A versatile supported cobalt(II) complex for heterogeneously catalysed processes: conventional vs. microwave irradiation protocols†
Fatemeh
Rajabi
*a,
Alina M.
Balu
b,
Fatemeh
Toreinia
a and
Rafael
Luque
*b
aDepartment of Science, Payame Noor University, P. O. Box: 19395–4697, Tehran, Iran. E-mail: f_rajabi@pnu.ac.ir; Fax: +982813344081; Tel: +982813336366
bDepartamento de Química Orgánica, Campus de Rabanales, Edificio Marie Curie, Ctra Nnal IV, Km 396, Universidad de Córdoba, E-14014, Córdoba, Spain. E-mail: q62alsor@uco.es; Fax: 0034 957212066; Tel: 0034057212065
First published on 20th July 2011
Abstract
A highly active and reusable Co(II) complex supported on SBA-15 has been employed as a versatile heterogeneous catalyst in a range of processes including the synthesis of 1,1-diacetates, the acetalisation of aldehydes and the selective oxidation of various alcohols. The results obtained by using conventional protocols have been compared to the use of microwave irradiation as an alternative reaction media. The highly active catalyst was successfully utilised in such heterogeneously catalysed processes, in which the improved rates of reactions (from hours to minutes), yields and even selectivities in some cases, demonstrated the usefulness of microwave-assisted protocols as alternative methodologies in organic synthesis. The reported catalyst is believed to be a potentially versatile option that can be extended to further catalytic systems.
Introduction
The immobilisation of metal complexes on porous supports is a very useful methodology in organic synthesis. Such processes render heterogenised catalysts that possess advantages as heterogeneous catalysts. They are easy to separate from the reaction mixture and generate minimal product contamination caused by metal leaching.1–3 Heterogenised catalysts can be also efficiently recycled many times without a significant loss in activity.2In particular, noble metals complexes including Rh, Ir, Ru, Au and Pd have been extensively immobilised on porous supports and employed as heterogeneous catalysts.1,2,4,5 However, related transition metal complexes of Fe, Cu or Co have been less investigated despite their potential activities and involvement in many (bio)catalysed processes.6–8 Among them, supported cobalt(II) complexes including Co-salen have been recently developed for oxidation reactions.9,10 Several porous materials have been employed as supports for such metal complexes including SBAs, M41S, metal oxides and clays.2,4,6,11,12
Following our previous studies on supported metal complexes on mesoporous materials1,6,10,13 and greener heterogeneously catalysed processes,14 herein we report the versatile use of a previously reported Co(II) complex supported on SBA-15 in a range of relevant heterogeneously catalysed processes including the preparation of 1,1 diacetates, the chemoselective O/O-acetalisation of various aldehydes under very mild conditions (typically low catalyst loadings and reactions carried out at room temperature) as well as the selective oxidation of alcohols and alkenes.
