A unique photo-activation mechanism by “in situ doping” for photo-assisted selective NO reduction with ammonia over TiO2 and photooxidation of alcohols over Nb2O5
Tetsuya
Shishido
*,
Kentaro
Teramura
and
Tsunehiro
Tanaka
Department of Molecular Engineering, Graduate School of Engineering, Kyoto University, 1 Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: shishido@moleng.kyoto-u.ac.jp; Fax: +81 75-383-2561; Tel: +81 75-383-2559
First published on 1st June 2011
Abstract
This paper reviews the recent development of the photocatalytic emission control reaction and photooxidation with molecular oxygen, specifically focusing on efforts based on revealing the reaction mechanism by the authors' group. TiO2 acts as an effective catalyst for the photo-assisted selective catalytic reduction of NO with NH3 in the presence of O2 (photo-SCR). Photooxidation of alcohols to carbonyl compounds proceeds selectively over Nb2O5 without organic solvents. Usually, both TiO2 and Nb2O5 work only in the ultraviolet (UV) region because of the limit of their bandgap energies. However, both photo-SCR over TiO2 and photooxidation of alcohols over Nb2O5 proceed even under visible light irradiation up to ca. 450 nm. This indicates that these two reactions take place by the different photo-activation mechanism from the classical electron transfer mechanism in semiconductor photocatalysis, that is, the formation of an excited electron in the conduction band and the positive hole in the valence band. A mechanistic study using UV-Vis, ESR, FT/IR, kinetic study, and DFT calculations revealed the reaction mechanisms of photo-SCR and photooxidation of alcohols, and that the surface complex consisting of the adsorbed molecule and catalyst plays an important role in the photo-activation step. The surface complex is converted to the photo-activated species even under visible light irradiation, because the direct electron transition from a donor level derived from the adsorbed molecule to the conduction band of a photocatalyst takes place and a photo-generated hole is trapped on the adsorbed molecule to form the photo-activated radical species. The effective wavelength is shifted to a longer wavelength by the formation of the donor level derived from the adsorbed molecule during a chemical reaction (called here “in situ doping”). This unique photo-activation mechanism by “in situ doping” gives us attractive ways for removing the limit of bandgap energy, and the utilization of visible light.
1. Introduction
As shown in Scheme 1, photocatalytic reactions on a semiconductor powder involve several steps. Photocatalysis is generally explained in terms of band theory (the classical electron transfer mechanism) accompanied by the interaction of reactants with the photo-generated electrons and holes, and is potentially available to make the catalytic reactions proceeding at low temperature. The band structure of the photocatalysts determines the utilizable light energy, oxidizability, and reducing ability. It has been considered that suppressing recombination between photo-generated electrons and holes in a photocatalyst is important to achieving the reaction, since the lifetime of the charge separation contributes to the photocatalytic activity. Therefore, a number of studies are related to the control of band structure and charge separation. On the other hand, little information about the adsorbed species and the intermediates in photocatalytic reactions is available. Photocatalytic reactions take place on the surface of the photocatalysts as well as the ordinary catalysts. The difference between the photocatalysts and the ordinary catalysts is just the driving force to activate the adsorbed reactants; the photocatalysts use the photo-energy and the ordinary catalysts use the thermal energy. Therefore, the kinetic interpretation and the knowledge of surface structure, surface property, and surface species during the photo-reaction are required to understand the photocatalysis; generally a catalytic reaction consists of several elementary steps and one or two of the elementary steps involves absorption of light in the case of a photocatalytic reaction. Hence, there is the same thermodynamic restriction in the photocatalysis with the ordinary catalysis. Evidently, it is necessary to consider the photo-activation mechanism in detail. The clarification of the reaction mechanism provides the beneficial information on the further improvement of the photocatalysis and a new insight of photocatalytic chemistry.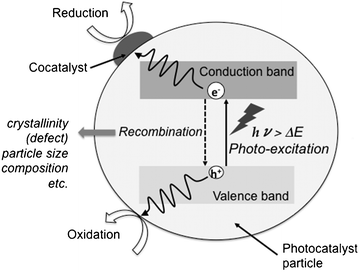 | ||
| Scheme 1 Model of reaction, charge separation, and recombination over photocatalyst. | ||
This article summarizes our recent work on the photo-activation mechanisms of NH3 over TiO2 in photo-SCR, and alcohols over Nb2O5 in the photooxidation with molecular oxygen. We show a detailed investigation of reaction mechanisms of photo-SCR over TiO2 and selective photooxidation of alcohols over Nb2O5 using in situ characterization, kinetic study and DFT calculations. A redshift of effective wavelength due to the direct electron transition from the donor level derived from the adsorbed molecule to the conduction band (“in situ doping”) is demonstrated.
