The importance of developing metal complexes with pronounced catalase-like activity
Atif
Mahammed
and
Zeev
Gross
*
Schulich Faculty of Chemistry, Technion-Israel Institute of Technology, Haifa 32000, Israel. E-mail: chr10zg@tx.technion.ac.il; Fax: +972-4-8295703; Tel: +972-4-8293954
First published on 23rd May 2011
Abstract
This critical survey reveals that in contrast with the large advances in the development of synthetic catalysts for decomposition of superoxide anion radical and peroxynitrite, most metal complexes reported until recently perform very poor when it comes to disproportionation of hydrogen peroxide, the reaction catalyzed by enzymes of the catalase family.
Introduction
The superoxide anion radical (O2˙−, Scheme 1), continuously generated by partial reduction of molecular oxygen during normal cellular metabolism,1 is the primary source for many reactive oxygen species (ROS) of which the most troublesome are hydrogen peroxide (H2O2) and peroxynitrous acid (HOONO, PN).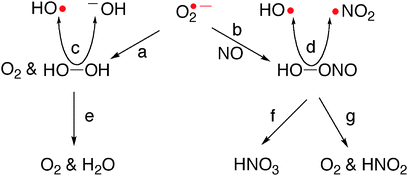 | ||
| Scheme 1 Desired and hazardous pathways that follow the generation of superoxide anion radical: (a) from two equivalents of O2˙−, either spontaneously with k = 5 × 105 M−1s−1 or SOD-catalyzed with kcat = 2 × 109 M−1s−1;2 (b) diffusion controlled rate;5 (c) catalyzed by low-valent metal ions;4 (d) about 30% of PN undergoes hemolytic cleavage, with k = 0.77 s−1 at 37 °C;5 (e) catalyzed by catalases, utilizing two equivalents of H2O2;2 (f) about 70% of PN undergoes spontaneous isomerization and iron(III) complexes of porphyrins and corroles are very fast acting catalysts of it;6,27b (g) from two equivalents of PN, via a catalytic pathway unique to manganese(III) corroles.27b,d | ||
Dismutation of O2˙− leads to H2O2 (path a, Scheme 1) either spontaneously and quite slow or via the very fast enzymatic process catalyzed by superoxide dismutases (SOD).2 The consequence is that wherever O2˙− is generated there is also formation of H2O2, which is stable at biological pH and easily crosses lipid membranes.3 Reduced transition metal ions such as iron(II) or copper(I) readily react with it to generate ˙OH (the very reactive and the most damaging ROS) in the metal catalyzed Haber–Weiss reaction (path c, Scheme 1).4 Accordingly, full detoxification of O2˙− may not be achieved by SOD alone, but only when it is coupled to catalases (CAT), the enzymes that catalyze the disproportionation of H2O2 to molecular oxygen and water (path e, Scheme 1).2d Any excess O2˙− that is not efficiently neutralized may combine with nitrogen oxide (NO, path b, Scheme 1) in a diffusion-controlled rate to form peroxynitrite (−OONO, PN−),5 which is either protonated to PN (pKa = 6.8) or reacts with CO2.6 These adducts decompose spontaneously, releasing HO˙ or CO3−˙, respectively, as well as NO2 (path d, Scheme 1).7 Importantly, there are no enzymes that catalytically decompose PN− and/or its adducts and all natural antioxidants do not react faster with them than the biomolecules (DNA, lipoproteins, enzymes, cellular components, etc.) that must be rescued from oxidation and nitration for preserving their natural functions.8 In fact, SOD are heavily and irreversibly damaged by PN,9 which further increases the amount of available O2˙− and the secondary ROS/RNS formed from it.
The most obvious resolution to the above described problems would be to supply native SOD and CAT as therapeutic agents for treating the numerous diseases that are characterized by an imbalance between the minute amounts of ROS/RNS required for normal life and the level of molecules or enzymes that may neutralize excessive amounts thereof.10 Attenuation of ROS-induced injury responses has, however, had mixed success.11 The principal limitations of using these proteins are their large size, the consequences of which are low cell permeability, a short circulating half-life, antigenicity and high-manufacturing costs.12 To overcome these limitations, a constantly increasing number of low molecular-weight catalytic antioxidants have been developed.13 The ideal catalytic antioxidant must be stable, non-toxic, and its size and charge may be exploited to target crucial cellular sites. It should also possess at least three distinct antioxidant properties, i.e., very fast and efficient scavenging of O2˙−, H2O2, and PN. While numerous catalytic antioxidants with SOD-like activity (SOD mimics) have been developed during the last decades, relatively little efforts have focused on development and optimization of CAT mimics for possible therapeutic use although H2O2 is produced in vivo during oxidative stress, is cytotoxic, and has a longer lifetime in biological medium than O2˙−. This concept article focuses on synthetic metal-based antioxidants that mimic CAT activity and the importance of developing multifunctional catalysts that may efficiently decompose all ROS/RNS to biologically benign species. Manganese(III) salens that display CAT activity (although typically with <10 catalytic turnovers) have been reported,14 but the following section is limited to the much more intensively investigated metalloporphyrins.
Catalase activity of metalloporphyrins
The porphyrin-based CAT mimics may be classified into three categories: (a) non-water soluble tetraarylporphyrins; (b) super-structured porphyrins; and (c) tetraarylporphyrins with water-solubilizing groups. As may be appreciated from Table 1, the catalytic activity of all these derivatives is quite poor. The simplest catalysts are the manganese(III) and iron(III) complexes of hydrophobic tetraarylporphyrins (1-Mn–4-Fe, Chart 1), which were examined regarding their ability to disproportionate H2O2 in organic solvents like acetonitrile, or in mixtures of water and organic solvents. 1-Mn decomposes H2O2 in a very low rate (TOF = 0.013 s−1),15 while the sterically hindered porphyrins work somewhat better, with TOF = 0.9 s−1 for 2-Mn and 3-Mn.16 The iron(III) porphyrins without sterically hindered meso-aryl substituents are virtually inactive H2O2 disproportionation catalysts and undergo extensive bleaching. Even 4-Fe, a highly potent catalyst for oxygen atom transfer to olefins, has been shown to form the deactivated diiron(III)-μ-oxo dimer and to undergo bleaching with production of only negligible amounts of O2.17Dimerization may be avoided by using tetraarylporphyrins with meso-mesityl groups and 2-Fe is a better catalyst indeed, but still with a very low TOF (0.003 s−1).18 Bleaching by H2O2 is a major problem for all iron(III) porphyrins and it is important to emphasize that all reported activities are hence based on initial rates rather than on true catalytic rates.| Catalyst | Initial TOF/s−1 | References |
|---|---|---|
| 1-Mn to 4-Fe (Chart 1) | ≤0.9 | 15–18 |
| Hangman porphyrin (A in Chart 2) | ≤1.7 | 18 |
| Superstructured porphyrins (B in Chart 2) | ≤0.3 | 16 |
| Rigidly linked porphyrin dimers (C in Chart 2) | 0 (≤5.4 in the presence of 0.15 M 1-methylimidazole) | 15 |
| Water soluble porphyrins | ≤0.03 for Mn porphyrins | 22 |
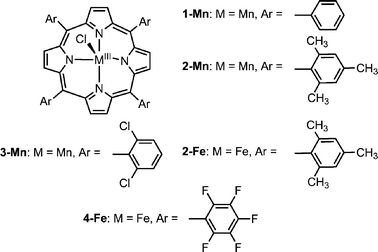 | ||
| Chart 1 | ||
A variety of superstructured porphyrins (some examples are shown in Chart 2) were synthesized for improving the catalytic activity of the corresponding metal complexes. Nocera and coworkers focused on porphyrins with substituted xanthene at one meso-C position that contains pendant groups with various proton-donating abilities (A in Chart 2) and checked the CAT activity of the corresponding chloroiron(III) complexes.18 The so-called Hangman porphyrin complexes bearing the most acidic pendants exhibit an initial rate constant (TOF = 1.7 s−1) that is almost 3 orders of magnitude greater than that of the baseline compound 2-Fe (TOF = 0.003 s−1). However, all of these Hangman complexes still undergo deactivation during catalysis. Meunier et al. prepared porphyrins with oxygen and nitrogen ligands covalently attached to the metal ion of the metalloporphyrin (B in Chart 2) in order to mimic in a more precise way the catalytic site of CAT. These ligands are part of a chain attached to two phenyl groups of the tetraarylporphyrin viaether or amide linkages, while the other side of the metalloporphyrin plane was protected by a hydrocarbon chain.16 They found the CAT-like activity to depend on the nature of the coordinated proximal ligand: Mn(–imidazole-prox) > Mn(–alk–O–prox) ≈ Mn(–Ph–O–prox) > Fe(–Ph–O–prox) ≈ Fe(–alk–O–prox). The manganese(III) porphyrins are generally more active than the iron(III) complexes, including the Fe(–Ph–O–prox) complex, which may be considered as a true model for CAT since it has a tyrosine oxygen atom attached to Fe(III). The TOF of manganese(III) derivatives are in the range of 0.3–0.4 s−1 and that of the iron(III) complexes only 0.02–0.07 s−1. The Naruta group prepared rigidly linked dinuclear manganese(III) porphyrin dimers (C in Chart 2) and checked their CAT-like activity bearing in mind that polynuclear manganese complexes play key roles in Mn-based CAT.15 For example, one subunit of Mn-CAT from Thermus thermophilus contains two Mn(III) ions separated by 3.6 Å in its enzymatic active center.19 All the rigidly linked manganese(III) porphyrin dimers showed almost no CAT activity, which however increased significantly in the presence of 1-methylimidazole. A dual role for coordinated imidazole was proposed in this system: as an accelerator of O–O homolysis and for stabilization of an intermediate Mn(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O complex. The biphenylene-linked porphyrin had absolutely no CAT activity even in the presence of 1-methylimidazole, even though the predicted metal–metal distance (3.6 Å) is similar to that of Mn-CAT. This was explained by formation of a binuclear μ-oxo dimer.15
O complex. The biphenylene-linked porphyrin had absolutely no CAT activity even in the presence of 1-methylimidazole, even though the predicted metal–metal distance (3.6 Å) is similar to that of Mn-CAT. This was explained by formation of a binuclear μ-oxo dimer.15
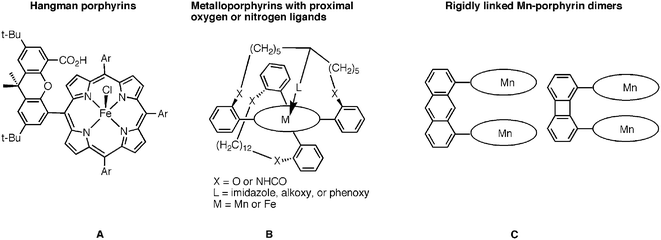 | ||
| Chart 2 | ||
All the porphyrins described so far are not water-soluble and their true relevance as CAT mimics is hence limited to fundamental science. On the other hand, many iron(III) complexes of tetraarylporphyrins with charged substituents are quite unstable with respect to the loss of iron at low pH, have a tendency to aggregate extensively, and they undergo dimerization at neutral and basic pH to binuclear μ-oxo-dimers. In addition, all reported water soluble iron(III) porphyrins undergo fast bleaching by excess of hydrogen peroxide in the absence of an oxidizable substrate (for example 5-Fe–7-Fe, Chart 3).20 This is even true for derivatives with the sterically hindered meso-aryls (for example 8-Fe and 9-Fe, Chart 3).21 The water-soluble manganese porphyrins are more stable, but decompose H2O2 very slowly. This statement holds for all the manganese complexes depicted in Chart 3 (10-Mn–15-Mn). The initialTOF of all of them is similar—about 0.03 s−1—and some of them were reported to undergo bleaching.22 Based on the above, it is not too surprising that there are very few literature reports regarding the usage of CAT mimics in either cellular or animal disease models, in sharp contrast with those that focused on SOD mimics and PN decomposition catalysts. One example is the work reported by Castello et al., who prepared a series of 23 glyoxylate manganese porphyrins and examined the potency of these lipophilic metalloporphyrins to inhibit, in vitro, rat brain mitochondrial H2O2 production.23 Only one particular derivative displayed reasonably good activity in that assay, with an IC50 of 17 nM and 32.4% relative to native CAT. It is important to emphasize at this stage that there are conceptual limitations in correlating data obtained in purely chemical systems with biological activity. Key variables include local intracellular concentrations of ROS/RNS and the contribution of metal reducing biomolecules.
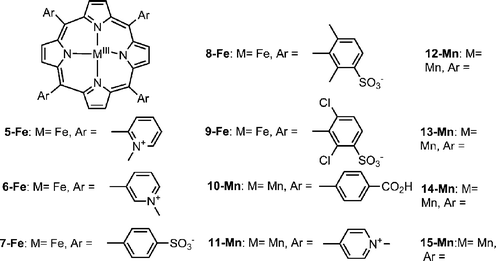 | ||
| Chart 3 | ||
Metallocorroles as catalytic antioxidants
Corroles share many properties with porphyrins, but they have also recently been shown to have unique characteristics that are advantageous in many fields, including medicine-relevant applications.24 One important discovery is that lipophilic corroles may easily be converted into amphiphilic derivatives of novel polarity (such as in 1C, Chart 4) and that the corresponding metal complexes spontaneously form tightly bound non-covalent conjugates with quite a variety of (natural and semi-synthetic) proteins.25 The intensively fluorescent gallium(III) complex 1-Ga is nowadays routinely used for imaging of cells, organs, and even whole animals; and it reduced tumor implanted into mice when conjugated with a breast cancer targeting protein.26 The manganese(III) and iron(III) complexes of the same corrole (1C-Mn and 1C-Fe, respectively) display SOD and CAT activity and are very potent catalysts for the catalytic decomposition of PN.27 Regarding the latter reaction, the catalytic rate of the iron(III) complex 1C-Fe was found to be exceptionally large, with indications that it acts as an isomerization catalyst. The manganese(III) corroles were identified as the first (and only so far) catalysts for disproportionation of PN to HNO2 and O2via a mechanism that does not involve any nitrating species.27b,d This advantage was demonstrated in a cellular model of diabetes: attenuation of intracellular nitration and subsequent death of insulin producing beta cells by managanese(III) corroles is unmatched by any other decomposition catalyst.28 Earlier investigations uncovered 1C-Fe as a very efficient antioxidant regarding the reaction between low-density lipoproteins (LDL) and PN; and oral administration of 1C-Fe to mice engineered to develop atherosclerosis revealed that it attenuated the development of atherosclerotic lesions more efficiently than any other compound reported to date.29 The latest development is that 1C-Fe also protects HDL (high-density lipoproteins, the “good cholesterol”) from becoming dysfunctional.30 The ability of corroles to decompose ROS/RNS was also found to be relevant in cellular models of degenerative disorder of the central and peripheral nervous system, where oxidative and nitrative stresses are involved.31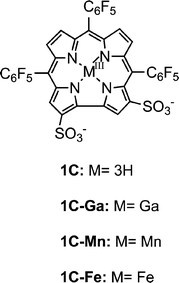 | ||
| Chart 4 | ||
The first clue for potent CAT-like activity of metallocorroles came from an investigation that focused on a biomimetic system for asymmetric sulfoxidation of sulfides, utilizing albumin-associated 1C-Fe and 1C-Mn as catalysts and hydrogen peroxide as the oxygen atom source.32 These investigations revealed that molecular oxygen was formed in a competitive reaction due to the disproportionation of H2O2 by the catalysts. This phenomenon was quantified (in the absence of sulfides and 0.027 mol% catalyst) by collecting 160 μmol O2 from 3.2 mmol H2O2, but a more focused investigation was published only most recently.27c The main findings were that the 1C-Fe-catalyzed decomposition of H2O2 produced O2 much too fast to be measured with the aid of a Clark electrode, as done for many other catalysts. In fact, applying the alternative method of monitoring the disappearance of the absorbance of H2O2 at 240 nm revealed that experiments needed to be done by the stopped flow technique since the reactions were complete within manual mixing time. A large range of H2O2 concentrations (0.9 mM–28.8 mM) was examined and its decay was first order at all concentrations of 1C-Fe (10–40 μM) at pH 7.4 and 37 °C. The first-order decay that was obtained in all cases and plots of thus obtained pseudo first order rate constants versus the catalyst concentrations allowed for the elucidation of a kcat value of 4300 M−1s−1. Identical values were obtained for various initial concentrations of H2O2 and/or 1C-Fe, consistent with non-saturation of a catalyst and a first order dependence on H2O2 concentration. A rate constant of 6400 M−1s−1 for decomposition of H2O2 by 1C-Fe was obtained at optimal concentration of buffer (200 mM phosphate, pH 7.4, 37 °C) and imidazole (3.2 mM) as an axial ligand. Calculations based on initial rates for decomposition of H2O2 under these conditions revealed a turnover frequency of >120 s−1, orders of magnitude larger than for metalloporphyrins. In addition, up to 103catalytic turnovers were obtained without significant catalyst bleaching. The latest demonstration about the superiority of corroles regarding CAT-activity comes from the Nocera group, who compared iron(IV) corroles with analogous iron(III) porphyrins.33 Simple (non-water-soluble) triarylcorrole complexes reacted 1–2 orders of magnitude faster than the corresponding tetraarylporphyrins, but the Hangman corroles were less efficient than Hangman porphyrins. This may however been attributed to the fact that the authors have performed their examinations with iron(IV) rather than with iron(III) corroles as pre-catalysts.
Conclusions
Table 2, which summarizes the highly significant differences in catalytic rate constants by which the iron/manganese complexes of corroles (1C-Fe and 1C-Mn) and porphyrins (11-Mn and 7-Fe) decompose O2˙−, H2O2, and PN, may serve for appreciating the need for developing more active catalysts for decomposition of hydrogen peroxide. The data clearly shows that the SOD activity of 1C-Fe is not outstanding, especially when keeping in mind that the rate for superoxide decomposition by advanced metalloporphyrins was increased to almost that of native SOD due to extensive research activity during the last decades.34 On the other hand, the PN decomposition rate of 1C-Fe matches that of even the most active metalloporphyrins; and the CAT-like activity (in terms of rate constant, catalytic turnovers, and stability) of the former exceeds that of the latter tremendously.| Catalyst | SOD activity kcat/M−1s−1 | Decomposition of PN kcat/M−1s−1 | CAT activity kcat/M−1s−1 | References |
|---|---|---|---|---|
| Data obtaineda by the cytochrome c assay;b by pulse radiolysis kinetics;c by stopped flow kinetics;d at pH 13;e at pH 9.5;f in the presence of ascorbate as co-reductant;g at initial rates and Clark electrode. | ||||
| 1C-Fe | 1.7 × 106a | 3.1 × 106c | 6400c | 27 |
| 3 × 106b | 46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600c,d 600c,d |
|||
| 1C-Mn | 4.8 × 105a | 8.6 × 104c | 5.6c,e | 27 |
| <1 × 105b | ||||
| 11-Mn | 3.8 × 106a | Not catalytic 3.3 × 104f | 20g | 27a, 31b |
| 5.6 × 106b | ||||
| 7-Fe | ∼6.0 × 105a | 8.6 × 105c | Rapid catalyst decomposition | 20 |
| SOD | 2 × 109 | 2 | ||
| CAT | 4 × 107 | 2 |
The biological/medicinal relevance of these findings is illustrated by the following examples, which convey one common message: SOD activity only is simply not enough for obtaining medicine-relevant effects by catalyticantioxidants. In fact, complexes that are able to decompose only superoxide may induce a toxic effect because they convert this rather non-reactive species to the more hazardous hydrogen peroxide. Kasugai et al. have actually used this feature for cancer therapy by using an iron(III) porphyrin for transforming basal concentrations of