For the three different processes, protocols run under room temperature conditions have been compared with microwave protocols. The reason for the comparison of conventional vs. microwave methodologies is related to the remarkable improvements on efficiency, rates of reactions and energy consumption as well as selectivities in some cases that have positioned microwave irradiation as one of the most useful and powerful tools as alternative media in organic synthesis.15,16
Experimental
The supported Co(II) complex on SBA-15 was prepared according to a previously reported protocol.13aSalicylaldehyde (2 mmol, 0.244g) was added to an excess of absolute MeOH, to which 3-aminopropyl (trimethoxy) silane (2 mmol, 0.352g) was subsequently added. The solution instantly became yellow due to imine formation. After 3 h, cobalt(II) acetate, Co(OAc)2·2H2O (1 mmol, 0.248g) was added to the solution, and the mixture was further stirred for 3 h to allow the new ligands to complex the cobalt. A colour change from pink to olive green is observed. Mesoporous SBA-15 (average pore diameter 6 nm, 3g) was activated by refluxing in concentrated hydrochloric acid (6 M) and then washed thoroughly with the deionized water and dried before undergoing chemical surface modification. This activation treatment readily hydrolyses the siloxane Si-O-Si bonds to Si-OH species, which will be key to anchor the cobalt complex. The complex and the activated silica were then mixed and the mixture stirred overnight. The solvent was removed using a rotary evaporator, and the resulting olive green solid dried at 80 °C overnight. The final product was washed with MeOH and water (to remove all physisorbed metal species) until the washings were colourless. Further drying of the solid product was carried out in an oven at 80 °C for 8 h (Scheme 1).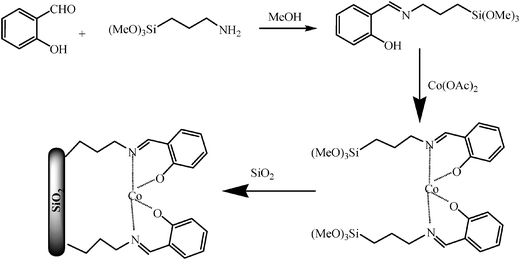 | ||
| Scheme 1 Structure of the supported Co(II) catalyst. | ||
The catalyst possesses a large surface area (450 m2 g−1), 3.6 nm pore size and 0.77 mL g−1pore volume. The cobalt loading was 0.3 mmol g−1. The material has been characterised by XRD, IR, N2 physisorption and UV-vis (see ESI†).
General procedure for the synthesis of 1,1-diacetates (RT protocol)
In a typical experiment, 5 mmol of aldehyde was reacted with 5.1 mmol of acetic anhydride in the presence of Co(II)/SBA-15 (0.5 mol%) for the required period of time at RT under continuous stirring (reaction monitored by TLC). After reaction completion, the final mixture was filtered off to recover the catalyst and washed with ethyl acetate. In the filtrate, the organic layers were subsequently washed with 5% NaHCO3 (2 × 5 mL) and brine (2 × 5 mL) and dried over Na2SO4. The solvent was eventually removed under vacuum to yield 1,1-diacetates as pure products. No byproducts were obtained in the reaction.General procedure for the synthesis of 1,1-diacetates (MW protocol)
5 mmol of aldehyde, 5.1 mmol of acetic anhydride and 0.5 mol% of the catalyst were placed on a pyrex microwave vial and microwaved for 1-5 min at 300 W in a CEM-DISCOVER microwave reactor. The resultant solution was then filtered off and the filtrate was analysed by GC and GC-MS using an Agilent 6890N GC model equipped, with a 7683B series autosampler, fitted with a DB-5 capillary column and an FID detector. The experiments were conducted on a closed vessel (pressure controlled) under continuous stirring. The microwave method was generally power-controlled where the samples were irradiated with different power outputs (normally settings at maximum power, 300 W). The product was isolated in a similar way to that of the conventional methodology described above.General procedure for the acetalisation of carbonyl compounds (RT protocol)
1,3-propanediol (1 mmol) was added to a solution of aldehyde (1 mmol) and Co(II)/SBA-15 (0.5 mol%) in a round-bottomed flask and the mixture was stirred at RT for some time. Upon reaction completion (monitored by TLC and GC), the reaction mixture was filtered off (to remove the catalyst) and washed with ethyl acetate. The solvent was subsequently removed through evaporation under reduced pressure to afford the crude products. These were purified (if needed) by flash chromatography to provide the pure acetals which were characterised by NMR (Bruker 500 MHz) and IR analysis.General procedure for the acetalisation of carbonyl compounds (MW protocol)