2. Photo-assisted selective reduction of NO with NH3 over TiO2 based catalysts
2.1 Low-temperature NH3-SCR systems
The growth of the global economy involved the environmental problems such as solid, air and water pollutions. These pollutions cause serious damage to human and nature. In order to solve these problems, many efforts are undergoing for the development of the environmental technology.NOx is one of the environmental pollutants and causes acid rain and photochemical smog. Therefore, it is desirable to remove NOx (de-NOx) from the stationary emission source and the mobile emission source. In the stationary emission source such as a thermal power station, an industrial boiler and a waste incinerator, NOx is conventionally removed from the exhaust gas by the selective catalytic reduction system with NH3 as a reductant (NH3-SCR) in the presence of the excess O2 over V2O5–WO3 (or V2O5–MoO3)/TiO2 catalyst.1–4 This technology was invented by three Japanese corporations (Hitachi Ltd., Babcock-Hitachi K. K., Mitsubishi Petrochemical Corp.).5 The reaction stoichiometry in the typical NH3-SCR is shown as follows;
| 4NO + 4NH3 + O2 → 4N2 + 6H2O | (1) |
| Catalyst | Reaction gas composition | Reaction conditions | Activity | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|
| NO (ppm) | NH3 (ppm) | O2 (ppm) | H2O (%) | T/K | SV/h−1 | X NO (%) | S N2 (%) | ||
| T: reaction temperature; SV: space velocity; XNO: NO conversion; SN2: N2 selectivity. | |||||||||
| MnO2–Al2O3 | 550 | 550 | 2 | 0 | 435 | 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
98 | 92 | 11 |
| MnO2–carbonized silica-alumina | 800 | 800 | 3 | 0 | 413 | 12000 |
94 | 91 | 13 |
| MnO2–NaY | 1000 | 1000 | 10 | 7 | 443 | 48000 |
88 | 93 | |
| MnO2–TiO2 | 2000 | 2000 | 2 | 0 | 393 | 8000 | 100 | 100 | 17 |
| MnO2–TiO2 | 400 | 400 | 2 | 11 | 448 | 50000 |
98 | 97 | |
| Fe–Mn oxide | 1000 | 1000 | 2 | 0 | 393 | 15000 |
100 | 100 | 19 |
| Fe–Mn oxide | 1000 | 1000 | 2 | 2.5 | 413 | 15000 |
98 | 100 | |
| MnO2–CeO2 | 1000 | 1000 | 2 | 0 | 393 | 42000 |
99 | 100 | 16 |
| MnO2–CeO2 | 1000 | 1000 | 2 | 19 | 393 | 42000 |
95 | 100 | |
| V2O5-sulfated carbon | 500 | 600 | 3 | 0 | 453 | 34000 |
92 | 100 | 15 |
Subsequently, several Mn-containing catalysts, which show the high activity with the durability to H2O at a low-temperature, are reported.13–15,17,18 Among the Mn-containing catalysts reported, MnO2–CeO2 shows the highest activity (95% NO conversion and 100% N2 selectivity in the presence of 19% H2O).17
2.2 Photo-SCR with NH3 over TiO2 photocatalysts
Over the past decades, the most attentions of the photocatalysis have been gathered to the water photolysis and the removal of harmful compounds over photocatalysts responding to visible light. In contrast, although photocatalysts have the advantage that the re-heating of the catalyst bed is unnecessary because of their possibility of application at low temperatures, the reports of the NH3-SCR reaction over photocatalysts (photo-SCR) were limited. Cant and Cole reported that NO reduction by NH3 to form N2 proceeds over TiO2 under photo-irradiation.20 The reaction was carried out in a closed system and the activity was very low. In contrast, we found that TiO2 and metal oxide promoted TiO2 are effective for the photo-SCR with NH3 in the presence of O2 proceeding at room temperature.21–29Table 2 shows the catalytic activity and physicochemical property of photo-SCR with NH3 over various TiO2 photocatalysts (JRC-TIO-1-13 supplied from Catalysis Society of Japan). JRC-TIO-11, a mixture of rutile and anatase phases, exhibited the highest activity among all TiO2 tested. JRC-TIO-8 and JRC-TIO-3 were the most active catalysts consisting of anatase or rutile single phases. There is poor correlation between the activity and crystal phase, crystallite size, and specific surface area. This indicates that these properties are not the important factors to determine the activity of photo-SCR.
| Catalysts | Sa/m2 g−1 | Phase | D c/Å | NO conv. (%) | N2 sel. (%) |
|---|---|---|---|---|---|
| A: anatase, R: rutile. Reaction conditions; NO: 1000 ppm, NH3: 1000 ppm, O2: 2%, Ar balance, GHSV = 32000 h−1. |
|||||
| JRC-TIO-1 | 71.1 | A | 187 | 36 | 100 |
| JRC-TIO-2 | 15.6 | A | 535 | 14.5 | 100 |
| JRC-TIO-3 | 45.6 | R | 219 | 53 | 100 |
| JRC-TIO-4 | 47.8 | R 29.4% | 382 | 35.5 | 100 |
| A 70.6% | 259 | ||||
| JRC-TIO-5 | 3–4 | R 92.4% | 2200 | 31 | 100 |
| A 7.6% | 1000 | ||||
| JRC-TIO-6 | 58.0 | R | 240 | 20 | 100 |
| JRC-TIO-7 | 108 | A | 197 | 35 | 100 |
| JRC-TIO-8 | 93.2 | A | 155 | 51.7 | 98.6 |
| JRC-TIO-9 | 95.2 | A | 197 | 31 | 100 |
| JRC-TIO-10 | 100 | A | 169 | 35.5 | 100 |
| JRC-TIO-11 | 76.6 | R 8.7% | 200 | 63 | 100 |
| A 91.3% | 153 | ||||
| JRC-TIO-12 | 98.7 | A | 159 | 41 | 100 |
| JRC-TIO-13 | 71.1 | A | 237 | 33 | 100 |
2.3 Mechanism of photo-SCR with NH3 over TiO2 photocatalysts
Fig. 1 shows the EPR spectra of TiO2. After evacuation at 673 K, the signals are derived from the Ti3+ species (Fig. 1(a)).30–33 There is little change in the EPR signal by the exposure of NH3 to TiO2 in the dark. On the other hand, the EPR signal changed drastically after photo-irradiation. New signals assignable to the NH2 radical34–38 were detected together with signals assigned to Ti3+. These new signals were quite stable even after more than 1 hour at 123 K without photo-irradiation. However, these signals immediately vanished by the exposure to NO in the dark whereas the intensity of signals due to Ti3+ species increased. This suggests that (1) the photo-generated electron is trapped on Ti4+ to form Ti3+ and the positive hole is captured by adsorbed NH3 species and is converted to an active NH2 radical, and that (2) NO in the gas phase attacks the NH2 radical on TiO2 rapidly. As both the NH2 radical and NO are doublet state species, it follows that the NH2 radical reacts with NO easily without irradiation. Moreover, the formation of a NH2NO intermediate was confirmed by FTIR spectroscopy after admittance of NO to TiO2 adsorbing NH3 under photo-irradiation (vide infra). As described above, the signals due to Ti3+ species increased in intensity after the introduction of NO. It seems that the electron transfer took place from the N atom of adsorbed NH3 to the Ti atom of TiO2 bulk. In other words, the photo-generated electron was trapped on the Ti atom and the photo-generated hole was captured by the NH2− species derived from adsorbed NH3. As a result, the NH2− species is converted to the active NH2 radical. On the other hand, the electron may move into the inside of TiO2 bulk as a stable free electron. Before the exposure to NO, recombination took place between a part of Ti3+ species and the NH2 radical. On the other hand, after the exposure to NO, the electron could not recombine because of losing an opponent (NH2 radical). The electron was localized and stabilized in the inside of TiO2, and the signals assigned to the Ti3+ species increased in intensity. | ||
