A new method to obtain β-glucan from Saccharomyces cerevisiaecells
Tatiana Felix
Ferreira
*a,
Leonardo Rodrigues
de Andrade
b,
Maria Alice Zarur
Coelho
a and
Maria Helena Miguez
da Rocha-Leão
a
aSchool of Chemistry, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil. E-mail: tatianafelix@ufrj.br; Fax: +55 21 25627622; Tel: +55 21 25627622
bInstitute of Biomedical Sciences, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
First published on 23rd May 2011
Abstract
In this work, a simple method was developed to increase cell wall porosity and to achieve partial hydrolysis of β-glucan. First, the cells were permeabilized using ethanol under agitation. Then the cells were treated with glass beads to increase cell wall porosity and subjected to an enzymatic treatment to degrade glycogen at 65 °C for 30 minutes. Finally, the cells were incubated at 57 °C for 2 hours to partially degrade β-glucan. Whole cells, permeabilized cells and permeabilized cells with partially hydrolyzed β-glucan were evaluated by optical microscopy and transmission electron microscopy. From the images of permeabilized cells with partially hydrolyzed β-glucan it was possible to visualize structures named “ghosts”, which are alike along microcapsules containing β-glucan. These structures can be used to aggregate β-glucan in food industrialized products.
Introduction
Yeasts are unicellular fungi and they are used in many industrial processes, generating millions of tons of waste biomass every year. Due to the numerous bioactive substances such as proteins and polysaccharide complexes, these cells have high nutritional value.5 So, many studies to obtain these bioactive compounds from yeast cell wall are being developed.The cell wall of yeast Saccharomyces cerevisiae contains three main components: glucan, a polymer β-1,3 and β-1,6 glucose (48–60%), mannoproteins (20–23%) and chitin, a polymer of β-1,4 N-acetylglucosamine (0.6–2.7%).2 Its structure is composed of modules consisting of an inner layer composed of β-1,3 glucan and chitin attached to the outer layer consisting of mannoproteins by β-1,6 glucan.6
Glucans are polymers of non-cyclical anhydroglucose united by β-1,3 glycosidic linkages and containing β-1,6 branches. The β-1,3 glucans have an estimated size of 1500 glucose residues, while β-1,6 glucans are smaller, with 150 to 200 residues.2 This polysaccharide is recommended in human diet because it presents innumerable bioactive properties, like immune stimulation and glucose deprivation in blood,1,10,13 which makes possible its use in the treatment of glycemia control and in increasing immunity. However, cell wall degradation and isolation of bioactive compounds are challenging processes. These are usually done through enzymatic processes using specific enzymes. With that, the disruption of yeast cell walls and fractionation of these polysaccharides make the process complex and expensive. So, in the present work was developed a simple method to obtain yeast cell wall containing partially hydrolyzed β-glucan.
The partial hydrolysis of β-glucan is important to make easy its absorption and consequently its bioactive action because when this polysaccharide is ingested in soluble or microparticle forms, it is more easily absorbed by the digestive system.9
Intracellular substances of low molecular weight are easily obtained after cell permeabilization with organic solvents and vigorous agitation.14 Under these conditions, plasma and organelle membranes are broken, ridding those barriers to cellular permeability. The obtained structures called “ghosts” can be compared to micrometric particles of encapsulated polymeric materials where the covering material is the cell wall and the nucleus is the intracellular macromolecule.15,16
The goal of this work was to establish a simple method to obtain β-glucan from S. cerevisiaecells. It was possible due to the production of “ghosts”, available from glucanase action, free of glycogen and trehalose, containing partially digested glucan and without any previous fractionation method of cellular macromolecules. These structures named “porous ghosts” can be used to add β-glucan in industrialized foods.