The mixture of 1 mmol of aldehyde, 1 mmol of 1,3-propanediol and 0.5 mol% of the catalyst was microwaved for 1–5 min at 300 W in a CEM-DISCOVER microwave reactor. The resultant solution was then filtered off and the filtrate was analysed by GC and GC-MS using an Agilent 6890N GC model, equipped with a 7683B series autosampler, fitted with a DB-5 capillary column and an FID detector. The experiments were conducted on a close vessel (pressure controlled) under continuous stirring. The microwave method was generally power-controlled where the samples were irradiated with different power outputs (normally settings at maximum power). The product was isolated in a similar way to that of the conventional methodology described above.General protocol for the selective oxidation of alcohols to carbonyl compounds (conventional heating and microwave irradiation)
1 mmol of benzyl alcohol, 4 mmol of H2O2 (50 wt% solution in water) and 2 mol% of Co(II)/SBA-15 were added to a round-bottomed flask and heated up under continuous stirring at 140 °C for 24–48 h. The progress of the reaction was monitored by sampling reaction aliquots from the reaction mixture and analysing them by GC and GC-MS using a similar instrument to that reported for the other catalytic experiments. The microwave reaction experiments were also carried out in the CEM-DISCOVER reactor under identical conditions reported above for the conventionally heated protocol, with the exclusive difference of the time of reaction (typically 0.5–1 h) and the power utilised (150–300 W) and temperature reached (100–130 °C, measured by an IR probe) and again analysed by GC/GC-MS.Reuses
The recycling of the catalyst in all reported reactions followed a similar procedure: Upon reaction completion, the catalyst was generally filtered off, thoroughly washed with MeOH/acetonitrile and oven-dried overnight at 80–100 °C prior to its reuse in another run. Comparatively, the recycled catalysts were conditioned for reutilisation by refluxing in methanol for 4–6 h and then dried overnight at 80–100 °C. The results obtained proved that the reflux in methanol as part of the catalytic reuse was not necessary in most cases.The metal content in the materials was also determined before and after the reaction (as well as in the reaction filtrate) using inductively coupled plasma (ICP) in a Philips PU 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 sequential spectrometer equipped with an Echelle monochromator (0.0075 nm resolution). The samples were digested in HNO3 and subsequently analyzed by ICP/MS.
000 sequential spectrometer equipped with an Echelle monochromator (0.0075 nm resolution). The samples were digested in HNO3 and subsequently analyzed by ICP/MS.
Characterisation of selected products
Results and discussion
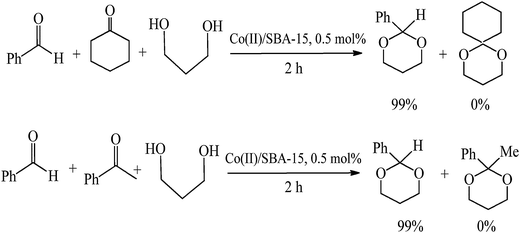
Synthesis of 1,1-diacetates from aldehydes
1,1-diacetates are synthetically useful as protecting groups for aldehydes due to their stability and simple conversion back into the parent aldehydes.17 These compounds are also important building blocks for the synthesis of dienes for Diels–Alder cycloaddition reactions17 and other reactions by introducing appropriate nucleophiles.18 The synthetic methodologies for 1,1-diacetates generally involved the use of acetic anhydride combined with strong protic acids19 or Lewis acid catalysts.17,20 However, these reported protocols have inherent drawbacks including low yields, long reaction times, waste generation and the employment of non-recoverable catalysts. Alternative methods have been recently developed to improve the green credentials of the reaction, employing mostly solid acids as heterogenous catalysts and more environmentally friendly reaction conditions.21 However, there are no reports to date of heterogenously catalysed Co(II) benign protocols at room temperature for this reaction.The results for the formation of 1,1 diacetates from different aldehydes (Scheme 1) are included in Table 1. The Co(II) catalysed protocol provided excellent yields of diacetates from a wide range of aldehydes including indole, tiophene and linear substrates at room temperature and relatively short times of reaction (typically 3–4 h) at room temperature, with improved reaction rates at slightly increased temperatures (quantitative conversion to product after 60 min, typically at 60–70 °C). Compared to this benign protocol at RT, the use of microwaves was found to significantly enhance the rates of reactions in the systems, with comparable yields to products obtained in very short times of reaction (1–2 min, Table 1) under very mild reaction conditions. The selectivity of the reaction follows the expected trends with 1,1-diacetates of aromatic derivatives preferentially formed from aldehydes in the presence of aliphatic and cyclic ketones (Scheme 2).