| Fig. 1 EPR spectra of TiO2 (a) after pretreatment, (b) after introduction of NH3 in the dark, (c) under photo irradiation and (d) after introduction of NO in the dark. | ||
Fig. 2 shows the time course of the N2 evolution rate of photo-SCR. NO conversion and N2 selectivity attained 100% and 96%, respectively, in the conventional fixed bed flow system (GHSV = 8000 h−1). The N2 evolution rate gradually increased at the initial stage and reached a steady rate at 1.5 h. However, when the reaction gas (a mixture of NO/NH3/O2) was passed in the dark for 0.5 h, and then photo-irradiation was started, the N2 evolution rate immediately jumped to the level of the steady rate.23,25 This clearly indicates that the induction period shown in Fig. 2 is the time for saturation of the adsorption equilibrium of the reactant molecule. When NH3 were passed for 1.5 h in the dark, then the gas was switched to a mixture of NO/O2 and the photo-irradiation was started, N2 was evolved. The N2 evolution rate gradually decreased and the total amount of evolved N2 was consistent with that of equilibrium adsorption of NH3 on TiO2. In contrast, when a mixture of NO/O2 was firstly passed and then switched to NH3, neither N2 nor N2O was formed. These results suggest that NH3 species adsorbed on the Lewis acid site is excited by photo-irradiation and reacts with NO in the gas phase to produce N2. Furthermore, this is supported by the fact that only 15N14N was evolved in the photo-SCR of 15NO with 14NH3 in the presence of O2.22
 | ||
| Fig. 2 Time course of N2 (circle) and N2O (square) in the photo-SCR with NH3 over TiO2 JRC-TIO-11, reaction conditions; GHSV = 8000 h−1, NO: 1000 ppm, NH3: 1000 ppm, O2: 2%, Ar balance. | ||
The adsorbed species and intermediates of photo-SCR were identified by in situ FT/IR spectra (Fig. 3). After NH3 adsorbed on TiO2, the bands (1136, 1215, and 1599 cm−1) due to adsorbed NH3 species on the Lewis acid site of TiO2 appeared.39–41 The bands at 1599 and 1215 cm−1 retained their intensity after evacuation (Fig. 3(b)) and exposure to NO in the dark (Fig. 3(c)). The bands due to adsorbed NH3 decreased gradually in intensity with irradiation time. On the other hand, the band at 1624 cm−1, which is assignable to the deformation vibration of H2O,42 grew. Furthermore, new bands between 1400 and 1600 cm−1 were observed and then disappeared. These new bands are assigned to the nitrosamide species (NH2NO) by comparing the FT/IR spectrum of TiO2 exposed to 14NO and NH3 to that exposed to 15NO and NH3.24
 | ||
| Fig. 3 FT-IR spectra of adsorbed species on TiO2 in the photo-SCR with NH3. (a) After introduction of NH3, (b) after evacuation, (c) after introduction of NO in the dark, (d) under photo irradiation for 10 min, (e) for 30 min, (f) for 60 min, and (g) for 120 min. | ||
These results indicate that the intermediate of photo-SCR is the nitrosamide species (NH2NO) and the nitrosamide species is decomposed to N2 and H2O. Moreover, it was confirmed that the Ti3+ species of TiO2 reduced by H2 was re-oxidized to the Ti4+ species by exposure to O2 easily even at room temperature using UV-Vis spectroscopy.27 On the basis of these results, we proposed the Eley–Rideal type mechanism as follows (Scheme 2);24–29 (1) the NH3 adsorbs on the Lewis acid site of TiO2, (2) the adsorbed NH3 species is excited by photo-irradiation, (3) the excited species (NH2 radical) reacts with NO in the gas phase to form the nitrosamide species (NH2NO), (4) the nitrosamide species is decomposed to N2 and H2O, and (5) the Ti3+ site is re-oxidized by molecular oxygen.
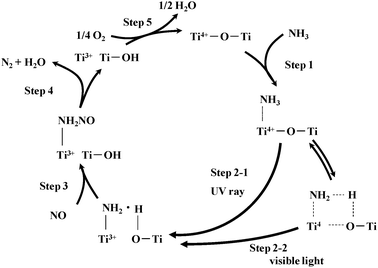 | ||
| Scheme 2 Reaction mechanism of photo-SCR with NH3 over TiO2. | ||
According to the dependencies of the partial pressure of NO, NH3, and O2, the reaction rate (r) of photo-SCR is expressed as [eqn (2)].
 | (2) |
2.4 Enhancement of activity by improving the lifetime of photo-activated species
An efficient charge separation promotes the chemical reactions competing with a recombination of the photo-generated electrons and holes. Einaga et al. reported that benzene as a model VOC (volatile organic compound) can be abated by total oxidation over TiO2 photocatalysts effectively.43–45 However, the specific activity of TiO2 is absolutely low. They reported that 120 ppm of benzene cannot be removed over TiO2, but 80 ppm of benzene is efficiently decomposed to CO2 and CO in the presence of H2O vapor. This would be caused by the insufficient lifetime of the charge-separated state over TiO2. It is widely thought that the photo-generated electrons and holes are consumed by recombination much more rapidly than by the photocatalytic reaction and the recombination is the main reason of too short lifetime of the charge-separated state and resulting in low activity of TiO2. Indeed, the half-life of the charge-separated state of TiO2 was estimated to be below 100 ps.46,47 Up to now, the limit of the extended lifetime of the charge-separated state is only several tens of ns despite the careful effort. Therefore, in the case of photo-SCR over TiO2, it seems that the half-life of NH2 radicals, which are formed by capturing photo-generated positive holes, is below 100 ps and that the rate-determining step of photo-SCR is the reaction of the NH2 radical with NO to form the nitrosamide species (NH2NO). However, as described above, the kinetic study indicated that the decomposition of the nitrosamide species (Step 4) is the rate-determining step of photo-SCR. This strongly suggests that the strongly adsorbed NH3 on the Lewis acid site of TiO2 lengthened the lifetime of the charge-separated state by trapping holes and consequently the recombination of the photo-generated electrons and holes is inhibited. Fig. 4 shows the decay curve of the NH2 radical signal recorded by EPR after irradiation stopped. All decay curves can be approximated to a hyperbolic curve, indicating that the NH2 radical is quenched by the secondary reaction between the NH2 radical and an electron. The half-life of the NH2 radical at reaction temperature of photo-SCR (323 K) is calculated to be 1.4 min by using the Arrhenius equation (Fig. 5). Since this half-life of the NH2 radical at 323 K is much longer than that of the charge-separated state of TiO2 (<100 ps), the concentration of the NH2 radical on the surface of TiO2 increased and the activity of photo-SCR is enhanced. To the best of our knowledge, the intermediate and active species having a longer lifetime such as the NH2 radical has not been reported. | ||
| Fig. 4 Decay curve of NH2 radical and the approximated curve (lines) at (a) 113 K, (b) 123 K, (c) 133 K and (d) 143 K. | ||
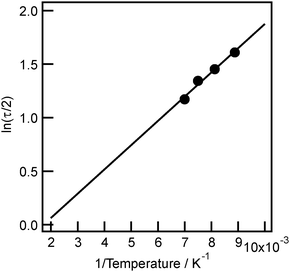 | ||
| Fig. 5 Arrhenius plots of the half-lives of NH2 radical at each temperature (dot) and the approximated line (line). | ||
2.5 Mechanism of formation of the NH2 radical over TiO2
Fig. 6 shows the apparent quantum efficiency of the photo-SCR over TiO2 as a function of the incident light (action spectrum) and a UV-Vis spectrum of TiO2. The band gap of this TiO2 is estimated to be 3.28 eV (photo-excitation energy is 385 nm). The action spectrum is in good agreement with the UV-Vis spectrum of TiO2 in the region of wavelength <385 nm. Although TiO2 is unable to absorb light at wavelengths >385 nm, photo-SCR proceeded under irradiation up to ca. 450 nm. The feature of this action spectrum is similar to the UV-Vis spectrum of N-doped TiO2.48 In order to reveal whether a new energy level derived from the adsorbed molecule is located between the HOMO and LUMO levels or not, density functional theory (DFT) calculations were employed. DFT calculations revealed that the N 2p electron donor level is located between O 2p and Ti 3d when NH2 species are formed on a TiO2 surface by the dissociative adsorption of NH3.27 | ||
| Fig. 6 Action spectrum of photo-SCR (dot) and UV-Vis spectrum of JRC-TIO-11 (line); reaction conditions of action spectrum: NH3: 1000 ppm, NO: 1000 ppm, O2: 2%, flow rate: 100 ml min−1. | ||
On the basis of these results, we conclude that the photo-activation of NH3 adsorbed on TiO2 to an NH2 radical occurs through two paths as shown in Fig. 7. One is the electron transition from the valence band consisting of O 2p orbitals to the conduction band consisting of Ti 3d orbitals of TiO2 under UV irradiation. The other is the direct electron transfer from N 2p of adsorbed NH3 to Ti 3d. This N 2p electron donor level formed between O 2p and Ti 3d enables the photo-SCR to proceed even under visible light irradiation (400–450 nm). It can be thought that the expansion of the effective wavelength of TiO2 by adsorption of NH3, called here, “in situ doping”, is one of the factors for high activity of TiO2 in the photo-SCR.
 | ||
| Fig. 7 Formation mechanism of NH2 radical over TiO2. | ||
Many researchers reported that the “pre-doped” or “pre-modified” photocatalysts with metals or ions such as N-doped TiO2 adsorb the visible light.48–51 However, there has been no report that the effective wavelength of photo-reaction is shifted to a longer wavelength by the formation of the donor level derived from the adsorbed molecule on the catalyst during a chemical reaction (“in situ doping”). Moreover, “in situ doping” was found not only in photo-SCR with NH3 over TiO2 but also in the photooxidation of alcohols over Nb2O5. The details of the photooxidation of alcohols over Nb2O5 are described in the next section.
3. Photooxidation of alcohols with molecular oxygen over Nb2O5 catalysts
3.1 Oxidation of alcohol with molecular oxygen
Catalytic alcohol oxidation to carbonyl compounds is one of the most important chemical transformations used in the industrial chemistry and in organic syntheses.52–54 Non-catalytic methods with stoichiometric, toxic, corrosive and expensive oxidants such as ClO−, dichromate, permanganate, and peroxy acids under stringent conditions of high pressure and/or temperature have been widely used for alcohol oxidations.52–55 In addition, these reactions are often carried out with high concentration of bases and environmentally unfriendly organic solvents. Therefore, much attention has been paid to the development of heterogeneous catalytic systems that use clean and atom efficient oxidants like molecular oxygen or H2O2 without organic solvents.55–66Recently, the aerobic oxidation of alcohols was successfully carried out by using heterogeneous catalysts such as tetrapropylammonium perruthenate (TPAP)/MCM-41,59Ru/CeO2,60Ru–hydrotalcite,61Ru/hydroxyapatite (Ru–HAP),62[RuCl2(p-cymene)]2/activated carbon,63Ru/Al2O3,64Pd–hydrotalcite which requires the addition of pyridine,65 and Pd or Pt on activated carbon.65,66 These systems require the use of organic solvents. Wu et al. reported on solvent-free aerobic oxidation of alcohols by Pd/Al2O3.67 However, the use of the noble metal, Pd, is an essential requirement. Despite the advantage of using heterogeneous catalysts without organic solvents or additives for oxidation of alcohols, few reports have appeared on the use of highly active solvent-free heterogeneous catalysts with only molecular oxygen as an oxidant.
In this respect, photoreactions are promising processes and the development of photocatalysts is a subject that is now receiving noticeable attention. TiO2 has been identified as one example of a practical and useful photocatalysts,68–71 and widely used in degradation of organic pollutants in air and water. However, in the most part of these reports, TiO2 is used in vapor phase oxidations at high temperature,67oxidation of only lower alcohols,69,70oxidation using solvents such as benzene71 and a low selectivity to partial oxidized products due to excess photo-activation of target products which leads to deep oxidation. Zhao et al.72,73 reported that the photooxidation of alcohols on TiO2 could be dramatically accelerated without any loss of selectivity by adsorption of Brønsted acid and this effect by Brønsted acid results from the decomposition of the relatively stable side-on peroxide promoted by the protons, which effectively clean the catalytic Ti–OH2 sites. However, this system requires the use of benzotrifluoride as a solvent.
Recently, we found that the photooxidation of alcohols to carbonyl compounds proceeded selectively at low temperature over Nb2O5 without organic solvents or any additives (Table 3).74–76 Various metal oxides (SiO2, MgO, Al2O3, ZrO2, V2O5, Ta2O5, MoO3, and WO3) showed no activity and the activity of ZnO was very low. TiO2 showed higher activity than Nb2O5, however, the Nb2O5 catalyst showed higher selectivity than TiO2 at the same conversion level.74Nb2O5 is suitable for selective oxidation. The photooxidation did not take place in the dark. Autooxidation proceeded when 1-phenylethanol, cyclohexanol and benzylalcohol were irradiated without the catalyst. This was due to the formation of radical species by the photo-decomposition of carbonyl compounds (Norrish Type I reaction) which were present as impurities in the alcohols (entries 1 to 3). The Nb2O5 catalyst improved the conversions and/or selectivities to carbonyl compounds greatly. The less reactive primary alcohol, 1-pentanol was also photooxidized over the Nb2O5 catalyst. The Nb2O5 catalyst was reusable and showed the same conversion and selectivity without any pretreatment as the catalyst as prepared.
| Entry | Substrate | Product | T/h | Conv. (%) | Sel. (%) |
|---|---|---|---|---|---|
| a Reaction conditions were as follows: alcohol (10 mL), Nb2O5 (100 mg), 323 K, under 0.1 MPa of O2, O2 flow rate (2 cm3 min−1): conversion and selectivity were determined by gas chromatography with an internal standard. b Figures in parentheses are the results of photochemical reaction without catalysts. | |||||
| 1 |

|

|
240 (72) | 99 (14) | 96 (69) |
| 2 |

|

|
168 (96) | 76 (46) | 64 (36) |
| 3 |

|
|
72 (72) | 67 (79) | 90 (43) |
| 4 |

|

|
168 (119) | 23 (2) | 85 (82) |
| 5 |

|

|
192 (121) | 18 (5) | 83 (81) |
| 6 |

|

|
84 (24) | 14 (0) | 92 (−) |
3.2 Mechanism of photooxidation of alcohol over Nb2O5
The adsorbed species and intermediates of photooxidation were identified by in situ FT/IR spectra of adsorbed cyclohexanol on Nb2O5. Fig. 8 shows the FT/IR spectra of adsorbed cyclohexanol on Nb2O5. The bands at 1467 and 1452 cm−1 were assigned to δs(CH2) and the bands at 1363 and 1347 cm−1 were assigned to ω(CH2), respectively. The new bands at 1091 and 1126 cm−1 appeared after the adsorption of cyclohexanol on Nb2O5. Therefore, these bands are assigned to the stretching mode of a C–O bond in the alcoholate species on the Nb2O5, because the formation of the alcoholate species by the adsorption of alcohol is usually accompanied by a shift of the stretching mode of a C–O bond to a higher wavenumber.77–79 The change in FT/IR spectra by UV irradiation (<390 nm) was shown in Fig. 9. The intensity of the band assigned to ν(C–O) (around 1090 cm−1) decreased as the irradiation time increased, whereas the bands assigned to ν(C = O) (1676 cm−1) and the symmetric-stretching of the carboxylic acid anion (1554 cm−1) gradually grew. This result indicates that the alcoholate species on Nb2O5 was excited by photons and oxidized to carbonyl compounds. Interestingly, the carbonyl compounds were formed even under visible light irradiation (>390 nm). | ||
| Fig. 8 FT-IR spectra of adsorbed cyclohexanol on Nb2O5. (a) Cyclohexanol was exposed to Nb2O5 for 1 h (physisorption + chemisorption), (b) evacuated for 2 h (chemisorption), (c) difference spectrum ((a)–(b): physisorption). Nb2O5 was evacuated at 773 K for 1 h and oxidized at 773 K with 10.7 kPa of O2 and then evacuated at 773 K for 1 h before FT-IR measurements. | ||
 | ||
| Fig. 9 FT-IR spectra of adsorbed species on Nb2O5 in the photo-reaction of adsorbed cyclohexanol with O2. (a) Cyclohexanol was exposed to Nb2O5 for 1 h and evacuated for 2 h, (b) under UV irradiation for 1, (c) 5, (d) 7, (e) 10, (f) 15 and (g) 30 min. Nb2O5 was evacuated at 773 K for 1 h and oxidized at 773 K with 10.7 kPa of O2 and then evacuated at 773 K for 1 h before FT-IR measurements. | ||
Fig. 10 shows the EPR spectra of Nb2O5. A broad EPR signal around g = 1.9 was observed at 123 K (Fig. 9(c)), when 1-pentanol was adsorbed on Nb2O5 under UV-irradiation. This broad signal at g = 1.9 was assignable to Nb4+80,81 and immediately disappeared by the exposure to O2 in the dark, indicating that Nb4+ was oxidized to Nb5+ rapidly even at 123 K. On the other hand, when 1-pentanol was adsorbed on Nb2O5 under UV-irradiation at 77 K, the EPR signal (g = 2.006, AH1 = 2.0 mT, AH2 = 4.4 mT) assigned to alkenyl radical species was observed (Fig. 11). These new signals were stable at 77 K without photo-irradiation, but disappeared at room temperature. The signal was restored by UV-irradiation at 77 K. Moreover, the signal did not change in the presence of O2 even under UV-irradiation (Fig. 11(f)).
 | ||
| Fig. 10 ESR spectra of Nb2O5 recorded at 123 K. (a) After pretreatment, (b) in the dark in the presence of 1-pentanol, (c) under irradiation for 5 h in the presence of an excess of 1-pentanol, (d) after introduction of O2. Nb2O5 was evacuated at 773 K for 1 h and oxidized at 773 K with 10.7 kPa of O2 and then evacuated at 773 K for 1 h before ESR measurements. | ||
 | ||
| Fig. 11 ESR spectra of Nb2O5 recorded at 77 K. (a) After pretreatment, (b) under irradiation, (c) in the dark in the presence of 1-pentanol, (d) under irradiation in the presence of 1-pentanol, (e) in the dark after the sample was heated up to RT and then cooled to 77 K, (f) under re-irradiation. Nb2O5 was evacuated at 773 K for 1 h and oxidized at 773 K with 10.7 kPa of O2 and then evacuated at 773 K for 1 h before ESR measurements, (g) alkenyl radical. | ||
This indicates that the alkenyl radical species does not react with O2. Therefore, it suggests that (1) the photo-formed electron is trapped on Nb5+ to form Nb4+ and the positive hole is captured by alcoholate species and is converted to an active alkenyl radical, (2) the active alkenyl radical is dehydrogenated to a carbonyl compound (the reduction of Nb5+ to Nb4+ takes place simultaneously), and (3) O2 in the gas phase re-oxidizes Nb4+ to Nb5+.
On the basis of these results, we proposed the reaction mechanism as shown in Scheme 3;75–77 (1) alcohol is adsorbed on Nb2O5 as alcoholate species, (2) alcoholate adsorbed on Nb2O5 is activated by transferring an electron to the conduction band reducing Nb5+ to Nb4+ and leaving a hole on alcoholate, (3) the formed alkenyl radical is converted to a carbonyl compound, (4) the product desorbs, and (5) the reduced Nb4+ sites are re-oxidized by the reaction with O2.
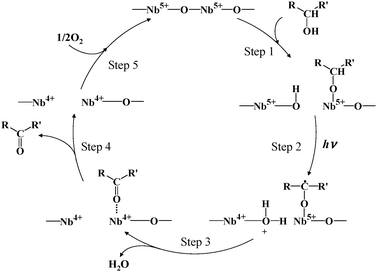 | ||
| Scheme 3 Reaction mechanism of alcohol photooxidation with molecular oxygen over Nb2O5. | ||
In this mechanism, oxygen anion radical species (O2− and O3−), which are formed by irradiation over TiO280–87 and often responsible for total oxidation, do not contribute to the photooxidation over Nb2O5. For instance, when Nb2O5 was irradiated in the presence of O2, no EPR signal due to oxygen anion radical species was observed. This presumably explains why the photooxidation of alcohol to a carbonyl compound proceeds selectively over the Nb2O5 catalyst.
We carried out the photooxidation of 1-pentanol under the various concentrations of 1-pentanol, O2 and the different light intensity to determine each reaction order. On the basis of these results, the reaction rate (r) of photooxidation of alcohol is expressed as [eqn (3)].
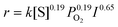 | (3) |
3.3 Mechanism of formation of the alkenyl radical over Nb2O5
Fig. 12 shows the apparent quantum efficiency of photooxidation of 1-pentanol as a function of the wavelength of the incident light (action spectrum) and a UV-Vis spectrum of Nb2O5. Although the Nb2O5 catalyst is not able to absorb visible light (>390 nm), the photooxidation of 1-pentanol took place under irradiation up to ca. 480 nm. This result is consistent with the change in the FT/IR spectra under visible light irradiation and a red shift of the effective wavelength of photo-reaction is similar to that of photo-SCR over TiO2. In the case of photo-SCR over TiO2, we found that the adsorbed NH3 was photo-activated by the direct electron transfer from the N 2p electron donor level formed between O 2p and Ti 3d of TiO2 to the conduction band.27 | ||
| Fig. 12 Action spectrum of photooxidation of 1-pentanol (dot) and UV-Vis spectrum of Nb2O5 (line). Reaction conditions of the action spectrum were as follows: 1-pentanol (10 ml), Nb2O5 (100 mg), 323 K, under 0.1 MPa of O2, O2 flow rate (2 cm3 min−1). | ||
In order to investigate the formation of a new energy level derived from the adsorbed molecule, DFT calculations were employed and showed that donor levels were generated between the HOMO and LUMO levels of Nb2O5 by adsorbed alcohol on Nb2O5 and that the electron transitions from the O 2p donor level derived from the adsorbed alcoholate species to the conduction band of Nb2O5 (Nb 4d orbitals) had lower energy than those from O 2p of Nb2O5 (the conduction band) to Nb 4d.75 On the basis of these results, we concluded that the photooxidation of alcohol over Nb2O5 takes place through the direct electron transfer from the O 2p orbital of adsorbed alcoholate species to the conduction band consisting of Nb 4d orbitals as shown in Fig. 13 (“in situ doping”). As a result of “in situ doping”, the photooxidation of alcohol proceeded even under visible light irradiation.
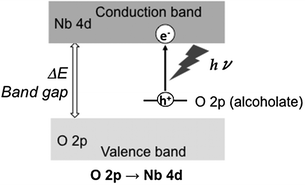 | ||
| Fig. 13 Formation mechanism of alkenyl radical over Nb2O5. | ||
4. Conclusions and outlook
By means of UV-Vis, ESR, FT/IR with the aid of kinetic study and DFT calculations, the detailed reaction mechanisms of photo-assisted selective reduction of NO with NH3 (photo-SCR) over TiO2 and photooxidation of alcohol with O2 over Nb2O5 were revealed and a unique photo-activation mechanism by “in situ doping” in both photo-SCR and photooxidation of alcohol was demonstrated.TiO2 acts as an effective catalyst for photo-SCR even at room temperature. In this photo-SCR system, the re-heating of the catalyst bed is unnecessary because of their possibility of application at low temperatures. Thus, the photo-SCR system can miniaturize the reactor. Moreover, in this photo-SCR system, the TiO2 photocatalyst can activate NH3 effectively even in the presence of excess O2. Indeed, we found that TiO2 acts as an effective catalyst for photo-assisted selective catalytic oxidation of NH3 (photo-SCO: 4NH3 + 3O2 → 2N2 + 6H2O).85–87 Although further improving the activity may be needed, it seems that this photo-SCO system can be used for removing unreacted NH3 in the SCR process and for removing NH3 from a small and isolated source such as daily firm.
Photooxidation of alcohols to carbonyl compounds proceeds selectively over Nb2O5 without organic solvents. Nb2O5 shows higher selectivity to a partial oxidation product than that of a commonly used TiO2 photocatalyst, and efficient conversion under a solvent-free condition.
In the case of photo-SCR over TiO2, a new electron donor (N 2p) was located between O 2p and Ti 3d by the adsorption of NH3 on TiO2. The direct electron transition from N 2p to Ti 3d took place to form NH2 radical species by visible light irradiation. As a result, the photo-SCR proceeded even under visible light irradiation. The high activity of TiO2 was caused by the expansion of the effective wavelength of TiO2 by adsorption of NH3 and the long lifetime of the NH2 radical. In the case of photooxidation of alcohol over Nb2O5, as well as the adsorbed NH3 on TiO2, the new electron donor level was generated between O 2p and Ti 3d by the adsorption of alcohol on Nb2O5. The direct electron transition from O 2p derived from alcohol to Ti 3d took place to form alkenyl radical species by photo-irradiation. Then, this alkenyl radical was immediately dehydrogenated to a carbonyl compound. As shown in the present review, the unique photo-activation mechanism by “in situ doping” gives us attractive ways for removing the limit of bandgap energy, and the utilization of visible light.
Acknowledgements
A part of this work was partially supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan.Notes and references
- S. Cho, Chem. Eng. Prog., 1994, 90, 39–45 Search PubMed.