superoxide to hydrogen peroxide and thus damaging the cancer cells.35 Levin and coworkers found no relationship between the (pure chemical) SOD activity of many metal complexes and their effect on a quite advanced model system for optic neuropathy, the protection of retinal ganglion cells from oxidative stress damage.36 We hypothesized that this result may reflect that the applied synthetic SOD mimics perform only half of the full mission, i.e., do not efficiently decompose the produced H2O2. Intriguingly, experiments performed on the same model and by the same research group, but with corroles instead of porphyrins, revealed that 1C-Fe displayed quite outstanding positive effects.31b Since its SOD activity is lower than that of the metalloporphyrins, this result strongly points towards the ability to decompose any H2O2 that is formed either spontaneously or by the catalyzed process. The last example comes from investigations that focused on treatment of cell lines with H2O2, with and without the presence of corrole- and porphyrin-based catalysts. Cell survival by the former was much more significant.28
In conclusion, we emphasize that a medicine-relevant catalytic antioxidant should have the ability to decompose O2˙−, PN, and H2O2 to biologically benign products very fast and without being irreversibly modified by these reactive species. The primary source of reactive oxygen and nitrogen species is O2˙−, for which full neutralization requires the simultaneous action of SOD and CAT. Quite a variety of metal-based catalytic antioxidants that perform very well as synthetic SOD mimics, as well as decomposition catalysts for PN, fail almost completely in terms of their CAT activity. While this account has not touched on the mechanism-based reasons for this phenomenon, it has highlighted the relatively new metallocorroles. Recently reported results show that they display CAT activity unmatched by any other synthetic catalyst, accompanied by excellent performance for PN decomposition and moderate SOD activity. The relevance of these features, elucidated from purely chemical systems, to medicine relevant applications is emphasized by in vitro and in vivo models of several diseases. We trust that continuous efforts on this family of compounds will eventually lead to new pharmaceutical entities.
Acknowledgements
The work on corrole metal complexes was supported by the Israel Science Foundation and the Herbert Irving Cancer and Atherosclerosis Research Fund.References
- M. Valko, D. Leibfritz, J. Moncol, M. T. T. Cronin, M. Mazur and J. Telser, Int. J. Biochem. Cell Biol., 2007, 39, 44 CrossRef CAS.
- (a) S. Goldstein, I. Fridovich and G. Czapski, Free Radical Biol. Med., 2006, 41, 937 CrossRef CAS; (b) L. M. Ellerby, D. E. Cabelli, J. A. Graden and J. S. Valentine, J. Am. Chem. Soc., 1996, 118, 6556 CrossRef CAS; (c) G. Bratosz, in Handbook of Environmental Chemistry, ed. T. Grune, Springer, Berlin, 2005, vol. 2, part O, pp. 109–149 Search PubMed; (d) ; For other pathways used by native glutathione peroxidases and the thioredoxin-assisted peroxidases to detoxify H2O2, see: B. J. Day, Biochem. Pharmacol., 2009, 77, 285 Search PubMed.
- S. G. Rhee, T.-S. Chang, W. Jeong and D. Kang, Mol. Cells, 2010, 29, 539 CrossRef CAS.
- (a) J. P. Kehrer, Toxicology, 2000, 149, 43 CrossRef CAS; (b) Myeloperoxidase also amplifies the oxidative potential of H2O2 by generating cytotoxic oxidants and diffusible radical species: E. A. Jaimes, C. Sweeny and L. Raij, Hypertension (Dallas), 2001, 38, 877 Search PubMed.
- G. Ferrer-Sueta and R. Radi, ACS Chem. Biol., 2009, 4, 161 CrossRef CAS.
- (a) R. Radi, A. Denicola, B. Alvarez, G. Ferrer-Sueta and H. Rubbo, in Nitric Oxide, Biology and Pathobiology, ed. L. J. Ignarro, Academic Press, San Diego, 2000, pp. 57–82 Search PubMed; (b) C. Szabo, H. Ischiropoulos and R. Radi, Nat. Rev. Drug Discovery, 2007, 6, 662 CrossRef CAS.
- P. Pacher, J. S. Beckman and L. Liaudet, Physiol. Rev., 2007, 87, 315 CrossRef CAS.
- (a) R. Bryk, P. Griffin and C. Nthan, Nature, 2000, 407, 211 CrossRef CAS; (b) C. Szabo, H. Ischiropoulos and R. Radi, Nat. Rev. Drug Discovery, 2007, 6, 662 CrossRef CAS; (c) J. Su and J. T. Groves, Inorg. Chem., 2010, 49, 6317 CrossRef CAS.
- (a) H. Ischiropoulos, L. Zhu, J. Chen, M. Tsai, J. C. Martin, C. D. Smith and J. S. Beckman, Arch. Biochem. Biophys., 1992, 298, 431 CrossRef CAS; (b) N. B. Surmeli, N. K. Litterman, A.-F. Miller and J. T. Groves, J. Am. Chem. Soc., 2010, 132, 17174 CrossRef CAS.
- J. M. McCord and M. A. Edeas, Biomed. Pharmacother., 2005, 59, 139 CrossRef CAS.
- S. G. Simonson, K. E. Welty-Wolf, H.-C. T. Huang, D. E. Taylor, S. P. Kantrow, M. S. Carraway, J. D. Crapo and C. A. Piantados, J. Appl. Physiol., 1997, 83, 550 CAS.
- S. Cuzzocrea, D. P. Riley, A. P. Caputi and D. Salvemini, Pharmacol. Rev., 2001, 53, 135 CAS.
- B. J. Day, Drug Discovery Today, 2004, 9, 557 CrossRef CAS.
- S. R. Doctrow, K. Huffman, C. B. Marcus, G. Tocco, E. Malfroy, C. A. Adinolfi, H. Kruk, K. Baker, N. Lazarowych, J. Mascarenhas and B. Malfroy, J. Med. Chem., 2002, 45, 4549 CrossRef CAS.
- Y. Naruta and K. Maruyama, J. Am. Chem. Soc., 1991, 113, 3595 CrossRef CAS.
- A. Robert, B. Loock, M. Momenteau and B. Meunier, Inorg. Chem., 1991, 30, 706 CrossRef CAS.
- I. D. Cunningham, T. N. Danks, K. T. A. O'Connell and O. W. Scott, J. Chem. Soc., Perkin Trans. 2, 1999, 2133 RSC.
- (a) J. Rosenthal, L. L. Chng, S. D. Fried and D. G. Nocera, Chem. Commun., 2007, 2642 RSC; (b) L. L. Chng, C. J. Chang and D. G. Nocera, Org. Lett., 2003, 5, 2421 CrossRef CAS.
- A. J. Wu, J. E. Penner-Hahn and V. L. Pecoraro, Chem. Rev., 2004, 104, 903 CrossRef CAS.
- (a) M. F. Zipplies, W. A. Lee and T. C. Bruice, J. Am. Chem. Soc., 1986, 108, 4433 CrossRef CAS; (b) V. Lepentsiotis, R. von Eldik, F. F. Prinsloo and J. J. Pienaar, J. Chem. Soc., Dalton Trans., 1999, 2759 RSC; (c) M. P. Jensen and D. P. Riley, Inorg. Chem., 2002, 41, 4788 CrossRef CAS; (d) R. F. Pasternack and W. R. Skowronek Jr., J. Inorg. Biochem., 1979, 11, 261 CrossRef CAS.
- K. Murata, R. Panicucci, E. Gopinath and T. C. Bruice, J. Am. Chem. Soc., 1990, 112, 6072 CrossRef CAS.
- (a) B. J. Day, I. Fridovich and J. D. Crapo, Arch. Biochem. Biophys., 1997, 347, 256 CrossRef CAS; (b) R. Kachadourian, C. A. Johnson, E. Min, I. Spasojevic and B. J. Day, Biochem. Pharmacol., 2004, 67, 77 CrossRef CAS; (c) I. Batini'c-Haberle, I. Spasojevi'c, P. Hambright, L. Benov, A. L. Crumbliss and I. Fridovich, Inorg. Chem., 1999, 38, 4011 CrossRef.
- P. R. Castello, D. A. Drechsel and B. J. Day, J. Pharmacol. Exp. Ther., 2007, 324, 970 CrossRef.
- I. Aviv-Harel and Z. Gross, Chem.–Eur. J., 2009, 15, 8382 CrossRef CAS; D. T. Gryko, J. Porphyrins Phthalocyanines, 2008, 12, 906 CrossRef CAS.
- (a) A. Mahammed, I. Goldberg and Z. Gross, Org. Lett., 2001, 3, 3443 CrossRef CAS; (b) A. Mahammed, H. B. Gray, J. J. Weaver, K. Sorasaenee and Z. Gross, Bioconjugate Chem., 2004, 15, 738 CrossRef CAS.
- H. Agadjanian, J. Ma, A. Rentsendorj, V. Valluripalli, J. Y. Hwang, A. Mahammed, D. L. Farkas, H. B. Gray, Z. Gross and L. K. Medina-Kauwe, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6105 CrossRef CAS.
- (a) M. Eckshtain, I. Zilbermann, A. Mahammed, I. Saltsman, Z. Okun, E. Maimon, H. Cohen, D. Meyerstein and Z. Gross, Dalton Trans., 2009, 7879 RSC; (b) A. Mahammed and Z. Gross, Angew. Chem., Int. Ed., 2006, 45, 6544 CrossRef CAS; (c) A. Mahammed and Z. Gross, Chem. Commun., 2010, 46, 7040 RSC; (d) Z. Okun, I. Goldberg and Z. Gross, Angew. Chem., Int. Ed., 2007, 465, 4320.
- Z. Okun, L. Kupershmidt, T. Amit, S. Mandel, O. Bar-Am, M. B. Youdim and Z. Gross, ACS Chem. Biol., 2009, 4, 910 CrossRef CAS.
- A. Haber, A. Mahammed, B. Fuhrman, N. Volkova, R. Coleman, T. Hayek, M. Aviram and Z. Gross, Angew. Chem., Int. Ed., 2008, 47, 7896 CrossRef CAS.
- A. Haber, M. Aviram and Z. Gross, Chem. Sci., 2011, 2, 295 RSC.
- (a) L. Kupershmidt, Z. Okun, T. Amit, S. Mandel, I. Saltsman, A. Mahammed, O. Bar-Am, Z. Gross and M. B. Youdim, J. Neurochem., 2010, 113, 363 CrossRef CAS; (b) A. Kanamori, M.-M. Catrinescu, A. Mahammed, Z. Gross and L. A. Levin, J. Neurochem., 2010, 114, 488 CAS.
- A. Mahammed and Z. Gross, J. Am. Chem. Soc., 2005, 127, 2883 CrossRef CAS.
- M. Schwalbe, D. K. Dogutan, S. A. Stoian, T. S. Teets and D. G. Nocera, Inorg. Chem., 2011, 50, 1368 CrossRef CAS.
- I. Batini'c-Haberle, J. S. Reboucas and I. Spasojevi'c, Antioxid. Redox Signaling, 2010, 13, 877 Search PubMed.
- (a) N. Kasugai, T. Murase, T. Ohse, S. Nagaoka, H. Kawakami and S. Kubota, J. Inorg. Biochem., 2002, 91, 349 CrossRef CAS; (b) S. Asayama, N. Kasugai, S. Kubota, S. Nagaoka and H. Kawakami, J. Inorg. Biochem., 2007, 101, 261 CrossRef CAS.
- C. R. Schlieve, C. J. Lieven and L. A. Levin, Invest. Ophthalmol. Visual Sci., 2006, 47, 3878 CrossRef.
| This journal is © The Royal Society of Chemistry 2011 |