| Entry | Substrate | Time (min) | Yield (%)c | TON | TOF (h−1) | |||
|---|---|---|---|---|---|---|---|---|
| RTa | MWb | RT | MW | RT | MW | |||
| a Reaction conditions: 5 mmol aldehyde, 5.1 mmol acetic anhydride, 0.5 mol% catalyst, RT. b 300W, 110–120 °C (maximum temperature reached) (monitored by IR). c If different, the MW yield is given in brackets. | ||||||||
| Blank | PhCHO | 360 | 60 | <5 | — | — | — | — |
| SBA-15 | PhCHO | 360 | 60 | <5 | — | — | — | — |
| 1 | PhCHO | 180 | 1 | 99 | 500 | 500 | 167 | 30000 |
| 2 | 4-ClC6H4CHO | 150 | 1 | 99(98) | 500 | 495 | 200 | >29000 |
| 3 | 4-NO2C6H4CHO | 180 | 1 | 99 | 500 | 500 | 167 | 30000 |
| 4 | 3-NO2C6H4CHO | 240 | 1.5 | 95(98) | 480 | 495 | 120 | 19700 |
| 5 | 2,4-Cl2C6H3CHO | 300 | 1 | 98(95) | 495 | 480 | 99 | >28000 |
| 6 | 4-FC6H4CHO | 270 | 1 | 92(89) | 465 | 450 | 103 | 26700 |
| 7 |

|
360 | 2 | 98 | 495 | 495 | 83 | >14800 |
| 8 |
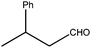
|
420 | 3 | 90(94) | 455 | 475 | 65 | >9400 |
| 9 |
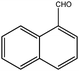
|
300 | 3 | 95(90) | 480 | 455 | 99 | >9000 |
| 10 |
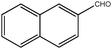
|
270 | 3 | 90(92) | 455 | 465 | 101 | >9200 |
| 11 |
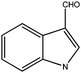
|
150 | 1 | 95(98) | 480 | 495 | 192 | >29000 |
| 12 |
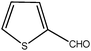
|
120 | 1 | 96 | 485 | 485 | 242 | >29000 |
| 13 | 4-OCH3C6H4CHO | 240 | 1.5 | 95(98) | 480 | 495 | 120 | 19700 |
| 14 | 4-CH3C6H4CHO | 210 | 1.5 | 98(99) | 495 | 500 | 141 | 20000 |
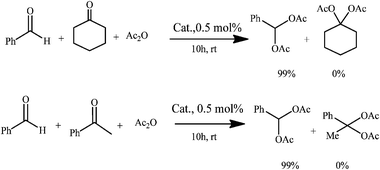 | ||
| Scheme 2 Chemoselectivity in the reaction of aldehydes and ketones with acetic anhydride to give 1,1-diacetates: aldehydesvs.ketones. | ||
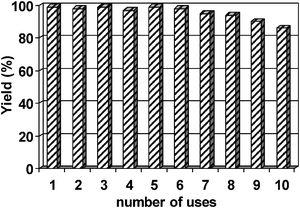
The cobalt catalyst was found to be highly stable and reusable under the investigated conditions (up to 12 runs) without any significant loss of its catalytic activity (Fig. 1). Indeed, ICP analysis of both reaction filtrate and catalyst showed no detectable Co leaching (<0.5 ppm) in the reaction filtrate upon reaction completion, with an almost identical Co content for both fresh and reused catalyst (0.30 vs. 0.29 mmol of Co per gram of catalyst for fresh and 12-time reused material, respectively) regardless of the methodology employed in the catalytic reaction. These findings were in good agreement with previously reported stability for the Co system13 and correlate well with the highly reusable nature of the material at room temperature and short time microwave irradiation.
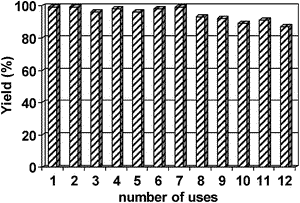 | ||
| Fig. 1 Reuses of Co(II)/SBA-15 catalyst in the 1,1-diacetate formation from benzaldehyde. Reaction conditions: 5 mmol of aldehyde, 5.1 mmol of acetic anhydride, 0.5 mol% of catalyst, RT, 3 h reaction. | ||
2.2. Chemoselective acetalisation of aldehydes
Another important process for the protection of aldehydes is the acetalisation of such carbonyl compounds with alcohols (e.g.1,3-propanediol). The formation of O–O-acetals through acetalisation is commonly employed in multi-step syntheses of organic molecules and/or natural products.22 These important compounds are useful as intermediates and high added value chemicals in polymers, cross-linking reagents and flavourings.19,23Acetals are normally prepared using a range of acid catalysts under different conditions.24,25 However, the use of a supported metal catalyst in this reaction has to date never been attempted.The results of the acetalisation of various aldehydes are summarised in Table 2. The methodology was amenable to a wide range of substituents and/or substrates, with excellent yields to products obtained in all cases (typically in 2–4 h) at room temperature. Similarly to that observed for the diacetate production, a temperature increase (from RT to 60–70 °C) will reduce times of reaction from many hours to minutes (45–60 min). Nevertheless, the reaction rates could be remarkably speeded up under microwave irradiation. Irradiating samples for a few minutes (1–5 min) was sufficient to provide comparable yields to those obtained after hours at room temperature. No significant differences were observed from the electron-donating/withdrawing substituents and/or aromatic or aliphatic derivatives either. Of note was the clearly marked directed selectivity to the reaction of aldehydes and linear primary alcohols (Scheme 3).
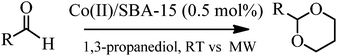
| Entry | Substrate | Time (min) | Yield (%)c | TON | TOF (h−1) | |||
|---|---|---|---|---|---|---|---|---|
| RTa | MWb | RT | MW | RT | MW | |||
| a Reaction conditions: 1 mmol substrate, 1 mmol 1,3-propanediol, 0.5 mol% g catalyst, RT. b microwave irradiation, 300 W, 120–130 °C (maximum temperature reached). c If different, the MW yield is given in brackets. | ||||||||
| Blank |
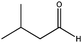
|
360 | 60 | <5 | — | — | — | — |
| SBA-15 |
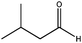
|
360 | 60 | <15 | 10 | 12 | <2 | 12 |
| 1 |
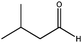
|
180 | 3 | 95(99) | 96 | 100 | 32 | 2000 |
| 2 |
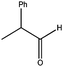
|
270 | 5 | 92(95) | 93 | 96 | 21 | 1150 |
| 3 |
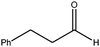
|
120 | 5 | 96(99) | 97 | 100 | 49 | >1200 |
| 4 |
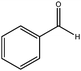
|
120 | 2 | 99 | 100 | 100 | 50 | 3000 |
| 5 |
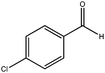
|
150 | 3 | 99 | 100 | 100 | 40 | 2000 |
| 6 |
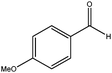
|
180 | 3 | 95(98) | 96 | 99 | 32 | >1900 |
| 7 |
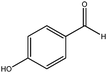
|
210 | 3 | 99 | 100 | 100 | 29 | 2000 |
| 8 |
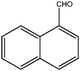
|
150 | 3 | 99 | 100 | 100 | 40 | 2000 |
| 9 |
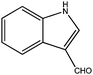
|
240 | 5 | 99 | 100 | 100 | 25 | 1200 |
 | ||
| Scheme 3 Selectivity in the acetalisation reaction: Aldehydesvs.ketones. | ||
The catalyst was also highly reusable under both investigated conditions (preserving over 90% of its initial activity after 10 uses, Fig. 2). ICP analysis of the final filtrate upon reaction completion as well as that of the fresh and reused catalyst showed similar results to those obtained for the formation of 1,1-diacetates from aldehydes at room temperature and short time microwave irradiation, confirming the stability of the catalyst under the investigated reaction conditions.