Materials and methods
Kinetics of amyloglucosidase action
Trehalose (1 mg mL−1), glucan (0.25 mg mL−1) and glycogen (1 mg mL−1) were incubated in the presence of amyloglucosidase (14 UI). Aliquots of 200 μL from each solution containing glycogen, glucan or trehalose were incubated with 50 μL of 3 M acetic acid, 700 μL of 0.2 M sodium acetate buffer (pH 4.8) and 50 μL of the enzymatic preparation at 37, 57, 65 and 75 °C, respectively. Samples were periodically taken for glucose quantification. In all these assays, free glucose was quantified by a glucose oxidase method (Diagnostic) in vitro. Saccharides and enzyme preparation (A7255-amyloglucosidase from Rhizopus sp.) herein used were purchased from SIGMA.An aliquot of 200 μL from solution containing a mixture of glycogen and glucan was incubated with 50 μL of 3 M acetic acid, 700 μL of 0.2 M acetate buffer (pH 4.8) and 50 μL of the enzymatic preparation at 65 °C/1 h and residual glucose was quantified. The temperature was reduced to 57 °C/2 h and afterwards glucose was quantified again. After that, the temperature was reduced to 37 °C and 20 h later the glucose was quantified once more. In parallel, an incubation assay was carried out at 57 °C/20 h and the released glucose was quantified.
Culture conditions
In this work, a diploid strain constructed by crossing reg1 (a his4 MAL2-8C MAL3 SUC3 CAT1-2d hex2-3/reg1) and hxk1hxk2 (alfa trp his4 hxk1 hxk2 GLK1 MAL2-8c) haploid strains4 was used. Cells were stocked in semi-solid medium at 4 °C. For proliferation, cells were inoculated in 500 mL flasks containing 100 mL of YPD medium, incubated at 28 °C in a rotatory shaker (160 oscillations min−1). After 18 h, an aliquot of 100 mg of cells (dry weight) was centrifuged and washed with sodium acetate buffer (75 mM, pH 4.8).Cell permeabilization
The cells (100 mg) were suspended in 5 mL sodium acetate buffer (75 mM, pH 4.8) with 0.2 mL of ethanol and subjected to vigorous agitation for 5 minutes.3 The permeabilized cells were washed with sodium acetate buffer (75 mM, pH 4.8) to release the trehalose. After that, the cells were subjected to different processes to increase the porosity: treatment with glass beads or with alkaline solution.Cell disruption with glass beads
An aliquot (750 μL) of the suspension previously prepared containing 15 mg of permeabilized cells was incubated with 1.5 g of 0.5 mm glass beads. The suspension was subjected to three agitation cycles with glass beads for 1 minute in vortex intercalating 1 minute in an ice bath. The extract of cells was separated and porous cells were washed with sodium acetate buffer (0.2 M, pH 4.8) to release the intracellular material, forming the structure called ghosts.Cell disruption with alkaline medium
An aliquot containing approximately 15 mg of permeabilized cells was incubated with 0.125 M Na2CO3 at 100 °C/3 h. The extract of cells was separated and porous cells were washed with sodium acetate buffer (0.2 M, pH 4.8) to release the intracellular material, forming a structure also called ghosts.Hydrolysis of saccharides in cell walls
An aliquot (200 μL) containing approximately 15 mg of ghosts was incubated with 50 μL of 3 M acetic acid, 700 μL of 0.2 M sodium acetate buffer (pH 4.8) and 50 μL of the enzymatic preparation (70% more concentrated) at 65 °C/30 minutes for all glycogen degradation (a small part of glucan was also degraded at this temperature). A sample was removed for glucose quantification and the temperature was reduced (57 °C) for glucan degradation. After one hour, the glucose was quantified again.Control assay
