. Bianchi, L. Balducci, R. Bortolo, R. D'Aloisio, M. Ricci, G. Spano, R. Tassinari, C. Tonini and R. Ungarelli, Adv. Synth. Catal., 2007, 349, 979 CrossRef CAS.
- B. Liptáková, M. Báhidsky and M. Hronec, Appl. Catal., A, 2004, 263, 33 CrossRef.
- S. Niwa, M. Eswaramoorthy, J. Nair, A. Raj, N. Itoh, H. Shoji, T. Namba and F. Mizukami, Science, 2002, 295, 105 CrossRef CAS.
- D. Bianchi, R. Bortolo, R. Tassinari, M. Ricci and R. Vignola, Angew. Chem., Int. Ed., 2000, 39, 4321 CrossRef CAS.
- M. Jian, L. Zhu, J. Wang, J. Zhang, G. Li and C. Hu, J. Mol. Catal. A: Chem., 2006, 253, 1 CrossRef CAS.
- J. Wang, C. Hu, M. Jian, J. Zhang and G. Li, J. Catal., 2006, 240, 23 CrossRef CAS.
- J. Zhang, Y. Tang, G. Li and C. Hu, Appl. Catal., A, 2005, 278, 251 CrossRef CAS.
- Y. Leng, H. Ge, C. Zhou and J. Wang, Chem. Eng. J., 2008, 145, 335 CrossRef CAS.
- K. Nomiya, K. Yagishita, Y. Nemoto and T. Kamataki, J. Mol. Catal. A: Chem., 1997, 126, 43 CrossRef CAS.
- R. Dimitrova and M. Spassova, Catal. Commun., 2007, 8, 693 CrossRef CAS.
- X. Gao and J. Xu, Appl. Clay Sci., 2006, 33, 1 CrossRef CAS.
- K. Lemke, H. Ehrich, U. Lohse, H. Berndt and K. Jähnisch, Appl. Catal., A, 2003, 243, 41 CrossRef CAS.
- Y. Masumoto, R. Hamada, K. Yokota, S. Nishiyama and S. Tsuruya, J. Mol. Catal. A: Chem., 2002, 184, 215 CrossRef CAS.
- L. D. Nguyen, S. Loridant, H. Launay, A. Pigamo, J. L. Dubois and J. M. M. Millet, J. Catal., 2006, 237, 38 CrossRef CAS.
- Y. Zhu, Y. Dong, L. Zhao and F. Yuan, J. Mol. Catal. A: Chem., 2010, 315, 205 CrossRef CAS.
- N. Das, H. Eckert, H. Hu, I. E. Wachs, J. F. Walzer and F. J. Feher, J. Phys. Chem., 1993, 97, 8240 CrossRef CAS.
- Y. W. Chen and Y. H. Lu, Ind. Eng. Chem. Res., 1999, 38, 1893 CrossRef CAS.
- C. W. Lee, W. J. Lee, Y. K. Park and S. E. Park, Catal. Today, 2000, 61, 137 CrossRef CAS.
- G. Mul, W. Wasylenko, M. S. Hamdy and H. Frei, Phys. Chem. Chem. Phys., 2008, 10, 3131 RSC.
- F. Tielens, M. Calatayud, S. Dzwigaj and M. Che, Microporous Mesoporous Mater., 2009, 119, 137 CrossRef CAS.
- K. M. Jinka, H. C. Bajaj, R. V. Jasra, E. A. Prasetyanto and S. E. Park, Top. Catal., 2009, 53, 238 CrossRef.
- G. Centi, S. Perathoner, F. Trifirb, A. Aboukais, C. F. Aissi and M. Guelton, J. Phys. Chem., 1992, 96, 2617 CrossRef CAS.
- M. Anpo, S. Higashimoto, M. Matsuoka, N. Zhanpeisov, Y. Shioya, S. Dzwigaj and M. Che, Catal. Today, 2003, 78, 211 CrossRef CAS.
- T. Sen, P. R. Rajamohanan, S. Ganapathy and S. Sivasanker, J. Catal., 1996, 163, 354 CrossRef CAS.
- N. K. Mal and A. V. Ramaswamy, Appl. Catal., A, 1996, 143, 75 CrossRef CAS.
- D. S. Bhange and V. Ramaswamy, Mater. Res. Bull., 2007, 42, 851 CrossRef CAS.
- J. Pitts, T. M. Thomas, A. W. Czanderna and M. Passler, Appl. Surf. Sci., 1986, 26, 107 CrossRef CAS.
- R. B. Shalvoy, P. J. Reucroft and B. H. Davis, J. Catal., 1979, 56, 336 CrossRef CAS.
- F. Tielens, M. Trejda, M. Ziolek and S. Dzwigaj, Catal. Today, 2008, 139, 221 CrossRef CAS.
- C. L. Zhang and Z. Y Wu, Acta Phys.–Chim. Sin., 1995, 11, 302 CAS.
- A. M. Prakash and L. Kevan, J. Phys. Chem. B, 2000, 104, 6860 CrossRef CAS.
- H. Berndt, A. Martin, A. Brückner, E. Schreier, D. Müller, H. Kosslick, G. Wolf and B. Lücke, J. Catal., 2000, 191, 384 CrossRef CAS.
- E. V. Kondratenko and M. Baerns, Appl. Catal., A, 2001, 222, 133 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cy00105a |
| This journal is © The Royal Society of Chemistry 2011 |