 | ||
| Fig. 2 Reuses of the Co(II)/SBA-15 catalyst in the chemoselective acetalisation of benzaldehyde. Reaction conditions: 1 mmol substrate, 1 mmol 1,3-propanediol, 0.5 mol% g catalyst, RT, 2 h reaction. | ||
2.3. Selective oxidation of alcohols using hydrogen peroxide
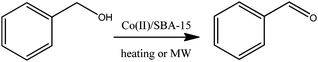
The selective oxidation of alcohols to carbonyl compounds including aldehydes and ketones has been the subject of many investigations in organic chemistry due to the versatility of the carbonyl group as building block. A wide range of alternative systems have been developed to replace the traditionally utilised stoichiometric reagents, hazardous peroxides as oxidating agents and energy intensive protocols that include the use of hydrogen peroxide and molecular oxygen as greener oxidants,10,12,26 various heterogeneous catalytic protocols with supported nanoparticles or supported metal complexes as catalysts27 as well as alternative reaction media (e.g. microwaves).14,28In this regard, we have previously reported a supported cobalt complex, which was reported to be highly active and selective in the aerobic oxidation of alcohols under relatively mild reaction conditions.10 Inspired by our previous approach to the oxidation of aromatic alcohols, we were prompted to study the utilisation of our versatile Co(II)/SBA-15 catalyst in the selective oxidation of benzyl alcohol with hydrogen peroxide (50 wt%) as a greener oxidant. The results shown in Table 3 demonstrate the reaction does not progress in the absence of heating as compared to previous acetate formation and acetalisation reactions. Temperatures > 120 °C were required to obtain moderate conversion values to products that were in any case achieved in long times of reaction (24 h and above). The selectivities to benzaldehyde obtained in most cases were not particularly great (<80%), with significant quantities of benzoic acid obtained as a byproduct from the over-oxidation of benzaldehyde. Interestingly, the use of microwave irradiation (MW) was found to have a remarkable effect in the rates of reaction of the oxidation, with excellent conversions and even improved selectivities to benzaldehyde achieved after 30–45 min of microwave irradiation, as compared to long times and incomplete conversions in the conventional heating method (Table 3), once again proving the usefulness of microwave-assisted methodologies in heterogeneous catalysed reactions.
| Entry | Run | Time of reaction (h) | Conv. (mol%) | Sbenz (mol %) | TON | TOF (h−1) |
|---|---|---|---|---|---|---|
| a Reaction conditions: 1 mmol benzyl alcohol, 4 mmol H2O2 (50 wt%), 2 mol% catalyst. Conventional heating (100–140 °C) or microwave irradiation (MW). | ||||||
| 1 | blank | 72 | — | — | — | — |
| 2 | SBA-15 | 72 | — | — | — | — |
| 3 | Heating, 100 °C | 48 | <30 | 70 | 5 | 0.1 |
| 4 | Heating, 120 °C | 48 | 55 | 60 | 11 | 0.2 |
| 5 | Heating 140 °C | 24 | 50 | 80 | 13 | 0.6 |
| 6 | MW, 150 W, 100 °C | 0.75 | 65 | 95 | 21 | 27 |
| 7 | MW, 300 W, 120–130 °C | 0.75 | 85 | 90 | 26 | 34 |
| Reuse | MW, 300 W, 120–130 °C | 1 | 85 | 95 | 27 | 27 |
Encouraged by the promising results obtained under microwave irradiation, the scope of the protocol was extended to a variety of substrates including aromatic, aliphatic and linear alcohols to aldehydes and ketones. The results included in Table 4 show good to excellent conversions in the systems could be achieved in reasonable times of reaction (typically 30–90 min) with good selectivities to target products, regardless of compounds, substituents and products (aldehydes or ketones) obtained.
| Entry | Substrate | Time (h) | Conv. (mol%) | Scarbonyl (mol%) | TON | TOF (h−1) |
|---|---|---|---|---|---|---|
| a Reaction conditions: 1 mmol substrate, 4 mmol H2O2 (50 wt%), 2 mol% catalyst, 300 W, 120–130 °C;. b reused recovered Co(II)/SBA-15 catalyst after 5 uses. | ||||||
| 1 |
|
0.75 | 80 | >95 | 26 | 35 |
| 2 |
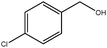
|
0.5 | 95 | >99 | 31 | 63 |
| 3 |
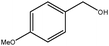
|
0.75 | 88 | >95 | 21 | 28 |
| 4 |
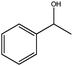
|
0.75 | 90 | >99 | 30 | 40 |
| 5 |
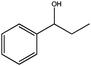
|
1 | >95 | >99 | 32 | 32 |
| 6 |
|
1.5 | 75 | >99 | 25 | 17 |
| 7 |
|
1 | 85 | >99 | 28 | 28 |
| 8 |
|
1 | 95 | >99 | 31 | 31 |
| 9 |

|
1.5 | 70 | >99 | 23 | 15 |
| reusedb |
|
1 | 90 | >99 | 30 | 30 |
The catalyst was also found to be highly reusable under the investigated reaction conditions (both under conventional heating and microwave irradiation), preserving over 90% of its initial activity after 5 uses (Table 3, entry reused). No significant Co(II) was detected by ICP of the reaction mixture upon filtration of the catalyst in the first uses, which was in good agreement with the almost negligible conversion increase (<5%) of the reaction filtrate after irradiating with microwaves for an additional hour. However, a minor discrepancy between the quantities of Co in the fresh (0.30 mmol g−1catalyst) and reused (0.25 mmol g−1) was found in the microwave-assisted oxidation process. This may imply a very minor leaching of Co(II) into solution each run (inferior to 0.5 ppm) due to the higher temperatures and longer times of reaction in the oxidation process (typically 1 h), which was however not sufficient to promote the oxidation reaction itself. In any case, the structure of the catalyst and its textural properties were preserved after 5 uses as clearly depicted in the ESI.†
Further investigations are currently ongoing in our laboratories to extend the application of this versatile system to related heterogeneously catalysed reactions including the (hydro)silylation of alcohols with hexamethyldisilazane (HMDS) and alkynes (e.g.phenyl acetylene) with different silanes including dichloromethyl silane.
3. Conclusions
A versatile and efficient Co(II) material has been proved to be an excellent heterogeneous catalyst for a range of processes including 1,1-diacetate formation, acetalisation of aldehydes and oxidation of alcohols both under conventional conditions and microwave irradiation. The catalyst was found to be highly active, selective and reusable under the investigated reaction conditions and provided good to excellent yields to products in the different heterogeneously catalysed processes. The comparison between conventional and microwave systems showed that, in general, microwave protocols predate conventional systems by significantly reducing times of reaction increasing, at the same time, the yields and even selectivities in some cases to the target products. In view of the demonstrated applications of the materials, we envisage a further catalytic extension to other processes including (hydro)silylations, alkylations and coupling reactions.Acknowledgements
Fatemeh Rajabi acknowledges Payame Noor University for partial funding. Alina Balu gratefully acknowledges funds from the research group FQM-162, Departamento de Quimica Organica and from Ministerio de Ciencia e Innovacion (Project CTQ 2008–01330/BQU) and Consejeria de Educacion y Ciencia, Junta de Andalucia (Projects P09-FQM-4781 and P10-FQM-6711), cofinanced with FEDER funds. Rafael Luque also gratefully acknowledges Ministerio de Ciencia e Innovación, Gobierno de España for the concession of a Ramon y Cajal contract (ref. RYC-2009-04199).References
- V. Caballero, F. M. Bautista, J. M. Campelo, D. Luna, R. Luque, J. M. Marinas, A. A. Romero, I. Romero, M. Rodriguez, I. Serrano, J. M. Hidalgo and A. Llobet, J. Mol. Catal. A: Chem., 2009, 308, 41–45 CrossRef CAS
.
-
(a) C. Gonzalez-Arellano, A. Abad, A. Corma, H. Garcia, M. Iglesias and F. Sanchez, Angew. Chem., Int. Ed., 2007, 46, 1536–1538 CrossRef CAS
-
(a) F. Fache, E. Schultz, M. L. Tommasino and M. Lemaire, Chem. Rev., 2000, 100, 2159–2231 CrossRef CAS
- A. Corma and H. Garcia, Adv. Synth. Catal., 2006, 348, 1391–1412 CrossRef CAS
-
(a) H. Huang, H. Wu, X. Liao and B. Shi, Catal. Commun., 2010, 11, 487–492 CrossRef
- R. Luque, S. K. Badamali, J. H. Clark, M. Fleming and D. J. Macquarrie, Appl. Catal., A, 2008, 341, 154–159 CrossRef CAS
-
(a) A. R. Silva, J. L. Figueiredo, C. Freire and B. De Castro, Catal. Today, 2004, 102, 154-159
- C. González-Arellano, R. Luque and D. J. Macquarrie, Chem. Commun., 2009, 1410–1412 RSC
-
(a) V. Mirkhani, M. Moghadam, S. Tangestaninejad, I. Mohammadpoor-Baltork and N. Rasouli, Catal. Commun., 2007, 9, 219–223
- F. Rajabi and B. Karimi, J. Mol. Catal. A: Chem., 2005, 232, 95–99 CrossRef CAS
-
(a) I. Kuzniarska-Biernacka, A. R. Silva, A. P. Carvalho, J. Pires and C. Freire, Catal. Lett., 2010, 134, 63–71 CrossRef CAS
- A. M. Balu, J. M. Hidalgo, J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, J. Mol. Catal. A: Chem., 2008, 293, 17–24 CrossRef CAS
-
(a) F. Rajabi, R. Luque, J. H. Clark, B. Karimi and D. J. Macquarrie, Catal. Commun., 2011, 12, 510–513 CrossRef CAS
- C. Gonzalez-Arellano, J. M. Campelo, D. J. Macquarrie, J. M. Marinas, A. A. Romero and R. Luque, ChemSusChem, 2008, 1, 746–760 CrossRef CAS
-
(a) C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250–6284 CrossRef CAS
-
(a)
Aqueous microwave assisted chemistry, ed. V. Polshettiwar and R. S. Varma, Royal Society of Chemistry, Cambridge, UK, 2010 Search PubMed
- A. R. Hajipour, L. Khazdooz and A. E. Ruoho, Catal. Commun., 2008, 9, 89–96 CrossRef CAS
-
(a) J. S. Yadav, V. B. Subba Reddy and P. Srihari, Synlett., 2001, 673 CAS
-
(a) T. J. Jin, G. Sun, Y. W. Li and T. S. Li, Green Chem., 2002, 4, 255–256 RSC
-
(a) C. Wang and M. Li, Synth. Commun., 2002, 32, 3469–3473 CrossRef CAS
-
(a) N. M. Nagy, M. A. Jakab, J. Konya and S. Antus, Appl. Clay Sci., 2002, 21, 213–216 CrossRef CAS
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS
-
(a)
H. Maarse, Volatile Compounds in Foods and Beverages, Marcel Dekker Inc, New York, 1991 Search PubMed
-
(a) B. Procuranti, L. Myles, N. Gathergood and S. J. Connon, Synthesis, 2009, 4082–4086 CAS
- C. Huo and T. H. Chan, Adv. Synth. Catal., 2009, 351, 1933–1938 CrossRef CAS
-
(a) F. Wang and W. Ueda, Catal. Today, 2009, 144, 358–361 CrossRef CAS
-
(a) T. Yasu-eda, S. Kitamura, N. Ikenaga, T. Miyake and T. Suzuki, J. Mol. Catal. A: Chem., 2010, 323, 7–15 CrossRef CAS
-
(a) P. J. Figiel, M. N. Kopylovich, J. Lasri, M. F. C. Guedes da Silva, J. J. R. Frausto da Silva and A. J. L. Pombeiro, Chem. Commun., 2010, 46, 2766–2768 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cy00101a |
| This journal is © The Royal Society of Chemistry 2011 |