Whole cells were suspended in 1 mL of 0.250 M Na2CO3 and incubated at 100 °C/90 minutes.12 After that, 15 mg of cells (dry weight) were incubated with 50 μL of 3 M acetic acid, 900 μL of 0.2 M sodium acetate buffer (pH 4.8) and 50 μL of the enzymatic preparation (70% more concentrated) at 65 °C/30 min for all glycogen degradation. The total hydrolysis of β-glucan was obtained at 57 °C/2 h and the total conversion of trehalose was made at 37 °C/22 h.Analytical methods
Cell growth was followed by optical density measurements at 570 nm in a DR4000 UV spectrophotometer (HACH) and the values were converted to mg mL−1 of cell dry weight, using a factor previously determined.The glucose formed by the hydrolysis of all substrates was always quantified by the glucose oxidase method.11
Transmission electron microscopy
The cells were fixed by immersion in 2.5% glutaraldehyde and 0.1 M sodium cacodylate buffer (pH 7.2) for 1 hour. Samples were then post-fixed in 1% OsO4 in the same buffer for 40 minutes in dark, dehydrated in 10, 35, 50, 70, 90, and 100% acetone series, embedded in PolyBed 812 resin and polymerized for 48 h at 60 °C.8 The blocks were sectioned using a diamond knife (Diatome) in an ultramicrotome. Ultrathin sections of 60 nm were picked up on 300 mesh copper grids and stained with 2% uranyl acetate for 20 minutes and 1% lead citrate for 1 minute. Sections were visualized using a Zeiss 900 transmission electron microscope operating at 80 kV and the images were acquired using a CCD camera (Zeiss) with a resolution of 1024×1024 pixels.Results and discussion
Kinetics of amyloglucosidase action
Saccharide hydrolyses were monitored by analysing free glucose during the enzymatic reaction. Fig. 1 shows that all glycogen were hydrolyzed at 37 °C and 57 °C. Nevertheless, amyloglucosidase hydrolyzed 60% of glycogen at 65 °C while this value was even lesser (only 40%) at 75 °C because of the fast inactivation of the enzyme.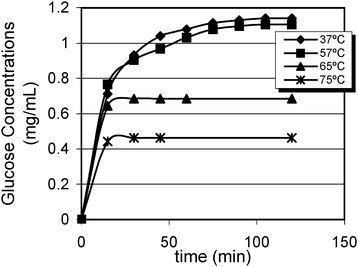 | ||
| Fig. 1 Kinetics of glycogen degradation at different temperatures. | ||
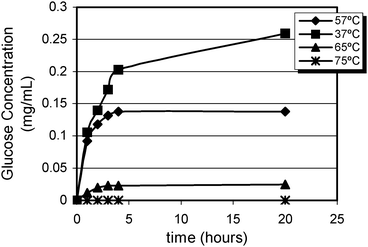
The results shown in Fig. 2 indicate that all trehalose were converted at 37 °C. However, at 57 °C trehalase hydrolyzed only 15% of total trehalose. At the highest temperatures, trehalase was inactivated and no free glucose was detected.
 | ||
| Fig. 2 Kinetics of trehalose hydrolysis at different temperatures. | ||
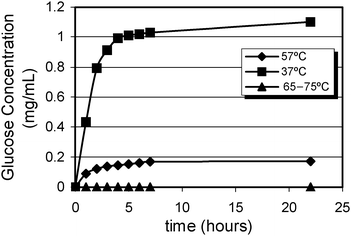
The values obtained for β-glucan hydrolysis are presented in Fig. 3. This polysaccharide was totally degraded at 37 °C, but 50% of hydrolysis was observed at 57 °C and at 65 °C it was reduced to only 9%. At 75 °C, glucanase was inactivated.
 | ||
| Fig. 3 Kinetics of β-glucan hydrolysis at different temperatures. | ||
The hydrolysis of a mixture of glycogen and glucan was also analyzed using two different procedures as described in the materials and methods section. The results are in agreement with those obtained for the solutions separately as demonstrated in Tables 1 and 2.
| Temperature/°C | T/h | % Hydrolysis |
|---|---|---|
| 65 | 1 | 60% Glycogen |
| 57 | 2 | 100% Glycogen |
| 50% β-Glucan | ||
| 37 | 20 | 100% Glycogen |
| 100% β-Glucan |
| Temperature/°C | T/h | % Hydrolysis |
|---|---|---|
| 57 | 20 | 100% Glycogen |
| 50% β-Glucan |
In the first procedure (Table 1) only 60% of glycogen was hydrolyzed in the first step (65 °C/1 h), but in the second step (57 °C/2 h) all glycogen and 50% of β-glucan were hydrolyzed. The total hydrolysis occurred in the last step (37 °C/20 h) of the procedure.
The other procedure (Table 2) had only one step (57 °C/20 h) where all glycogen and 50% of β-glucan were hydrolyzed. The results showed that it is possible to control β-glucan hydrolysis by controlling the time of enzyme action at 57 °C. Incubation for two hours at 57 °C is enough to degrade all glycogen and about half of β-glucan. This strategy was applied to degrade all glycogen and part of glucan in Saccharomyces cerevisiaecells to obtain partially hydrolyzed β-glucan.
Hydrolysis of saccharides in cell walls
On the basis of the results obtained in previous experiments, a procedure to allow the access of amyloglucosidase, glucanase and trehalase to their respective substrates was established within the cells. Initially, the cells were permeabilized using ethanol and washed for trehalose release. After that, two different treatments were used to increase cell porosity, forming structures called ghosts. The ghosts were incubated at 57 °C/2 h with the enzymatic preparation for total degradation of glycogen and partial degradation of β-glucan. The supernatant obtained in the permeabilization step was subjected to the enzymatic treatment for total hydrolysis of trehalose.It was verified that after these procedures, the cells were free of all glycogen and more than 90% of trehalose (Table 3). Moreover, the cells subjected to treatment with glass beads had yet approximately 60% of glucan (Table 3), and in the cells treated with alkali solution, the percentage of glucan was approximately 93%, indicating that only 7% was hydrolyzed (Table 3). The composition of these saccharides in the cells obtained in a control assay is also shown in Table 3.
| Glycogen | β-Glucan | Trehalose | |
|---|---|---|---|
| Whole cells | 0.130 | 0.120 | 0.150 |
| Cells treated with glass beads | 0.130 | 0.074 | 0.014 |
| Cells treated with alkaline solution | 0.130 | 0.112 | 0.014 |
The results showed that the treatment with glass beads is more efficient to increase cell porosity and allow the glucanase action.
Cell ultrastructure
Cell changes were studied throughout the different stages of the tested treatments by transmission electron microscopy (TEM). From the images of the whole cell it is possible to visualize the cell wall surrounding its membrane with a dense cytoplasm, and nucleus (Fig 4a).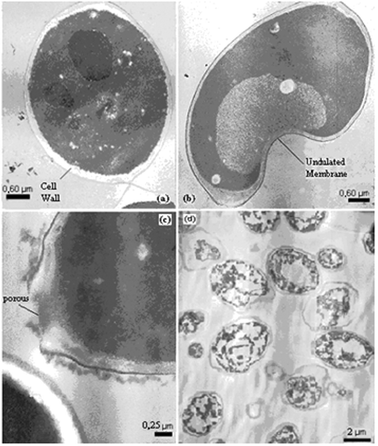 | ||
| Fig. 4 TEM images of (a) whole cell (b) cell after permeabilization (c) cell wall porous and (d) cell ghosts. | ||
Cell permeabilization was carried out through a plasmatic membrane. From the images obtained after the permeabilization it is possible to see an undulated membrane and some loss of cytoplasmic material indicated by the electron-lucent areas (Fig. 4b).
After cell permeabilization, micronutrients could be isolated from the cells, however many macromolecules and organelles remained. In order to remove such structures and to improve cell hydrolysis, a treatment with glass beads was performed. After this treatment, it is possible to visualize pores in the cell wall, as shown in Fig. 4c.
The step of enzymatic treatment reduced the cell wall width. Fig. 4d shows a cell practically without intracellular material and also a cell wall with reduced width, i.e. β-glucan partially hydrolyzed.
Conclusions
In this work it is proposed a simple method to increase S. cerevisiae cellular porosity using a unique commercial enzymatic preparation that is unpurified with other hydrolytic enzymes such as glucanases and trehalase. Moreover, the proposed method can increase β-glucan solubility from S. cerevisiaecells using a permeabilization technique followed by an adequate hydrolytic action of enzymes present in the commercial amyloglucosidase preparation obtained from Rhizopus sp. The microstructures obtained, named “porous ghosts”, have reduced content of intracellular material, glycogen and trehalose, and they contain an important natural β-glucan fiber with a cell wall reduced shape. These whole porous ghosts may be used as additives in food industry or as raw material for β-glucan soluble fibers production.This method is more practical compared with traditional methods because it is possible to obtain the desired product after three simple steps: permeabilization with ethanol, disruption with glass beads and enzymatic hydrolysis. The first two are pretty fast and spend little energy because there is no heating but only agitation. The last step was performed at 57 °C for 2 hours. Magnani et al.7 also proposed a methodology for extraction of β-D-glucan from Saccharomyces cerevisiae but their method has five hard-working steps. In the first step was performed the autolysis and hot water treatment. It is a long step that involves agitation in a water bath at 55 °C for 24 hours, heating to 121 °C in an autoclave for 4 hours. Then the sonication was performed followed by lipid extraction, which was conducted using isopropanol and petroleum ether. The use of these solvents in the extraction process prevents the product from being applied in food or drugs industry. This step takes a little over 2 hours. So, the proteolysis of the cell wall was performed at 55 °C for 5 hours followed by dialysis for 48 hours.
This method also represents the possibility of reusing the biomass sub-product from some industrial processes for ethanol production and baking. It is important to explain that this work was developed with S. cerevisiae because this microorganism is present in a large number of industrial bioprocesses. But it is possible to obtain β-glucan from other yeast species because this polysaccharide is present in the cell wall of most of the yeasts.
Notes and references
- J. A. Bohn and J. N. BeMiller, Carbohydr. Polym., 1995, 28, 3–14 CrossRef CAS.
- L. F. Fleuri and H. H. Sato, Quim. Nova, 2005, 28(5), 871–879 Search PubMed.
- C. Gancedo, Eur. J. Biochem., 1973, 61, 479–482 Search PubMed.
- R. Jiang and M. Carlson, Genes Dev., 1996, 10, 3105–3115 Search PubMed.
- F. M. Klis, P. Mol, K. Hellingwerf and S. Brul, FEMS Microbiol. Rev., 2002, 26, 239–256 CrossRef CAS.
- P. N. Lipke and R. Ovalle, J. Bacteriol., 1998, 180(15), 3735–3740 CAS.
- M. Magnani, C. M. Calliari, F. C. Macedo Jr, M. P. Mori, I. M. S. Cólus and R. J. H. Castro-Gomez, Carbohydr. Polym., 2009, 78, 658–665 Search PubMed.
- A. Moino, S. B. Alves, R. B. Lopes, P. M. Oliveira, J. Neves, R. M. Pereira and S. A. Vieira, Sci. Agric. (Piracicaba, Braz.), 2002, 59(2), 267–273 Search PubMed.
- Y. K. Park, M. Ikegaki, S. M. Alencar and C. L. Aguiar, Cienc. Tecnol. Aliment. (Campinas, Braz.), 2003, 23(3), 312–316 Search PubMed.
- A. C. Pelizon, R. Kaneno, A. M. V. C. Soares, D. A. Meira and A. Sartori, Physiol. Res. (Prague, Czech Repub.), 2005, 54, 557–564 Search PubMed.
- E. Raabo and T. C. Terkildsent, Scand. J. Clin. Lab. Invest., 1960, 12, 402–407 Search PubMed.
- M. H. M. Rocha-Leão, M. A. Z. Coelho and O. Q. F. Araújo, Braz. J. Chem. Eng., 2003, 20(3), 241–250 Search PubMed.
- G. D. Ross, V. Vetvicka, J. Yan, Y. Xia and J. Vetvickova, Immunopharmacology, 1999, 42, 61–74 CrossRef CAS.
- R. Serrano, J. M. Gancedo and C. Gancedo, Eur. J. Biochem., 1973, 34, 479–482 Search PubMed.
- M. P. Szostak, A. Hensel, F. O. Eko, R. Klein, T. Auer, H. Mader, A. Haslberger, S. Bunka, G. Wanner and W. Lubitz, J. Biotechnol., 1996, 44, 161–170 Search PubMed.
- M. Takeuchi, H. Miyamoto, Y. Sako, H. Komizu and A. Kusumi, Biophys. J., 1998, 74, 2171–2183 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |