A review of NOx storage/reduction catalysts: mechanism, materials and degradation studies
Gang
Liu
and
Pu-Xian
Gao
*
Department of Chemical, Materials and Biomolecular Engineering & Institute of Material Science, University of Connecticut, Storrs, CT 06269-3136, USA. E-mail: puxian.gao@ims.uconn.edu; Fax: +1 860 486 4745; Tel: +1 860 486 9213
First published on 7th April 2011
Abstract
The catalytic removal of nitrogen oxides (NOx) from lean-burn exhaust emissions is one of the major challenges in environmental catalysis. Among the NOx emission control technologies, NOx storage/reduction (NSR) is currently regarded as one of the most practical technologies for lean-burn gasoline and diesel vehicles. This review gives a comprehensive overview of NSR technology, including the NSR reaction mechanisms, degradation mechanisms and NSR catalyst developments. The NSR reaction and degradation mechanisms will be addressed based on a typical NSR catalyst such as Pt/BaO/Al2O3, along with the concurrent new NSR catalyst developments for enhancing the NSR performance and alleviating their sulfur poisoning and thermal degradation.
1. Introduction
Conventional gasoline engines operate near stoichiometric conditions with an air to fuel ratio of 14.7 (for petrol).1,2 Lean-burn gasoline and diesel engines operate under lean-burn conditions with an air to fuel ratio higher than the stoichiometric combustion ratio (usually in the range of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 to 65∶1).3–5 Lean-burn engines are attracting more and more attention than conventional gasoline engines due to their higher fuel efficiency and lower CO2 emission.3–8 However, under lean-burn conditions, the nitrogen oxides (NOx) exhaust emissions cannot be efficiently reduced over the classical three-way catalysts in the presence of excessive O2.3,6,9
∶1 to 65∶1).3–5 Lean-burn engines are attracting more and more attention than conventional gasoline engines due to their higher fuel efficiency and lower CO2 emission.3–8 However, under lean-burn conditions, the nitrogen oxides (NOx) exhaust emissions cannot be efficiently reduced over the classical three-way catalysts in the presence of excessive O2.3,6,9
The NOx emissions have multi-fold hazards for the atmosphere, environment and human health, due to the formation of fine particles, ozone smog, acid rain and eutrophication.10 In the US, the NOx emissions from mobile vehicles contribute almost half of all NOx produced, and therefore, rigorous regulations were introduced for reducing NOx emissions from mobile vehicles.11
To meet the more and more stringent NOx emission regulations, three technologies, including direct decomposition of NOx, selective catalytic reduction (SCR) of NOx and NOx storage/reduction (NSR), have been developed for the control of lean-NOx emissions.9,12 The direct decomposition of NOx is thermodynamically favorable at temperatures below 900 °C, but the activation energy is relatively too high.13Cu-ZSM-5 has been extensively studied as the most promising catalyst for direct NOx decomposition in 1990s.14 However, its poor activity with ∼10% of NOx decomposed together with a low thermal stability has hindered its use in practice.15,16SCR technologies have been extended to urea/ammonia-SCR,17 hydrocarbon-SCR18 and plasma-assisted SCR.19 Among them, urea/ammonia-SCR is a widely commercialized technology for NOx removal in stationary sources and heavy-duty vehicles. However, the adoption of urea/ammonia-SCR technology to lean-burn engines results in complex exhaust after treatment system and enforcement difficulties. The NSR technology was first developed by Toyota researchers in 1995,20–22 which does not require an additional reducing agent. The NOx emissions are first trapped in the storage materials of NSR catalysts under lean conditions and then reduced by reducing agents under rich conditions. Although NSR technology is regarded as the most practical technology for lean-burn gasoline and diesel vehicles, the role of every component in the NSR performance and the inter-component relationship are still not very clear.9,23 Meanwhile, the state-of-the-art NSR catalyst (Pt/BaO/Al2O3) is weakly resistant to sulfur poisoning and thermal treatment.9
In this work, we review the NSR reaction mechanism and address the role of each component in a typical NSR catalyst (Pt/BaO/Al2O3) during the whole reaction process. Meanwhile, NSR degradation mechanisms, especially on sulfur poisoning and thermal degradation, are addressed. Concerning the existing problems of typical NSR catalysts, the latest developments of new NSR catalysts are also reviewed. Finally, major conclusions and some research directions are presented.
2. Mechanism of NSR reactions
NSR catalysts are generally comprised of precious metals, NOx storage components and support metal oxides, with Pt/BaO/Al2O3 as the most typical NSR catalyst. NSR catalysts work under cyclic operations with periodic switches between lean and rich conditions.24,25Fig. 1 shows a representative profile of NOx storage/reduction during lean and rich cycles. When an engine runs in the lean-burn cycle, NOx is trapped in the storage component of the NSR catalyst. Upon engine runs in the rich-burn cycle, the released NOx from storage components is reduced to N2 on precious metal. Usually a lean-burn cycle lasts 1–2 minutes followed by a 3–5 second rich-burn operation. Researchers also explored the NSR catalyst performance with long rich regeneration periods (for example, 1500 seconds).26,27 Under such conditions, the outlet NOx concentration during cyclic operation decreased, however, with higher fuel consumption.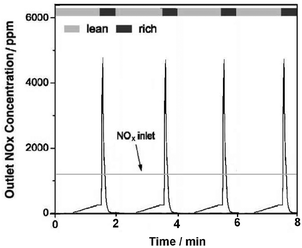 | ||
| Fig. 1 A representative profile of the NOx breakthrough and release during the lean-rich cycles. | ||
The NSR reaction mechanism during lean and rich cycles can be assumed to contain the following five steps:9,23
(I) NO oxidation to NO2;
(II) NOx (NO and NO2) sorption on the surface of alkali and/or alkaline-earth adsorption sites in the form of nitrites or nitrates;
(III) reducing agents (such as H2, CO, hydrocarbons) evolution from the rich exhaust gas;
(IV) NOx release from the nitrite or nitrate sites;
(V) NOx reduction to N2.
Steps I and II occur during lean-burn cycles and Steps III, IV and V happen during rich-burn cycles. Fig. 2 schematically illustrates the possible mechanism of NOx storage/reduction in a typical Pt/BaO/Al2O3 catalyst. First, under lean-burn conditions, NO is oxidized to NO2 on the precious metal Pt. After that, the NO and NO2 are adsorbed on the surface of BaO by the formation of barium nitrites or nitrates. Under rich-burn conditions, the released NOx from barium nitrites or nitrates are reduced to N2 on the Pt surface by H2, CO, and/or hydrocarbons from the rich exhaust gas.
 | ||
| Fig. 2 An illustration of the possible mechanism of the NOx storage/reduction on a typical Pt/BaO/Al2O3 catalyst. | ||
In this section, we discussed the five-step NSR mechanism. Despite a general agreement on the NSR mechanism currently, the understanding of each step in the NSR mechanism is still not very clear, especially the last three steps regarding the NSR catalysts regeneration. More systematic and detailed experimental and modeling research is expected to help better understand the NSR mechanism in order to achieve more efficient NSR catalysts.
3. Roles of each component in the Pt/BaO/Al2O3 catalyst during NSR reactions
As discussed in Section 2, there are five steps in a cycle of NOx storage/reduction reactions. Each step is critical for efficient operation, with each component of the Pt/BaO/Al2O3 catalyst holding important functions during the NSR reactions. In the following section, the roles of each catalyst component in different NSR reaction steps will be discussed individually. The effect of gas composition will be discussed on the functions of each component in different NSR reaction steps as well.3.1 Roles of Pt
Pt plays several key roles in the NOx storage/reduction reaction steps, such as (I) NO oxidation under lean-burn conditions, (II) NOx storage under lean-burn conditions, and (III) NOx reduction under rich-burn conditions. Although different NOx trapping components and support metal oxides will affect the functions of Pt in the NSR reactions, in this section, we will focus on the functions of Pt on BaO/Al2O3 in the NSR reactions. | ||
| Fig. 3 Variation of steady-state NO conversion with catalyst temperature during NO oxidation on the Pt/BaO/Al2O3 catalyst (inlet NO = 528 ppm, O2 = 5%). Reproduced from ref. 31 with permission from Elsevier. | ||
Considering the reversible adsorption and desorption of NO, O2 and NO2, Bhatia et al.31 referred to the microkinetic modeling studies of NO oxidation by Xu et al.32 and assumed that the NO oxidation happened by the following steps:
Step 1: NO + Pt ↔ NO–Pt
Step 2: O2 + 2Pt ↔ 2O–Pt
Step 3: NO–Pt + O–Pt ↔ NO2–Pt + Pt
Step 4: NO2–Pt ↔ NO2 + Pt
Interestingly, Xu et al. suggested that the O2 adsorption/dissociation on Pt (Step 2) was the rate-determining step, while the density functional theory (DFT) calculation on NO oxidation to NO2 by Smeltz et al. suggested33 the dissociation of atomic O from the O–Pt was the rate determining step (Step 3). However, Mulla et al.34 and Bhatia et al.31 were convinced that O2 adsorption was the rate-determining step based on the microkinetic analysis using the global model. The actual NO oxidation mechanism still remains unclear and a detailed elementary kinetic model should be developed to explain the NO oxidation reactions.
Pt in Steps 1 to 4 refers to a vacant Pt site and oxides formation (PtO and PtO2), which has been certified to decrease the activity of Pt for the NO oxidation.30,31,35Fig. 4 shows the NO conversion results on Pt/Al2O3 with various pretreatments. Here, the absence of NOx trapping component (BaO) would decrease the storage of NO2. The NO conversion was the highest when the catalyst was pretreated with reductive gas, whereas the lowest with NO2 pretreatment. Olsson and Fridell30 also investigated the poisoning effect of NO2 and O2 on the NO oxidation on Pt/Al2O3 and Pt/BaO/Al2O3 catalysts. From the XPS analysis of the Pt/Al2O3 and Pt/BaO/Al2O3 catalysts after different pretreatments (Fig. 5 and Table 1), the Pt was further oxidized to Pt oxides by NO2 other than O2.30 Due to the highly oxidizing nature of NO2, NO2 can be an effective source of atomic oxygen. The chemisorbed oxygen on Pt prevents the adsorption of NO, thus inhibiting the NO oxidation.36,37 The presence of BaO enhances the formation and stability of less reactive Pt oxides.30 Meanwhile, BaO also reduces the Pt exposed surface area by its physical blockage of Pt sites or formed nitrate species.9 These two reasons result in the lower NO oxidation on Pt/BaO/Al2O3 than that on Pt/Al2O3.
 | ||
| Fig. 4 Comparison of transient NO conversion for various pretreatments on the Pt/Al2O3 catalyst (inlet NO = 500 ppm, O2 = 5%; catalyst temperature = 198 °C). Reproduced from ref. 31 with permission from Elsevier. | ||
 | ||
| Fig. 5 XPS Pt 4f spectra for different pretreatments: (a) Pt/Al2O3 pretreated in NO2, (b) Pt/BaO/Al2O3 pretreated in NO2, (c) Pt/Al2O3 pretreated in O2, and (d) Pt/BaO/Al2O3 pretreated in O2. The energy scale is adjusted by setting Al 2s at 119.3 eV. Reproduced from ref. 30 with permission from Elsevier. | ||
The effect of Pt dispersion on the NO oxidation has been widely investigated.30,31,38,39 The strong impact of the Pt particle size on the NO oxidation rate suggests that NO oxidation is a structure sensitive reaction. As the Pt dispersion decreases, the Pt activity for NO oxidation increases. The reason for this may be that the larger Pt particles form less surface Pt oxides, thus are less active for NO oxidation.
Büchel et al.46 applied a two-nozzle flame spray pyrolysis method to prepare two NSR catalysts with Pt on either Al- or Ba-components without altering the Al2O3 or BaCO3 crystal sizes, Al/Ba weight ratio and Pt dispersion. Fig. 6 shows a set of transmission electron microscopy (TEM) images of Pt–Al–Ba (Pt preferentially deposited on Al2O3) and Al–Ba–Pt (Pt preferentially deposited on BaCO3) and the corresponding energy-dispersive X-ray (EDX) spectra of the two catalysts. The influence of Pt location on BaCO3 or Al2O3 has been studied during NOx storage/reduction. They found that Pt on Al2O3 exhibited a better NO oxidation activity which was the limiting step for the overall NOx storage process at low temperature (<300 °C). So, the Pt–Al–Ba catalyst performs better for NOx storage than the Al–Ba–Pt catalyst at <300 °C. However, at high temperature (>350 °C), the location of Pt barely affected the NOx storage performance.

When using hydrocarbons as reductants, two general mechanisms have been reported for the NOx reduction on NSR catalysts. One mechanism is based on the NO decomposition on Pt sites,48–50 and the other is based on the direct reaction between NO2 and reductants.51,52 Previous studies indicated that different types of hydrocarbon lead to different NOx reduction mechanisms.53,54 Due to the appearance of different reductants in the rich-burn cycle, it is difficult to distinguish between the two mechanisms. As summarized by Burch,55 the subtle changes in operating conditions would also affect the NOx reduction mechanism. Therefore, multiple NOx reduction mechanisms may exist during the NOx reduction on NSR catalysts.
No matter through which mechanism NOx is reduced, the research results indicate that NOx reduction occurs on the precious metal (Pt for the Pt/BaO/Al2O3 catalyst). In addition, the similar Pt particle size effect for NO oxidation was found for NOx reduction.56 Large Pt particles were found to be more efficient for NOx reduction than small ones since larger Pt particles tend to suppress the formation of Pt oxides and expose the Pt metal as the NOx reduction catalyst.
Apart from hydrocarbons as reductants for the NOx reduction, other reductants, such as H2 and CO, are also investigated for achieving high NOx reduction.57,58 H2 is more efficient for NOx reduction than CO, especially at low temperatures (<150 °C), due to the CO's poisoning effect on Pt sites as a result of the strong adsorption of CO on Pt sites. However, at high temperatures (>350 °C), CO and hydrocarbons have similar activities as H2 for NOx reduction.
3.2 Roles of BaO
BaO is the most commonly studied NOx trapping component among metal oxides,59–62 with a major contribution to the overall NOx trapping capacity. Besides the NOx storage step, BaO also plays some roles in the NO oxidation, NOx release and NOx reduction steps. Although different metal oxide supports and precious metal components affect the functions of BaO in the NSR reactions, in this section, we still focus on the general functions of BaO of the Pt/BaO/Al2O3 catalyst in the NSR reactions.In the nitrite route, it is proposed that NO is oxidized on Pt sites and directly trapped by nearby BaO sites to form Ba-nitrites. The Ba-nitrites are finally oxidized to Ba-nitrates. In the nitrate route, it is proposed that NO is oxidized to NO2 on Pt sites. NO2 spills over to the BaO site to form Ba-nitrate with evolution of NO. Fig. 7 shows the two different mechanisms for NO oxidation and the followed NOx trapping on the Pt/BaO/Al2O3 catalyst. The Ba loading affects which route is dominating during the NOx trapping step.67 When Ba loading is high or low, there are more or less Ba sites that are close to the Pt sites, which lead to the predominance of the nitrite route ornitrate route.
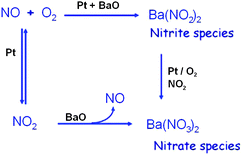 | ||
| Fig. 7 Two different pathways for the NO oxidation and the NOx trapping on the Pt/BaO/Al2O3 catalyst. Reproduced from ref. 67 with permission from Elsevier. | ||
According to the literature, BaO is more effective to trap NO2 than to trap NO. Therefore, the effect of NOx composition and temperature on the BaO trapping capacity is actually related to its effect on the NO oxidation.68 At high temperature, no matter whether NO or NO2 is used as the NOx precursor, similar NOx trapping capacity is observed on the Pt/BaO/Al2O3 catalyst. This is because NO can be easily oxidized to NO2 at high temperature. However, at low temperature, the NOx trapping capacity of BaO is low when using NO as a NOx precursor due to the limited NO oxidation into NO2.
With the NOx trapping BaO, the H2O and CO2 in the exhaust could react to produce barium hydroxide and carbonate. Lietti et al.69,70 found that BaO, Ba(OH)2 and BaCO3 coexist at the catalyst surface at 360 °C and the NOx trapping occurs at the BaO sites first, then the Ba(OH)2 and finally the BaCO3. The NOx trapping order on different Ba-based components is derived from the sequence of the evolved H2O and CO2, as shown in Fig. 8. The H2O and CO2 evolutions are due to the decompositions of Ba(OH)2 and BaCO3, respectively. As shown in Fig. 8a, in the first cycle, NOx slip was detected after about 50 s and CO2 evolution was observed at the same time with NOx slip. This reveals that the BaO reacts with NOx in the first 50 s, with no displaced product evolution. Then, BaCO3 is decomposed to form the Ba(NO3)2. In the first cycle, there is barely H2O evolution during the whole testing time. This is because the tested Pt/Ba/Al2O3 catalyst was pretreated in dry air. So there is no surface hydroxyl species and Ba(OH)2 on the catalyst surface. As shown in Fig. 8b, in the second cycle, NOx slip was detected after about 250 s and CO2 evolution was again observed coincident with NOx slip. Moreover, H2O evolution was found in this cycle after about 125 s, which is ahead of CO2 evolution and NOx slip. In each cycle, after the catalyst was trapped with NOx, the catalyst was regenerated with H2 in balance with He. This regeneration process produces some Ba(OH)2. So there is H2O evolution in the second cycle. From the results shown in Fig. 8b, it is obvious that Ba-based components decomposed in the order of Ba(OH)2 < BaCO3. Since there is no new BaCO3 produced during the testing (no CO2 in the regeneration process) and much of the BaCO3 has been decomposed during the first two cycles, the CO2 evolution is very small in the third cycle.
 | ||
| Fig. 8 A sequence of the evolution of displaced H2O and CO2 during the NOx storage at 350 °C on a previously unused (fresh) Pt/BaO/Al2O3 catalyst. The data set in (b) was obtained in the cycle immediately following the data shown in (a). (c) was obtained immediately after the cycle depicted in (b). The lean phase contained 1000 ppm NO, 3% O2, and a balance of He. The regeneration phase prior to each of the data sets contained 2000 ppm H2 in a balance of He. Reproduced from ref. 69 with permission from Elsevier. | ||
The thermodynamic stability of BaCO3 is higher than Ba(OH)2. Different from BaO, in order to trap NOx, Ba(OH)2 and BaCO3 must decompose first and then react with NOx. Therefore, the presence of H2O and CO2 in the exhaust gas phase will affect the rate of NOx trapping. Epling et al.71,72 studied the effect of CO2 and H2O on the NOx trapping capacity of NSR catalysts and they found that the presence of H2O and CO2 during the lean-burn phase decreases the NOx trapping capacity. In addition, CO2 decreases more NOx trapping capacity than H2O. Moreover, Hodjati et al.40,71 found that the presence of H2O reduces the negative effect of CO2 on decreasing NOx trapping capacity, since H2O reduces the amount of BaCO3 at the catalyst surface through the equilibrium reaction (BaCO3 + H2O ⇔ Ba(OH)2 + CO2).
The presence of CO also decreases the NOx trapping capacity either through competitive adsorption on Pt sites or viaoxidation to CO2 for subsequent BaCO3 formation, or by reducing the NO2 amount via the CO + NO2 → CO2 + NO reaction.42
The presence of O2 shows a positive effect on the increase of NOx trapping capacity. As O2 concentration increases, NOx trapping capacity increases.63,73 This is attributed to more efficient oxidation of NO into NO2, which facilitates formation of thermodynamically more stable nitrates.74
Apart from the reductant gases and O2, the presence of other gases, such as H2O and CO2, also affects the NOx release during the rich-burn cycle. The presence of H2O reduces the NOx release, while CO2 increases the NOx release.9,75
3.3 Roles of Al2O3
Al2O3 is widely used as the support for NSR catalysts due to its high surface area and high thermal stability. The important role of Al2O3 in NSR catalysts is to help in dispersing noble metals and NOx trapping materials. Al2O3 can also adsorb a little amount of NOx by forming Al-nitrate species.76 However, due to Al-nitrate's low thermal stability and small amount, the NOx trapping by Al2O3 is usually neglected.In the above Section 3, we reviewed the roles of each component of a typical NSR catalyst (Pt/BaO/Al2O3) in different NOx storage/reduction reaction steps. The gas composition effect on each component's functions during different NSR reaction steps was also detailed. Clear understanding of the roles of each component of the Pt/BaO/Al2O3 catalyst in the NSR process is very important to further increase NSR efficiency of this type of NSR catalyst and, the development of more efficient NSR catalysts. However, there are still debates on how Pt/Ba proximity, Pt and Ba loadings, Pt particle morphology, and the Pt/BaO, Pt/Al2O3 and BaO/Al2O3 interactions affect the NOx storage/reduction activity. In addition, the lean-rich operating conditions have a strong influence on the catalytic activity of NSR catalyst. Further understanding of these questions will help to understand the Pt/BaO/Al2O3 catalyst and the NSR mechanism.
4. Deactivation mechanism of NSR catalysts
Sulfur poisoning, thermal degradation and carbon deposition are the primary deactivation mechanisms that affect the application of NSR catalysts. In the following section, we will discuss in detail how these three deactivation mechanisms influence the key components in the typical Pt/BaO/Al2O3 NSR catalyst and the NSR performance.4.1 Sulfur poisoning
In typical lean exhaust, sulfur is mainly present in the form of SO2. During the lean-burn cycle, SO2 can poison not only the basic NOx trapping component but also the metal oxide support. In addition, during the rich-burn cycle, sulfur can also accumulate on the precious metal component, and therefore decrease its NOx reduction and the following NO oxidation activities remarkably.Surprisingly, Amberntsson et al.79,80 observed that the introduction of SO2 at 400 °C enhanced the NO oxidation ability on Pt/BaO/Al2O3 during the lean-burn cycle. This was attributed to the inhibition of the Pt oxides formation in the presence of SO2. As reviewed previously, Pt metal is considered the active sites for the NO oxidation. Less Pt oxides contribute to a higher NO oxidation ability.
Engstrom et al.81 found that the NO oxidation activity was remarkably decreased at the beginning of the lean-burn cycle when high concentration sulfur was introduced during the rich-burn cycle. This is because a large amount of PtS was produced during the rich conditions. Therefore, at the beginning of the lean-burn cycle, there were not enough Pt sites for the NO oxidation, since a large amount of PtS needs to decompose to release Pt sites. In addition, no matter whether the SO2 was introduced during lean or rich conditions, the NOx reduction was dramatically inhibited by the presence of sulfur. The low NOx reduction was attributed to the blockage of Pt sites by PtS formed during the rich conditions.
When SO2 was introduced during the lean-burn cycle, BaSO4 was observed. In addition, if the SO2 dose is small, BaSO4 is primarily observed on the surface86,87 and, if the SO2 dose increases continuously, bulk BaSO4 is also observed.78,87,88 Meanwhile, H2O89 and CO290 evolution were detected when introducing SO2 to the NSR catalyst. This reveals that SO2 also displaces the common NOx trapping precursors, Ba(OH)2 and BaCO3. Interestingly, when SO2 was introduced during the rich-burn cycle, no sulfur sorption on Ba or a Ba–S interaction was observed.78
Two mechanisms for SO2 deactivation over the Pt/BaO/Al2O3 catalyst have been proposed.9 Under lean conditions, SO2 is oxidized on Pt sites to SO3, which is subsequently adsorbed by the Ba trapping components to form surface BaSO4. As the sulfation continues, the surface BaSO4 migrates into the bulk phase. Under rich conditions, the deactivation is initially related to the Pt poisoning. As discussed in Section 4.1.1, PtS forms on the Pt sites, which blocks the Pt surface and hinders the NOx reduction and NO oxidation. However, ultimately the deactivation is still primarily due to the loss of NOx trapping sites by the formation of sulfates.
4.2 Desulfation
As discussed in the above sections, sulfur will poison the key components of the NSR catalyst, especially the Pt and BaO. Therefore, periodic sulfur removal from the NSR catalyst surface is required in order to recover the catalyst surface and keep an acceptable NSR performance.The desulfation is usually carried out at high temperature (>600 °C) under rich conditions. H2 has been demonstrated to be more effective for desulfation than CO and other hydrocarbons.20,93,94 However, further research by Liu and Anderson95 revealed that in the absence of Pt, the addition of H2 during the desulfation process actually hinders sulfur removal from the catalyst surface. They proposed that H2 needs to be dissociated on Pt to reduce sulfate species subsequently.
The rich feed composition also influences desulfation efficiency. It has been reported that the presence of CO296 and H2O97 has some positive effect on desulfation. During the desulfation process, BaSO4 will transform to BaS, which is also very stable and difficult to be recovered. However, with addition of CO2 into the rich feed, BaCO3 will form and decrease the chance to form BaS. As studied by Mahzoul et al.,97 the presence of H2O in the rich feed can lower the desulfation temperature. The first proposed effect of H2O could be the same with CO2, in favor of the formation of less stable Ba(OH)2 instead of BaS. The second proposed function of H2O is the easy transformation of PtS to PtO in the presence of H2O by the reaction: PtS + H2O → PtO + H2S.98
Apart from operating conditions that influence the desulfation efficiency, NSR catalyst formulation also plays significant roles. As reported in the literature, Pt reduces the onset temperature for desulfation,20 while Rh in the NSR catalyst not only reduces the sulfur deactivation, but also increases the desulfation efficiency.80 As for NOx trapping components, the addition of Li, Na and/or K to BaO-based NSR catalysts will reduce the desulfation temperature.92 This is probably due to the formation of the less stable sulfates with the added alkali metals. The oxide support composition also affects the sulfur resistance and the desulfation process. For example, the Pt/BaO/CeO2 shows better sulfur resistance than the Pt/BaO/Al2O3 catalyst.99 Furthermore, the desulfation over Pt/BaO/CeO2 proceeds at relatively mild conditions compared to those over Pt/BaO/Al2O3. This will be discussed in more detail in Section 5.
4.3 Thermal degradation
As discussed in the previous subsections, sulfate formation at the NOx trapping sites leads to a decrease of NOx trapping capacity, and hence degrades the NSR catalyst performance. Therefore, periodical sulfur removal from the NSR catalyst surface (desulfation) at high temperature (>600 °C) is needed to recover NSR catalyst performance to an acceptable level. Thermal degradation of NSR catalysts is primarily caused by this high temperature desulfation treatment. In addition, during a rich-burn cycle, the oxidation of hydrocarbon, CO and H2, generates heat at the catalyst surface and results in thermal degradation as well. | ||
| Fig. 9 XRD patterns of the catalysts aged at 950 °C in 1% H2, 0.006% O2, 0.05% O2, 0.5% O2, and 6.7% O2. Reproduced from ref. 100 with permission from Springer. | ||
 | ||
| Fig. 10 Representative TEM images and the corresponding particle size distribution diagrams of (a) fresh catalyst, (b) catalyst aged at 950 °C in 1% H2, and (c) catalyst aged at 950 °C in 6.7% O2. Reproduced from ref. 100 with permission from Springer. | ||
As discussed previously, the activity of NO oxidation and NOx reduction on Pt increases with the increasing Pt particle size. However, Li87 and Parks9et al. reported that the activities of NO oxidation and NOx reduction on Pt decreased after a long-term thermal treatment at high temperature owing to a significant Pt surface area loss.
 | ||
| Fig. 11 XRD patterns of Pt–Ba/Al2O3 catalysts treated for 24 h at (a) 550 °C, (b) 850 °C, (c) 950 °C, (d) 1050 °C, and (e) BaAl2O4 powder. Reproduced from ref. 102 with permission from Springer. | ||
X-Ray photoelectron spectroscopy (XPS)102 and Fourier transform infrared (FT-IR) spectra103 were also applied to confirm the reaction between Ba and Al2O3 at high temperature. It was revealed that the Ba and Al could exist as isolated BaO dispersed on Al2O3 in the fresh catalyst, but after aging above 800 °C, BaO and Al2O3 interact with each other to form stable BaAl2O4.
Szailer et al.103 investigated the formation of BaAl2O4 as a function of Ba loading on Al2O3. When the Ba loading was less than 8 wt%, there was no BaAl2O4 observed, even with increasing aging temperature to 1000 °C. However, when the Ba loading increased to 20 wt%, the formation of BaAl2O4 was observed at 800 °C. When the loading of BaO is low, only a thin layer of BaO forms on the Al2O3 surface. Therefore, without bulk BaO, no BaAl2O4 forms.
Most of the researchers concluded that BaAl2O4 decreases the NOx trapping capacity. As reported by Fekete et al. and Jang et al.102,104 (Fig. 12), the NOx conversion efficiency of the Pt–Ba/Al2O3 catalyst decreased from 86 to 55% after aging at 750 °C for 24 h. On the other hand, BaAl2O4 was reported as a potential NOx trapping component. However, further detailed studies revealed that the nitrates formed on the BaAl2O4 surface cannot be easily regenerated without the presence of Pt. Meanwhile, the temperature window for NOx sorption on BaAl2O4 is very narrow, usually below 350 °C, since the nitrates formed on the BaAl2O4 surface are not stable above 350 °C. Therefore, according to the literature, the formation of BaAl2O4 decreases NOx trapping capacity for NSR catalysts during the real cyclic operation.
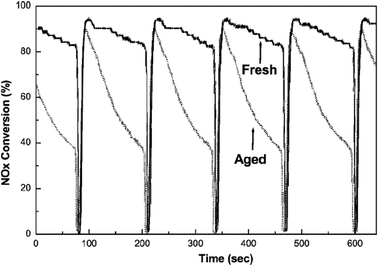 | ||
| Fig. 12 NOx conversion efficiency during the lean (120 s)–rich (6 s) cycle test for fresh (full line) and furnace aged (dotted line) Pt–Ba/Al2O3 catalyst at 450 °C (gas composition: rich = 4 vol% CO + 1.3 vol% H2 + 444 vppm C3H8 + 889 vppm C3H6 + 750 vppm NO + 10 vol% CO2 + 10 vol% H2O in N2 balance; lean = 8.0 vol% O2 + 444 vppm C3H8 + 889 vppm C3H6 + 750 vppm NO + 10 vol% CO2 + 10 vol% H2O in N2 balance). Reproduced from ref. 102 with permission from Springer. | ||
4.4 Carbon deposition
Carbon deposition on Pt sites by the decomposition of CO and hydrocarbons (C2H4, C3H6) is another deactivation mechanism that may lead to a progressive decay in NSR performance. Carbon can be deposited on Pt sites according to the following reactions: CO + Pt → Pt–CO and Pt–CO + Pt → Pt–C + Pt–O.23 In addition, over Pt sites, C2H4 can be decomposed to carbon by these reactions: C2H4 + Pt → Pt–C2H4 and Pt–C2H4 + 4O → CO2 + H2O + Pt–C.105 Although Pt–Ba/Al2O3 is a potential soot oxidation catalyst, the remained carbon on the Pt sites hinders the NO oxidation and NOx reduction.In Section 4, we reviewed the degradation mechanisms of the Pt/BaO/Al2O3 catalyst, especially focusing on the sulfur poisoning and thermal degradation of the key components of the Pt/BaO/Al2O3 catalyst, and the influence of these degradations on the NOx storage/reduction process. The studies of the degradation mechanisms of the Pt/BaO/Al2O3 catalyst will direct the development of more efficient and stable NSR catalysts.
5. Development of NSR catalysts
The Pt/BaO/Al2O3 catalyst is the first generation NSR catalyst and significant NOx emission control has been achieved with this catalyst. However, to meet the more stringent NOx emission standards and realize broad implementation of this NSR technology, it is very urgent to develop low-cost, highly efficient and durable NSR catalysts. The development of next generation NSR catalysts is mainly focused on the improvements of precious metal components for higher NO oxidation and NOx reduction activity, NOx trapping components for higher NOx storage capacity and a wide storage window, and novel metal oxide supports with higher tolerance of sulfur poisoning and higher thermal stability. In the following section, we will give a detailed review of the recent development of NSR catalysts.5.1 Precious metals
Pt is the most commonly used precious metal in the NSR catalysts. As discussed in the previous sections, Pt plays several key roles in the NOx storage/reduction cycles. However, in the literature, Pd and Rh were also used as the substitute of Pt metal for NSR catalysts. To combine the merits of different precious metals for the NOx storage/reduction reactions, bimetallic systems were also reported.Salasc et al.109 compared the NOx storage/reduction activity between Pd/BaO/Al2O3 and Pt/BaO/Al2O3 using lean-burn exhausts containing NO, O2, C3H6 and N2. As shown in Fig. 13, at 300 °C, the outlet NOx concentration over Pd/BaO/Al2O3 is lower than that over Pt/BaO/Al2O3. Therefore, Pd/BaO/Al2O3 shows a higher NOx storage capacity and better NOx reduction activity than Pt/BaO/Al2O3. Further XPS analysis revealed that the convertibility of Pd2+ ↔ Pd during the lean and rich conditions contributes to its high NSR activity.108,110 At 300 °C, the NOx reduction during the rich cycle over Pt/BaO/Al2O3 is not complete possibly due to the self-poisoning of the reaction by adsorption of NO or C3H6 derived species (CO, carbonaceous species) onto Pt sites.111 However, when increasing operating temperature to 400 °C, BaO prevents the self-poisoning of Pt sites and at this temperature, Pt/BaO/Al2O3 shows slightly better NSR activity than Pd/BaO/Al2O3. Su et al.111 also got similar results at relatively low temperatures when using hydrocarbons as reducing agents.
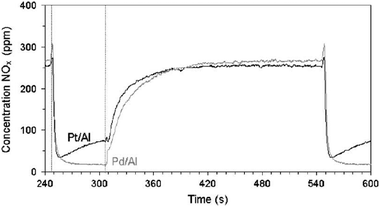 | ||
| Fig. 13 Outlet NOx (NO + NO2) concentration during a transient experiment for the Pt/Ba/Al2O3 and Pd/Ba/Al2O3 catalysts at 300 °C (gas composition: rich = 300 ppm NO + 900 ppm C3H6 in N2 balance; lean = 300 ppm NO + 900 ppm C3H6 + 8% O2 in N2 balance). Reproduced from ref. 109 with permission from Elsevier. | ||
The NOx storage/reduction activity of monometallic Rh/Ba/Al2O3 catalyst was also studied by Breen112 and Abdulhamid113et al. Their studies showed that Rh has the highest NOx reduction activity among Pt, Pd and Rh, although the total NOx conversion on Rh is lower than the other two metal-based NSR catalysts. The low NSR activity of Rh/Ba/Al2O3 is due to its low NOx storage capacity as a result of its low NO oxidation activity.
As reported by Amberntsson et al.,79,114 the NOx trapping capacity and NO oxidation activity is lower for Pt–Rh/BaO/Al2O3 catalysts than Pt/BaO/Al2O3 catalysts. However, NOx reduction activity over the Pt–Rh/BaO/Al2O3 catalyst is better than that on the Pt/BaO/Al2O3 catalyst. Due to a higher NOx reduction activity on Rh regardless of sulfur poisoning, the combination of Pt and Rh into the NSR catalysts exhibits a better NOx storage/reduction performance than the monometallic Pt/BaO/Al2O3 catalyst.
Recently, Wang et al.115 prepared a Pt/Co/Ba/Al2O3 catalyst by an impregnation method. The Pt/Co/Ba/Al2O3 catalyst showed better NSR activity and higher N2 selectivity than the conventional Pt/Ba/Al2O3 catalyst. The studies revealed that the addition of Co not only accelerates nitrites/nitrates formation on Ba sites, but also improves NOx adsorption on Al sites. The intimate contact of Co with Ba/Al provides more active sites for NO adsorption, oxidation and desorption. In addition, the synergistic effect of Pt and Co may accelerate the NOx reduction.
5.2 NOx trapping components
BaO or Ba is the first investigated NOx storage material. However, the biggest drawback using BaO as NOx storage material is its poor resistance to sulfur poisoning toward synthesis of thermodynamically more stable BaSO4 than Ba(NO3)2. Therefore, other alkaline and alkali metals have been extensively studied as NOx storage materials for NSR catalysts.Kustov and Makkee116 studied the NOx storage/release performance on Ba/Al2O3, Sr/Al2O3, Ca/Al2O3 and Mg/Al2O3 using FT-IR spectroscopy coupled with mass spectrometry (MS). Fig. 14 shows the FT-IR spectra upon adsorption of 800 ppm NO in 20% O2 in the He mixture on Al2O3, Ba/Al2O3, Sr/Al2O3, Ca/Al2O3 and Mg/Al2O3, respectively. The maximum NOx adsorption is observed on pure Al2O3 after ∼5 minutes of contact. For supported Ba, Sr, Ca and Mg systems, most of the NOx is adsorbed within the first 20–30 min and the complete NOx sorption takes about 1–2 h. After the NOx adsorption was saturated, the flow of NO was stopped and desorption occurred in air with increasing temperature from 200 to 600 °C. The desorbed species were detected by MS, with NOx desorbed amount and storage capacity listed in Table 2. The NOx desorbed from Al2O3 is very little, which is not shown in Table 2. It is obvious that the NOx storage capacity increases from Ba to Ca with increasing basicity of the alkaline earth materials. For the Mg/Al2O3 system, the NOx storage is very low, which may be due to low thermal stability of trapped nitrates at a storage temperature of 200 °C. Except the Mg/Al2O3 system, most of the adsorbed NOx are released above 500 °C. However, for Ba and to some extent for Sr, trapped nitrates even will be stable at temperatures above 600 °C. The high thermal stability of Ba- and Sr-nitrates will decrease their NOx release amount at relatively low temperature and decrease the cyclic NOx storage activity.
 | ||
| Fig. 14 FT-IR spectra upon adsorption of 800 ppm NO in 20% O2 in He mixture on (a) Al2O3, (b) Ba/Al2O3, (c) Sr/Al2O3, (d) Ca/Al2O3, and (e) Mg/Al2O3 at 200 °C. Reproduced from ref. 115 with permission from Elsevier. | ||
| Sample | T bulk/°C | T surface/°C | NOx desorbed/μmol g−1 | Storage capacity (%) |
|---|---|---|---|---|
| Ba(NO3)2/Al2O3 | 610 | >600 | 114 | 5.7 |
| Sr(NO3)2/Al2O3 | 505 | 600 | 330 | 16.5 |
| Ca(NO3)2/Al2O3 | 300–450 | 500–600 | 498 | 24.9 |
| Mg(NO3)2/Al2O3 | 200–350 | 400 | 122 | 6.1 |
To increase the Ba-based system's storage activity at low temperature, a Ba–Mg-based system was investigated as a promising NOx storage system.117 When Ba was partially replaced by Mg, the Pt/Ba–Mg/Al2O3 catalyst showed better NOx storage capacity than the Pt/Ba/Al2O3 catalyst at 200 °C, while at higher temperatures (300 and 400 °C) the trend was reversed. Therefore, the optimization of Ba and Mg composition in the NSR catalyst is important in order to obtain better NOx capacity at a wide temperature window. Meanwhile, the Ba–Mg system exhibited high resistance to the deactivation by SO2, which may be due to a synergistic effect between Ba and Mg and a better interaction with the Al2O3 support.
Apart from the above discussed alkaline metals, alkali metal, such as K, is also widely used as a NOx storage component in NSR catalysts. Toops et al.118 studied the NOx trapping capacity on the Pt/K/Al2O3 catalyst at 250 °C. They found that the addition of K to Pt/Al2O3 increases the trapping capacity from 2.3 μmol NOx m−2 to 6.2 μmol NOx m−2. Without K, the NOx is primarily trapped on Al2O3 in the form of nitrates with monodentate, chelating and bridged forms. However, with the presence of K, the NOx is primarily trapped by K in a free nitrate ion. Büchel et al.119 found that their flame synthesized Pt/K/Al2O3 can achieve over 80% NOx conversion at a temperature range of 300–400 °C. At 400 °C almost no NOx exhaust was detected in the 50 fuel lean/rich cycles. This superior performance resulted from a good K distribution in the catalyst and the amorphous nature of K species, which were obtained by the novel flame spray synthesis method. However, at temperatures higher than 400 °C, the NOx conversion decreased to around 60%. The sudden drop of NOx conversion is probably due to the partial crystallization of K2CO3. Meanwhile, this performance decrease cannot be recovered upon operation at low temperature (300 °C) again. This research also revealed that the novel catalyst synthesis method is very important to enhance the catalyst NSR performance.
5.3 Metal oxide supports
The metal oxide support performs a significant role in the NSR catalysts. It not only helps in dispersing the precious metals and the NOx storage materials but also helps in increasing the stability, sulfur resistance and NOx storage/reduction activity of the NSR catalysts. Al2O3 has been widely used as a support for NSR catalysts. However, as discussed previously, BaO will react with Al2O3 to form spinel BaAl2O4 at temperature over 600 °C, which has been considered as one of the degradation pathways for the Pt/BaO/Al2O3 catalyst.102 Recognizing the importance of the NSR catalyst support, different single oxide and mixed oxide supports have been developed as an alternative of Al2O3 for NSR catalysts.Kwak et al.123 compared the sulfur resistance and thermal resistance of Pt/BaO/CeO2 and Pt/BaO/Al2O3 catalysts. As shown in Fig. 15, after sulfation with SO2 at 250 °C, the NOx uptake on Pt/BaO/CeO2 decreased from 60% to 43%, while that decreased from 48% to 23% on Pt/BaO/Al2O3. After desulfation at 600 °C, the NOx uptake on Pt/BaO/CeO2 recovered to 46%, and recovered to 29% on Pt/BaO/Al2O3. The Pt/BaO/CeO2 catalyst showed better sulfur resistance and regeneration performance than the Pt/BaO/Al2O3 catalyst. To figure out the positive effect of CeO2, the Pt/BaO/CeO2 and Pt/BaO/Al2O3 catalysts after desulfation were characterized by XPS and TEM (Fig. 16 and 17). As shown in Fig. 16, after desulfation, there is less sulfur left on Pt/BaO/CeO2 than that on the Pt/BaO/Al2O3 catalyst. Therefore it was concluded that for the Pt/BaO/Al2O3 catalyst, BaSO4 is mostly transformed to BaS with little sulfur actually removed after desulfation. While for Pt/BaO/CeO2, the better sulfur resistance is not due to the sulfur trapped on CeO2, but the small-sized Pt on Pt/BaO/CeO2, which suppresses SO2 oxidation into SO3, and therefore hinders the formation of BaSO4. As shown by the TEM images (Fig. 17) of these catalysts after desulfation at 600 °C, it is clear that agglomeration of the Pt particles was observed on the Pt/BaO/Al2O3 catalyst, while no obvious Pt sintering effect on the Pt/BaO/CeO2 catalyst was observed, which may be another reason why Pt/BaO/CeO2 performs better than Pt/BaO/Al2O3 after high temperature desulfation.
 | ||
| Fig. 15 Histogram of NOx uptake (%) for Pt/Ba/Al2O3 and Pt/Ba/CeO2 catalysts after sulfation and desulfation. Reproduced from ref. 122 with permission from Elsevier. | ||
 | ||
| Fig. 16 XPS data of sulfur 2p data for fresh and after desulfation at 600 °C of Pt/Ba/Al2O3 (a and c) and Pt/Ba/CeO2 (b and d). Reproduced from ref. 122 with permission from Elsevier. | ||
 | ||
| Fig. 17 TEM images of Pt/Ba/CeO2 (a) and Pt/Ba/Al2O3 (b) catalysts after desulfation at 600 °C. Reproduced from ref. 122 with permission from Elsevier. | ||
Other explanations121 suggest that high water-gas shift activity of Pt/CeO2 increases the H2 concentration during the rich conditions, which helps remove sulfur at relatively low temperature from the unstable cerium sulfate.
Besides CeO2, ZrO2 is also investigated as an alternative support of Al2O3 for NSR catalysts. Piacentini et al.124 studied the NOx trapping capacity on ZrO2 and Al2O3 based NSR catalysts. They found that Pt/BaCO3/ZrO2 presented a higher NOx trapping capacity than Pt/BaCO3/Al2O3 at low Ba loading. While at higher Ba loading, the NOx trapping capacity on the two tested catalysts is similar.
Strobel et al.125 synthesized a series of Pt/Ba/CexZr1–xO2catalysts by a two-nozzle flame spray pyrolysis method. They found that the support composition (CexZr1–xO2) strongly affected the NOx reduction activity of Pt. Higher Ce content favored the formation of Pt oxides, therefore lowered its NOx reduction activity and the total NOx conversion efficiency (Fig. 18). The NOx storage capacity of the Pt/Ba/CexZr1–xO2catalysts before and after CO2 exposure was also evaluated, as summarized in Table 3. It is obvious that complete BaCO3 recovery was achieved on CeO2, whereas BaCO3 was not reformed on ZrO2 and only partly reformed on Ce–Zr mixed oxide. Therefore, as discussed above, further optimization of the CexZr1–xO2 support is favorable to get a better NSR performance.
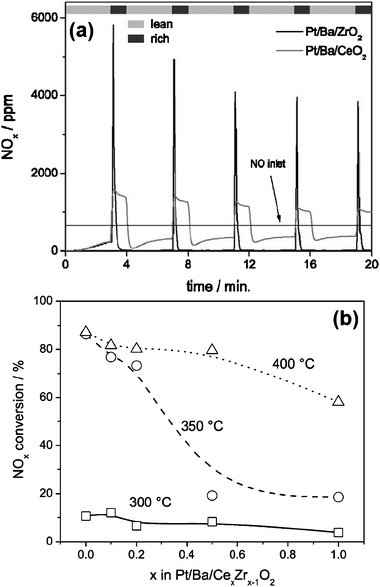 | ||
| Fig. 18 (a) Outlet NOx concentration during a transient experiment for the as-prepared Pt/Ba/ZrO2 and Pt/Ba/CeO2 catalysts at 350 °C, and (b) NOx concentration as a function of temperature and support composition during the fifth lean-rich cycle (compare Fig. 21a). Reproduced from ref. 125 with permission from Elsevier. | ||
| Sample | Annealing a | NOx stored/mg gcat−1 | NOx storage capacityb (%) |
|---|---|---|---|
| a 1 h at 800 °C in 10% O2/He followed by 1 h in 20% CO2/He. b Relative amount of Ba involved in the storage process assuming complete Ba(NO3)2 formation. | |||
| Pt/Ba/CeO2 | No | 51.8 | 77 |
| Yes | 62.2 | 92 | |
| Pt/Ba/Ce0.5Zr0.5O2 | Yes | 32.5 | 48 |
| Pt/Ba/ZrO2 | No | 40.9 | 61 |
| Yes | 6.6 | 10 |
In addition, the sulfur resistance and regeneration ability of the Pt/Ba/CexZr1–xO2catalysts was also evaluated by some researchers.126Pt/Ba/CexZr1–xO2catalysts showed better sulfur resistance than Pt/Ba/Al2O3 catalysts. Meanwhile, the sulfates elimination under desulfation conditions was more efficient on Pt/Ba/CexZr1–xO2 than on the Pt/Ba/Al2O3 catalyst.
Imagawa et al.128,129 synthesized a nano-composite containing Al2O3 and ZrO2–TiO2 solid solution (AZT), which was used as the support for Pt/Rh/Ba/K/AZT NSR catalysts. As shown in Fig. 19, the particle size of ZrO2–TiO2 in the nano-composite was smaller than that in the physically mixed oxide at all the measured temperatures. The typical TEM images of the AZT nano-composite and physically mixed AZT after thermal treatment at 900 °C were shown in Fig. 20. It was believed that Al2O3 particles act as a diffusion barrier to ZrO2–TiO2 particles in the nano-composite oxide to prevent the agglomeration of ZrO2–TiO2 particles. As shown in Fig. 21, after a thermal aging test at different temperatures, the AZT nano-composite based Pt/Rh/Ba/K/AZT catalyst had a larger amount of NOx storage and less amount of NOx release during the lean-rich cycle when compared with that of the physically mixed AZT based Pt/Rh/Ba/K/AZT catalyst. Especially at 400 °C, the NOx storage capacity in the AZT nano-composite based Pt/Rh/Ba/K/AZT catalyst was about twice that of the physically mixed AZT based Pt/Rh/Ba/K/AZT catalyst. In addition, better NOx storage performance after sulfur aging was observed on the AZT nano-composite based Pt/Rh/Ba/K/AZT catalyst (Fig. 22). The monolithic AZT inhibited the solid phase reaction of K with support materials and a high percentage of active K was maintained for NOx storage. Meanwhile, it was suggested that Ba could help prevent sulfur poisoning on K and facilitate the NOx storage.
 | ||
| Fig. 19 Average particle size of ZrO2–TiO2 determined by XRD dataversus thermal treatment temperature. (●) Nano-composite of Al2O3 and ZrO2–TiO2; (○) physically mixed Al2O3 and ZrO2–TiO2. Reproduced from ref. 127 with permission from Elsevier. | ||
 | ||
| Fig. 20 FE-TEM micrographs of samples after thermal treatment at 900 °C, (a) nano-composite of Al2O3 and ZrO2–TiO2; (b) physically mixed Al2O3 and ZrO2–TiO2. Reproduced from ref. 127 with permission from Elsevier. | ||
 | ||
Fig. 21 (a) NOx storage performance versus reaction temperature after thermal aging test: (●) Cat. A, Pt/Rh/Ba/K/AZT catalyst using nano-composite of Al2O3 and ZrO2–TiO2 as a support; (○) Cat. B, Pt/Rh/Ba/K/AZT catalyst using physically mixed Al2O3 and ZrO2–TiO2 as a support. (b) NOx concentration profile in the outlet gas at 400 °C under the lean-rich cycle after thermal aging test: Cat. A (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ), Cat. B ( ), Cat. B (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ). Reproduced from ref. 127 with permission from Elsevier. ). Reproduced from ref. 127 with permission from Elsevier. | ||
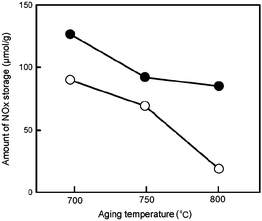 | ||
| Fig. 22 NOx storage after sulfur aging as a function of aging temperature. (●) Monolithic AZT catalyst and (○) monolithic physically mixed catalyst. Reproduced from ref. 128 with permission from Elsevier. | ||
As reported in the literature, other mixed metal oxides, such as MgO–Al2O3,130MgO–CeO2,131MnOx–CeO2132 and Al2O3–CexZr1–xO2,133 were also evaluated as supports for NSR catalysts, which exhibited some superior performance compared to the Al2O3 support.
In Section 5, we reviewed the recent development of NSR catalysts, focused on the improvement of precious metals, NOx storage materials and metal oxide supports. The newly developed NSR catalysts possessed improved NSR activity and/or good resistance to sulfur poisoning and thermal degradation compared with the Pt/BaO/Al2O3 catalyst. The future development of low-cost, highly efficient and durable NSR catalysts can be achieved by utilizing the relatively low-cost other PGMs or non-PGMs and suitable perovskite oxides, the mixed alkali and/or alkaline earth metal compounds and novel metal oxide supports as substitutes of Pt, BaO and Al2O3, respectively.
6. Conclusions and outlook
In this review, we first addressed the NOx storage/reduction mechanism, and then based on the Pt/BaO/Al2O3 catalyst, we surveyed the roles of each component (precious metal Pt, NOx storage material BaO and support Al2O3) in the NSR reactions. The deactivation mechanisms of the Pt/BaO/Al2O3 catalyst, especially the sulfur poisoning and thermal degradation, were extensively reviewed. Finally, recent developments of NSR catalysts were addressed in detail, concentrating on the improvements over precious metals, NOx storage materials, and metal oxide supports.Despite the first generation NSR catalyst (Pt/BaO/Al2O3) being quite successful in NOx emission control, new generation NSR catalysts with low-cost, high efficiency and durability are urgently needed to meet the ever rigorous NOx emission regulations and develop the NSR technology. Currently, there is a consensus on the five-step NSR mechanism (Section 2), however, the understanding of each step of the NSR mechanism is far from clear, especially the last three steps regarding the regeneration of NSR catalysts. A further investigation of the NSR mechanism for a better understanding should be carried out while in search for more efficient NSR catalysts. In addition, more efficient NOx trapping materials and support materials need to be developed to alleviate the sulfur poisoning and thermal degradation, which are two big problems that affect the long-term stability of the NSR catalysts. Furthermore, the development of new materials (e.g. perovskite oxide-based materials8,134) as alternatives of precious metals is very promising to reduce the cost of NSR catalysts. Finally, a novel synthesis method and a novel structured NSR catalyst (e.g. 3-D structured catalysts135–137) would help to increase the NSR activity, sulfur poisoning resistance, and thermal stability.
Acknowledgements
The authors are grateful for the financial support from the Uconn New Faculty start-up funds, Honda Initiation Grant, and the DOE/National Energy Technology Laboratory (Award number: DE-EE0000210).References
- J. G. Nunan, H. J. Robota, M. J. Cohn and S. A. Bradley, J. Catal., 1992, 133, 309 CrossRef CAS.
- B. H. Engler, D. Lindner, E. S. Lox, A. S. Sindlinger and K. Ostgathe, Stud. Surf. Sci. Catal., 96, 441 Search PubMed.
- R. M. Heck and R. J. Farrauto, Catalytic Air Pollution Control, Van Nostrand-Reinhold, New York, 1995 Search PubMed.
- L. Li, J. Chen, S. Zhang, F. Zhang, N. Guan, T. Wang and S. Liu, Environ. Sci. Technol., 2005, 39, 2841 CrossRef CAS.
- R. D. Clayton, M. P. Harold and V. Balakotaiah, Appl. Catal., B, 2008, 84, 616 CrossRef CAS.
- F. Basile, G. Fornasari, A. Grimandi, M. Livi and A. Vaccari, Appl. Catal., B, 2006, 69, 58 CrossRef CAS.
- Q. Wang, J. H. Sohn and J. S. Chung, Appl. Catal., B, 2009, 89, 97 CrossRef CAS.
- J. E. Parks II, Science, 2010, 327, 1584 CrossRef.
- W. S. Epling, L. E. Campbell, A. Yezerets, N. W. Currier and J. E. Parks II, Catal. Rev. Sci. Eng., 2004, 46, 163 CrossRef.
- P. S. Monks, et al. , Atmos. Environ., 2009, 43, 5268 CrossRef CAS.
- F. Klingstedt, K. Arve, K. Eranen and D. Y. Murzin, Acc. Chem. Res., 2006, 39, 273 CrossRef CAS.
- Z. Liu and S. I. Woo, Catal. Rev. Sci. Eng., 2006, 48, 43 CrossRef CAS.
- Y. Yokomichi, T. Yamabe, T. Kakumoto, O. Okada, H. Ishikawa, Y. Nakamura, H. Kimura and I. Yasuda, Appl. Catal., B, 2000, 28, 1 CrossRef CAS.
- M. Iwamoto and H. Yahiro, Catal. Today, 1994, 22, 5 CrossRef CAS.
- L. Čapek, J. Dědeeček, B. Wichterlová, L. Cider, E. Jobson and V. Tokarová, Appl. Catal., B, 2005, 60, 147 CrossRef CAS.
- J. Dědeeček, L. Čapek and B. Wichterlová, Appl. Catal., A., 2006, 307, 156 CrossRef CAS.
- H. L. Fang and H. F. M. DaCosta, Appl. Catal., B, 2003, 46, 17 CrossRef CAS.
- V. Houel, P. Millington, R. Rajaram and A. Tsolakis, Appl. Catal., B, 2007, 73, 203 CrossRef CAS.
- J. Li, R. Ke, W. Li and J. Hao, Catal. Today, 2007, 126, 272 CrossRef CAS.
- S. Matsumoto, Y. Ikeda, H. Suzuki, M. Ogai and N. Miyoshi, Appl. Catal., B, 2000, 25, 115 CrossRef CAS.
- K. Yamazaki, T. Suzuki, N. Takahashi, K. Yokota and M. Sugiura, Appl. Catal., B, 2001, 30, 459 CrossRef CAS.
- H. Hirata, I. Hachisuka, Y. Ikeda, S. Tsuji and S. Matsumoto, Top. Catal., 2001, 16/17, 145 CrossRef.
- S. Roy and A. Baiker, Chem. Rev., 2009, 109, 4054 CrossRef CAS.
- Y. Li, S. Roth, J. Dettling and T. Beutel, Top. Catal., 2001, 16/17, 139 CrossRef.
- W. S. Epling, A. Yezerets and N. W. Currier, Catal. Lett., 2006, 110, 143 CrossRef CAS.
- H. Abdulhamid, E. Fridell and M. Skoglundh, Top. Catal., 2004, 30/31, 161 CrossRef.
- W. S. Epling, A. Yezerets and N. W. Currier, Appl. Catal., B, 2004, 74, 117.
- L. Olsson, H. Persson, E. Fridell, M. Skoglundh and B. Andersson, J. Phys. Chem. B, 2001, 105, 6895 CrossRef CAS.
- L. Olsson, B. Westerberg, H. Persson, E. Fridell, M. Skoglundh and B. Andersson, J. Phys. Chem. B, 1999, 103, 10433 CrossRef CAS.
- L. Olsson and E. Fridell, J. Catal., 2002, 210, 340 CrossRef CAS.
- D. Bhatia, R. W. McCabe, M. P. Harold and V. Balakotaiah, J. Catal., 2009, 266, 106 CrossRef CAS.
- J. Xu, V. Balakotaiah and M. P. Harold, Appl. Catal., B, 2009, 89(1–2), 73 CrossRef CAS.
- A. D. Smeltz, R. B. Getman, W. F. Schneider and F. H. Ribeiro, Catal. Today, 2008, 136, 84 CrossRef CAS.
- S. S. Mulla, N. Chen, W. N. Delgass, W. S. Epling and F. H. Ribeiro, Catal. Lett., 2005, 100(3–4), 267 CrossRef CAS.
- R. L. Muncrief, P. Khanna, K. S. Kabin and M. P. Harold, Catal. Today, 2004, 98, 393 CrossRef CAS.
- J. Segner, W. Vielhaber and G. Ertl, Isr. J. Chem., 1982, 22, 375 CAS.
- D. H. Parker and B. E. Koel, J. Vac. Sci. Technol., A, 1990, 8(3), 2585 CrossRef CAS.
- R. D. Clayton, M. P. Harold, V. Balakotaiah and C. Z. Wan, Appl. Catal., B, 2009, 90, 662 CrossRef CAS.
- E. Xue, K. Seshan and J. R. H. Ross, Appl. Catal., B, 1996, 11, 65 CrossRef CAS.
- S. Hodjati, P. Bernhardt, C. Petit, V. Pitchon and A. Kiennemann, Appl. Catal., B, 1998, 19, 209 CrossRef CAS.
- S. Hodjati, K. Vaezzadeh, C. Petit, V. Pitchon and A. Kiennemann, Catal. Today, 2000, 59, 323 CrossRef CAS.
- S. Erkfeldt, E. Jobson and M. Larsson, Top. Catal., 2001, 16/17, 127 CrossRef.
- F. Rodrigues, L. Juste, C. Potvin, J. F. Tempere, G. Blanchard and G. Djega-Mariadassou, Catal. Lett., 2001, 72, 59 CrossRef CAS.
- N. W. Cant and M. J. Patterson, Catal. Today, 2002, 73, 271 CrossRef CAS.
- P. Fanson, M. Horton, W. Delgass and J. Lauterbach, Appl. Catal., B, 2003, 46, 393 CrossRef CAS.
- R. Büchel, R. Strobel, F. Krumeich, A. Baiker and S. E. Pratsinis, J. Catal., 2009, 261, 201 CrossRef.
- O. Bailey, D. Dou, G. Denison, SAE Technical Paper Series, 972845.
- A. Obuchi, A. Ohi, M. Nakamura, A. Ogata, K. Mizuno and H. Ohuchi, Appl. Catal., B, 1993, 2, 71 CrossRef.
- L. Olsson, E. Fridell, M. Skoglundh and B. Andersson, Catal. Today, 2002, 73, 263 CrossRef CAS.
- R. Burch, J. Breen and F. Meunier, Appl. Catal., B, 2002, 39, 283 CrossRef CAS.
- R. Burch, P. Millington and A. Walker, Appl. Catal., B, 1994, 4, 65 CrossRef CAS.
- H. Hamada, Y. Kintaichi, M. Inaba, M. Tabata, T. Yoshinari and H. Tsuchida, Catal. Today, 1996, 29, 53 CrossRef CAS.
- R. Burch and T. C. Watling, Catal. Lett., 1997, 43, 19 CrossRef CAS.
- T. Maunula, J. Ahola and H. Hamada, Appl. Catal., B, 2000, 26, 173 CrossRef CAS.
- R. Burch, 7th DOE Cross-Cut Lean Exhaust Emissions Reductions Simulations (CLEERS), Detroit, MI, June 2004.
- D. James, E. Fourre, M. Ishii and M. Bowker, Appl. Catal., B, 2003, 45, 147 CrossRef CAS.
- J. E. Parks, S. Huff, J. A. Pihl, J. S. Choi and B. H. West, SAE Technical Paper Series, 2005, 2005-01-3876.
- J. A. Pihl, J. Parks and C. S. Daw, SAE Technical Paper Series, 2006, 2006-01-3441.
- H. Arai and M. Machida, Catal. Today, 1994, 22, 97 CrossRef CAS.
- M. Machida, N. Masuda and T. Kijima, J. Mater. Chem., 1999, 9, 1369 RSC.
- K. Eguchi and T. Hayashi, Catal. Today, 1998, 45, 109 CrossRef CAS.
- K. Eguchi, T. Kondo, T. Hayashi and H. Arai, Appl. Catal., B, 1998, 16, 69 CrossRef CAS.
- H. Mahzoul, J. F. Brilhac and P. Gilot, Appl. Catal., B, 1999, 20, 47 CrossRef CAS.
- S. Hodjati, C. Petit, V. Pitchon and A. Kiennemann, Appl. Catal., B, 2000, 27, 117 CrossRef CAS.
- F. Prinetto, G. Ghiotti, I. Nova, L. Lietti, E. Tronconi and P. v. Forzatti, J. Phys. Chem. B, 2001, 105, 12732 CrossRef CAS.
- P. Schmitz and R. Baird, J. Phys. Chem. B, 2002, 106, 4172 CrossRef CAS.
- P. Forzatti, L. Castoldi, I. Nova, L. Lietti and E. Tronconi, Catal. Today, 2006, 117, 316 CrossRef CAS.
- E. Fridell, M. Skoglundh, B. Westerberg, S. Johansson and G. Smedler, J. Catal., 1999, 183, 196 CrossRef CAS.
- L. Lietti, P. Forzatti, I. Nova and E. Tronconi, J. Catal., 2001, 204, 175 CrossRef CAS.
- I. Nova, L. Castoldi, L. Lietti, E. Tronconi and P. Forzatti, Catal. Today, 2002, 75, 431 CrossRef CAS.
- W. S. Epling, G. Campbell and J. Parks, Catal. Lett., 2003, 90, 45 CrossRef CAS.
- W. S. Epling, C. H. F. Peden and J. Szanyi, J. Phys. Chem. C, 2008, 112(29), 10952 CrossRef CAS.
- N. Takahashi, et al. , Catal. Today, 1996, 27, 63 CrossRef CAS.
- J. Anderson, A. Paterson and M. Fernandez-Garcia, Stud. Surf. Sci. Catal., 130, 1331 Search PubMed.
- G. Lutkemeyer, R. Weinowski, G. Lepperhoff, M. Brogan, R. Brisley, A. Wilkins, SAE Technical Paper Series, 962046.
- B. Westerberg and E. Fridell, J. Mol. Catal. A: Chem., 2001, 165, 249 CrossRef CAS.
- E. Fridell, H. Persson, L. Olsson, B. Westerberg, A. Amberntsson and M. Skoglundh, Top. Catal., 2001, 16/17, 133 CrossRef.
- C. Sedlmair, K. Seshan, A. Jentys and J. Lercher, Catal. Today, 2002, 75, 413 CrossRef CAS.
- A. Amberntsson, M. Skoglundh, M. Jonsson and E. Fridell, Catal. Today, 2002, 73, 279 CrossRef CAS.
- A. Amberntsson, M. Skoglundh, S. Ljungstron and E. Fridell, J. Catal., 2003, 217, 253 CAS.
- P. Engstrom, A. Amberntsson, M. Skoglundh, E. Fridell and G. Smedler, Appl. Catal., B, 1999, 22, L241 CrossRef CAS.
- P. Engström, A. Amberntsson, M. Skoglundh, E. Fridell and G. Smedler, Appl. Catal., B, 1999, 22, L241 CrossRef CAS.
- K. Wilson, C. Hardacre, C. J. Baddeley, J. Lüdecke, D. P. Woodruff and R. M. Lambert, Surf. Sci., 1997, 372, 279 CrossRef CAS.
- J. A. Anderson, Z. Liu and M. F. Garcia, Catal. Today, 2006, 113, 25 CrossRef CAS.
- M. Happel, A. Desikusumastuti, M. Sobota, M. Laurin and J. Libuda, J. Phys. Chem. C, 2010, 114, 4568 CrossRef CAS.
- C. Courson, A. Khalfi, H. Mahzoul, S. Hodjati, N. Moral, A. Kiennemann and P. Gilot, Catal. Commun., 2002, 3, 471 CrossRef CAS.
- J. Li, J. Theis, W. Chun, C. Goralski, R. Kudla, W. Watkins, R. Hurley, SAE Technical Paper Series, 2001-01-2503.
- E. Fridell, M. Skoglundh, SAE Technical Paper Series, 2004-01-0080.
- J. De Wilde and G. Marin, Catal. Today, 2000, 62, 319 CrossRef.
- J. Breen, M. Marella, C. Pistarino and J. Ross, Catal. Lett., 2002, 80, 123 CrossRef CAS.
- H. Mahzoul, L. Limousy, J. Brilhac and P. Gilot, J. Anal. Appl. Pyrolysis, 2000, 56, 179 CrossRef CAS.
- S. Matsumoto, Y. Ikeda, H. Suzuki, M. Ogai and N. Miyoshi, Appl. Catal., B, 2000, 25, 115 CrossRef CAS.
- J. Theis, J. Li, J. Ura, R. Hurley, SAE Technical Paper Series, 2002-01-0733.
- Y. Takahashi, Y. Takeda, N. Kondo, M. Murata, SAE Technical Paper Series, 2004-01-0580.
- Z. Liu and J. A. Anderson, J. Catal., 2004, 228, 243 CrossRef CAS.
- S. Poulston and R. Rajaram, Catal. Today, 2003, 81, 603 CrossRef CAS.
- H. Mahzoul, P. Gilot, J. F. Brilhac and B. Stanmore, Top. Catal., 2001, 16/17, 293 CrossRef.
- L. Limousy, H. Mahzoul, J. F. Brilhac, F. Garin, G. Maire and P. Gilot, Appl. Catal., B, 2002, 42, 237.
- D. H. Kim, J. H. Kwak, J. Szanyi, X. Wang, G. Li, J. C. Hanson and C. H. F. Peden, J. Phys. Chem. C, 2009, 113, 21123 CrossRef CAS.
- G. Graham, H. W. Jen, W. Chun, H. Sun, X. Pan and R. McCabe, Catal. Lett., 2004, 93, 129 CrossRef CAS.
- P. Wynblatt and N. A. Gjostein, in Progress in Solid State Chemistry, ed. J. O. McCaldin and G. Somorjai, Pergamon Press, Oxford, 1975, vol. 9, ch. 2 Search PubMed.
- B. H. Jang, T. H. Yeon, H. S. Han, Y. K. Park and J. E. Yie, Catal. Lett., 2001, 77, 21 CrossRef CAS.
- T. Szailer, J. H. Kwak, D. H. Kim, J. Szanyi, C. Wang and C. H. F. Peden, Catal. Today, 2006, 114, 86 CrossRef CAS.
- N. Fekete, R. Kemmler, D. Voigtlander, B. Krutzsch, E. Zimmer, G. Wenninger, W. Strehlau, J. A. A. van den Tillaart, J. Leyrer, E. S. Lox and W. Muller, SAE, 1997, 970746.
- C. M. L. Scholz, B. H. W. Maes, M. H. J. M. de croon and J. C. Schouten, Appl. Catal., A., 2007, 332, 1 CrossRef CAS.
- Z. Hu, C. Z. Wan, Y. K. Lui, J. Dettling and J. J. Steger, Catal. Today, 1996, 30, 83 CrossRef CAS.
- J. Noh, O. Yang, D. H. Kim and S. I. Woo, Catal. Today, 1999, 53, 575 CrossRef CAS.
- P. Bera, K. C. Patil, V. Jayaram, G. N. Subbanna and M. S. Hegde, J. Catal., 2000, 196, 293 CrossRef CAS.
- S. Salasc, M. Skoglundh and E. Fridell, Appl. Catal., B, 2002, 36, 145 CrossRef CAS.
- S. Roy and M. S. Hegde, Catal. Commun., 2008, 9, 811 CrossRef CAS.
- Y. Su, K. S. Kabin, M. P. Harold and M. D. Amiridis, Appl. Catal., B, 2007, 71, 207 CrossRef CAS.
- J. P. Breen, R. Burch, C. Fontaine-Gautrelet, C. Hardacre and C. Rioche, Appl. Catal., B, 2008, 81, 150 CrossRef CAS.
- H. Abdulhamid, E. Fridell and M. Skoglundh, Appl. Catal., B, 2006, 62, 319 CrossRef CAS.
- A. Amberntsson, E. Fridell and M. Skoglundh, Appl. Catal., B, 2003, 46, 429 CrossRef CAS.
- X. Wang, Y. Yu and H. He, Appl. Catal., B, 2010, 100, 19 CrossRef CAS.
- A. L. Kustov and M. Makkee, Appl. Catal., B, 2009, 88, 263 CrossRef CAS.
- F. Basile, G. Fornasari, A. Gambatesa, M. Livi and A. Vaccari, Catal. Today, 2007, 119, 59 CrossRef CAS.
- T. J. Toops, D. B. Smith, W. S. Epling, J. E. Parks and W. P. Partridge, Appl. Catal., B, 2005, 58, 255 CrossRef CAS.
- R. Büchel, R. Strobel, A. Baiker and S. E. Pratsinis, Top. Catal., 2009, 52, 1799 CrossRef.
- J. Kašpar, P. Fornasiero and M. Graziani, Catal. Today, 1999, 50, 285 CrossRef CAS.
- G. Jacobs, L. Williams, U. Graham, G. A. Thomas, D. E. Sparks and B. H. Davis, Appl. Catal., A., 2003, 252, 107 CrossRef CAS.
- M. A. Peralta, V. G. Milt, L. M. Cornaglia and C. A. Querini, J. Catal., 2006, 242, 118 CrossRef CAS.
- J. H. Kwak, D. H. Kim, J. Szanyi and C. H. F. Peden, Appl. Catal., B, 2008, 84, 545 CrossRef CAS.
- M. Piacentini, M. Maciejewski, T. Bürgi and A. Baiker, Top. Catal., 2004, 30/31, 71 CrossRef.
- R. Strobel, F. Krumeich, S. E. Pratsinis and A. Baiker, J. Catal., 2006, 243, 229 CrossRef CAS.
- E. C. Corbos, X. Courtois, N. Bion, P. Marecot and D. Duprez, Appl. Catal., B, 2008, 80, 62 CrossRef CAS.
- K. Yamamoto, R. Kikuchi, T. Takeguchi and K. Eguchi, J. Catal., 2006, 238, 449 CrossRef CAS.
- H. Imagawa, T. Tanaka, N. Takahashi, S. Matsunaga, A. Suda and H. Shinjoh, J. Catal., 2007, 251, 315 CrossRef CAS.
- H. Imagawa, N. Takahashi, T. Tanaka, S. Matsunaga and H. Shinjoh, Appl. Catal., B, 2009, 92, 23 CrossRef CAS.
- S. Roy, N. V. Vegten and A. Baiker, J. Catal., 2010, 271, 125 CrossRef CAS.
- C. N. Costa and A. M. Efstathiou, J. Phys. Chem. C, 2007, 111, 3010 CrossRef CAS.
- M. Machida, D. Kurogi and T. Kijima, J. Phys. Chem. B, 2003, 107, 196 CrossRef CAS.
- L. F. Liotta, A. Macaluso, G. E. Arena, M. Livi, G. Centi and G. Deganello, Catal. Today, 2002, 75, 439 CrossRef CAS.
- C. H. Kim, G. Qi, K. Dahberg and W. Li, Science, 2010, 327, 1624 CrossRef CAS.
- D. L. Jian, P. X. Gao, W. J. Cai, B. S. Allimi, S. P. Alpay, Y. Ding, Z. L. Wang and C. Brooks, J. Mater. Chem., 2009, 19, 970 RSC.
- W.J. Cai, P. Shimpi, C. Brooks and P.X. Gao, 2010, submitted.
- G. Liu, Y.B. Guo and P.X. Gao, 2011, in preparation.
| This journal is © The Royal Society of Chemistry 2011 |