- P. Forzatti and L. Lietti, Heterog. Chem. Rev., 1996, 3, 33–51 CrossRef CAS.
- F. Nakajima, Catalysis, 1990, 32, 236–239 Search PubMed.
- S. Wood, Chem. Eng. Prog., 1994, 90, 32–38 Search PubMed.
- A. Kato, S. Matsuda, F. Nakajima, M. Imanari and Y. Watanabe, J. Phys. Chem., 1981, 85, 1710–1713 CrossRef CAS.
- K. Otto, M. Shelef and J. T. Kummer, J. Phys. Chem., 1970, 74, 2690–2696 CrossRef CAS.
- F. Janssen, F. Van den Kerkhof, H. Bosch and J. Ross, J. Phys. Chem., 1987, 91, 5931–5934 CrossRef CAS.
- F. Janssen, F. Van den Kerkhof, H. Bosch and J. Ross, J. Phys. Chem., 1987, 91, 6633–6637 CrossRef CAS.
- B. Duffy, H. Curryhyde and N. Cant, J. Phys. Chem., 1994, 98, 7153–7161 CrossRef.
- U. Ozkan, Y. Cai and M. W. Kumthekar, J. Catal., 1994, 149, 375–389 CrossRef CAS.
- U. Ozkan, Y. Cai and M. W. Kumthekar, J. Phys. Chem., 1995, 99, 2363–2371 CrossRef CAS.
- V. Parvulescu, P. Grange and B. Delmon, Catal. Today, 1998, 46, 233–316 CrossRef CAS.
- R. Long and R. Yang, J. Catal., 2002, 207, 158–165 CrossRef CAS.
- L. Singoredjo, R. Korver, F. Kapteijn and J. Moulijn, Appl. Catal., B, 1992, 1, 297–316 CrossRef CAS.
- M. Richter, A. Trunschke, U. Bentrup, K. Brzezinka, E. Schreier, M. Schneider, M. Pohl and R. Fricke, J. Catal., 2002, 206, 98–113 CrossRef CAS.
- E. Garcia-Bordeje, J. Pinilla, M. Lazaro, R. Moliner and J. Fierro, J. Catal., 2005, 233, 166–175 CrossRef CAS.
- G. Qi and R. Yang, Chem. Commun., 2003, 848–849 RSC.
- D. Pena, B. Uphade and P. Smirniotis, J. Catal., 2004, 221, 421–431 CrossRef CAS.
- L. Gang, B. Anderson, J. van Grondelle and R. van Santen, Catal. Today, 2000, 61, 179–185 CrossRef CAS.
- N. Cant and J. R. Cole, J. Catal., 1992, 134, 317–330 CrossRef CAS.
- T. Tanaka, K. Teramura and T. Funabiki, Phys. Chem. Chem. Phys., 2000, 2, 2681–2682 RSC.
- T. Tanaka, K. Teramura, T. Yamamoto, S. Takenaka, S. Yoshida and T. Funabiki, J. Photochem. Photobiol., A, 2002, 148, 277–281 CrossRef CAS.
- T. Tanaka, K. Teramura, K. Arakaki and T. Funabiki, Chem. Commun., 2002, 2742–2743 RSC.
- K. Teramura, T. Tanaka and T. Funabiki, Langmuir, 2003, 19, 1209–1214 CrossRef CAS.
- K. Teramura, T. Tanaka, S. Yamazoe, K. Arakaki and T. Funabiki, Appl. Catal., B, 2004, 53, 29–36 CrossRef CAS.
- K. Teramura, T. Tanaka and T. Funabiki, Chem. Lett., 2003, 32, 1184–1185 CrossRef CAS.
- S. Yamazoe, K. Teramura, Y. Hitomi, T. Shishido and T. Tanaka, J. Phys. Chem. C, 2007, 111, 14189–14197 CrossRef CAS.
- S. Yamazoe, T. Okumura, K. Teramura and T. Tanaka, Catal. Today, 2006, 111, 266–270 CrossRef CAS.
- S. Yamazoe, Y. Masutani, Y. Hitomi, T. Shishido and T. Tanaka, Appl. Catal., B, 2008, 83, 123–130 CrossRef CAS.
- P. Meriaudreau, M. Che and C. K. Jorgensen, Chem. Phys. Lett., 1970, 5, 226–228 CrossRef.
- C. Hauser and P. Naccache, Chem. Phys. Lett., 1971, 8, 45–48 CrossRef CAS.
- R. F. Howe and M. Gratzel, J. Phys. Chem., 1985, 89, 4495–4499 CrossRef CAS.
- D. Hurum, A. G. Agrios, K. A. Gray, T. Rajh and M. C. Thurnauer, J. Phys. Chem. B, 2003, 107, 4545–4549 CrossRef CAS.
- S. N. Forer, E. L. Cochran, V. A. Bowers and C. K. Jen, Phys. Rev. Lett., 1958, 1, 91–94 CrossRef CAS.
- E. F. Vansant and J. H. Lunsford, J. Phys. Chem., 1972, 76, 2716–2718 CrossRef CAS.
- O. I. Brotikovskii, G. M. Zhidomirov, V. B. Kazanskii, A. I. Mashchenko and B. N. Shelmimov, Kinet. Katal., 1971, 12, 616–619.
- S. Nagai, Bull. Chem. Soc. Jpn., 1973, 46, 1144–1148.
- N. Shimamoto, K. Hatano, T. Katsu and Y. Fujita, Bull. Chem. Soc. Jpn., 1975, 48, 18–21 CrossRef CAS.
- G. Ramis, G. Busca, F. Bregani and P. Forzatti, Appl. Catal., 1990, 64, 259–278 Search PubMed.
- M. C. Kung and H. H. Kung, Catal. Rev., 1985, 27, 425–460.
- C. C. Chuang, J. S. Shiu and L. J. Lin, Phys. Chem. Chem. Phys., 2000, 2, 2629–2633 RSC.
- E. Ito, Y. J. Mergler, B. E. Nieuwenhuys, H. P. A. Calis, H. vanBekkum and C. M. vandenBleck, J. Chem. Soc., Faraday Trans., 1996, 92, 1799–1806 RSC.
- H. Einaga, S. Futamura and T. Ibusuki, J. Jpn. Pet. Inst., 1999, 42, 363–364 Search PubMed.
- H. Einaga, S. Futamura and T. Ibusuki, Phys. Chem. Chem. Phys., 1999, 1, 4903–4908 RSC.
- H. Einaga, S. Futamura and T. Ibusuki, Appl. Catal., B, 2002, 38, 215–225 CrossRef CAS.
- S. Ikeda, N. Sugiyama, B. Pal, G. Macri, L. Palmisano, H. Noguchi, K. Uosaki and B. Ohtani, Phys. Chem. Chem. Phys., 2001, 3, 267–273 RSC.
- S. Ikeda, N. Sugiyama, S. Murakai, H. Kominami, Y. Kera, H. Noguchi, K. Uosaki, K. Torimoto and B. Ohtani, Phys. Chem. Chem. Phys., 2003, 5, 778–783 RSC.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS.
- S. Sato, Chem. Phys. Lett., 1986, 123, 126–128 CrossRef CAS.
- H. Luo, T. Takata, Y. Lee, J. Zhao, K. Domen and Y. Yan, Chem. Mater., 2004, 16, 846–849 CrossRef CAS.
- M. Miyauchi, A. Nakajima, T. Watanabe and K. Hashimoto, Chem. Mater., 2002, 14, 4714–4720 CrossRef CAS.
- R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, New York, 1981 Search PubMed.
- M. Hudlicky, Oxidations in Organic Chemistry, ACS Monograph Series, American Chemical Society, Washington, DC, 1990 Search PubMed.
- C. L. Hill, Advances in Oxygenated Process, ed. A. L. Baumstark, JAI, London, 1998, vol. 1, p.1 Search PubMed.
- R. C. Larock, Comprehensive Organic Transformations, VCH, New York, 1989 Search PubMed.
- R. A. Sheldon, I. W. C. E. Arends and A. Dijksman, Catal. Today, 2000, 57, 157 CrossRef CAS.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS.
- N. Srinivas, V. R. Rani, M. R. Kishan, S. J. Kulkarni and K. V. Raghavan, J. Mol. Catal. A: Chem., 2001, 172, 187–191 CrossRef CAS.
- A. Bleloch, B. F. G. Johnson, S. V. Ley, A. Price, D. S. Shephard and A. W. Thomas, Chem. Commun., 1999, 1907–1908 RSC.
- F. Vocanson, Y. P. Guo, J. L. Namy and H. B. Kagan, Synth. Commun., 1998, 398, 1907–1908.
- T. Matsushita, K. Ebitani and K. Kaneda, Chem. Commun., 1999, 265–266 RSC.
- (a) K. Yamaguchi, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2000, 122, 7144–7145 CrossRef CAS; (b) K. Mori, S. Kanai, T. Hara, T. Mizugaki, K. Ebatani, K. Jitsukawa and K. Kaneda, Chem. Mater., 2007, 19, 1249–1256 CrossRef CAS.
- E. Choi, C. Lee, Y. Na and S. Chang, Org. Lett., 2002, 4, 2369–2371 CrossRef CAS.
- K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2002, 41, 4538–4540 CrossRef CAS.
- N. Kakiuchi, Y. Maeda, T. Nishimura and S. Uemura, J. Org. Chem., 2001, 66, 6620–6623 CrossRef CAS.
- T. Mallat and A. Baiker, Catal. Today, 1994, 19, 247–283 CrossRef CAS.
- H. Wu, Q. Zhang and Y. Wang, Adv. Synth. Catal., 2005, 347, 1356–1357 CrossRef CAS.
- U. R. Pillai and E. S. Demessie, J. Catal., 2002, 211, 434–444 CrossRef CAS.
- J. Chen, D. F. Ollis, W. H. Rulkens and H. Bruning, Water Res., 1999, 33, 661–668 CrossRef CAS.
- (a) D. S. Muggli, J. T. Mccue and J. L. Falconer, J. Catal., 1998, 173, 470–483 CrossRef CAS; (b) J. L. Falconer and K. A. M. Bair, J. Catal., 1998, 179, 171–178 CrossRef CAS.
- F. H. Hussein, G. Pattenden, R. Rudham and J. J. Russell, Tetrahedron Lett., 1984, 25, 3363–3364 CrossRef CAS.
- M. Zhang, Q. Wang, C. Chen, L. Zang, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2009, 48, 6081–6084 CrossRef CAS.
- Q. Wang, M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2010, 49, 7976–7979 CrossRef CAS.
- T. Ohuchi, T. Miyatake, Y. Hitomi and T. Tanaka, Catal. Today, 2007, 120, 233–239 CrossRef CAS.
- T. Shishido, T. Miyatake, K. Teramura, Y. Hitomi and T. Tanaka, J. Phys. Chem. C, 2009, 113, 18713–18718 CrossRef CAS.
- S. Furukawa, Y. Ohno, T. Shishido, K. Teramrau and T. Tanaka, Catalysts and Catalysis, 2011, 53, 132–134 Search PubMed.
- L. H. Little, A. V. Kiselev and V. I. Lygin, Infrared Spectra of Adsorbed Species, Academic Press Inc., London, 1966 Search PubMed.
- G. Socrates, Infrared Characteristic Group Frequencies: Table and Charts, Wiley, New York, 1994 Search PubMed.
- V. Z. Fridman, A. A. Davydov and K. Titievsky, J. Catal., 2004, 222, 545–557 CrossRef CAS.
- M. Sugantha and U. V. Varadaraju, J. Solid State Chem., 1994, 111, 33–40 CrossRef CAS.
- C. Veríssimo, F. M. S. Garriodo, O. L. Alves, P. Calle, A. Martínez-Juárez, J. E. Iglesias and J. M. Rojo, Solid State Ionics, 1997, 100, 127–134 CrossRef CAS.
- P. Meriaudeau and J. C. Vedrine, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 472–480 RSC.
- A. R. Gonzalez-Elipe, G. Munuera and J. Soria, J. Chem. Soc., Faraday Trans. 1, 1979, 75, 748–761 RSC.
- Y. Nakaoka and Y. Nosaka, J. Photochem. Photobiol., A, 1997, 110, 299–305 CrossRef CAS.
- S. Yamazoe, T. Okumura and T. Tanaka, Catal. Today, 2007, 120, 220–225 CrossRef CAS.
- S. Yamazoe, T. Okumura, Y. Hitomi, T. Shishido and T. Tanaka, J. Phys. Chem. C, 2007, 111, 11077–11085 CrossRef CAS.
- S. Yamazoe, Y. Hitomi, T. Shishido and T. Tanaka, Appl. Catal., B, 2008, 82, 67–76 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |