An investigation of the nature and reactivity of the carbonaceous species deposited on mordenite by reaction with methanol†
L. J.
France
a,
D. C.
Apperley
b,
E. J.
Ditzel
c,
J. S. J.
Hargreaves
*a,
J. P.
Lewicki
d,
J. J.
Liggat
d and
D.
Todd
d
aWestCHEM, School of Chemistry, Joseph Black Building, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: Justin.Hargreaves@glasgow.ac.uk; Fax: +44-141-330-4888
bDepartment of Chemistry, Durham University, Durham DH1 3LE, UK
cHull Research and Technology Centre, BP Chemicals Ltd., Hull HU12 8DS, UK
dWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow G1 1XL, UK
First published on 7th June 2011
Abstract
An investigation of the nature of the carbonaceous species deposited upon mordenite by reaction with methanol has been undertaken. The nature of the species has been shown to be a strong function of both temperature and time on stream. Upon reaction at 300 °C a range of alkyl and aromatic species, consistent with the development of an active hydrocarbon pool, are evident and time on stream studies have shown that these are developed within 5 min. Upon reaction at 500 °C, a narrower range of hydrogen deficient aromatic species is evident. Thermal volatilisation analysis (TVA), not previously applied to the study of coked zeolites, is shown to be complementary to the more commonly applied C analysis, 13C MAS NMR and TGA techniques.
Introduction
The conversion of methanol to hydrocarbons over acidic zeolites and silicoaluminophosphates is well documented, e.g.ref. 1–3. Currently there is a degree of consensus that activity in zeolite and related systems is associated with the presence of an organic co-catalyst, the hydrocarbon pool, which is established under reaction conditions.4–7 Although specific differences in the nature of the “hydrocarbon pool mechanism” are described in the literature, it is generally agreed that the active co-catalyst species involves alkylated aromatic derivatives. To date, whilst mordenite is active for methanol to hydrocarbon reactions, it has been far less well studied than the ZSM-5 or SAPO-34 systems. However, the influence of the acid site density upon the efficacy of mordenite has been recently documented for methanol to olefin reactions and deactivation, and facilitated formation of polyaromatic hydrocarbons, was reported to be enhanced by higher site densities.8 The difference in product distribution between mordenite and SAPO-34, in that the latter material has higher selectivity to lower olefins (C2H4 and C3H6) whilst C4–C5olefins are favoured over mordenite, was related to differences in their pore sizes favouring the formation of alkyl aromatic species containing larger alkyl groups in mordenite than is the case for SAPO-34.8 The coking and aging of mordenite has been compared with other zeolite systems (ZSM-5 and Offretite) for methanol conversion.9 Intracrystalline coking was evident in the mordenite system where a relatively rapid loss of activity was reported. A related system was investigated for the selective transformation of a dimethylether–water mixture into light olefins10 in which intermediate methylbenzenes were alkylated or slowly transformed into coke.In view of the application of mordenite, it is of general interest to identify the carbonaceous species deposited by reaction with methanol at elevated temperatures from both the perspective of the generation of active reaction species (the active hydrocarbon pool) and also possible deactivation pathways. In the present study, our primary focus has been an investigation of the nature of the deposited species rather than the determination of reaction product distributions. To this end, we have employed a wide range of complementary techniques, including thermal volatilisation analysis which, to our knowledge, has not previously been applied to this type of work.
Experimental
Methanol coking was undertaken using a fixed bed microreactor. 0.3 g of ammonium form mordenite (Zeolyst, SiO2/Al2O3 20) housed in a quartz reactor tube was activated under 60 ml min−1 of Ar (BOC 99.999%) at 500 °C for 1 h to yield the proton form. The material was then allowed to cool to reaction temperature. Following this, 0.03 ml min−1methanol (Sigma-Aldrich 99.8%) introduced from a Knauer K501 HPLC pump was vaporised and fed into a 30 ml min−1 Ar stream which was then passed over the sample. Temperature control was achieved using a tube furnace and was regulated using a thermocouple housed in a silica thermowell which was placed centrally within the mordenite bed. All reactor lines were maintained at ca. 150 °C throughout the reaction using heating tape .Nitrogen physisorption isotherms were measured at liquid nitrogen temperature using a Micromeritics Gemini BET machine. Prior to analysis, samples were degassed overnight using helium at 110 °C to remove adsorbed moisture and gases.
Solid state NMR 13C and 1H experiments were carried out using a Varian VNMRS spectrometer equipped with a 9.40 T magnet, operating at 100.56 MHz for 13C and 399.88 MHz for 1H. Spectral referencing was undertaken with respect to neat tetramethylsilane. A 4 mm (rotor o.d.) magic angle spinning probe was used for 13C enriched samples and a 6mm (rotor o.d.) magic angle spinning probe for un-enriched samples.
Cross polarisation spectra of un-enriched samples were obtained using a 1 s delay, a 1 ms contact time, a sample spin rate of 6.8 kHz and ‘TOSS’ spinning sideband suppression. 1700 to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 repetitions were accumulated.
000 repetitions were accumulated.
Direct polarisation spectra of 13C enriched samples were obtained following a 90° pulse (4.0 μs duration) with a 120 s recycle delay, at a spin rate of 14.0 kHz. 352 to 3600 repetitions were accumulated.
C elemental analysis was undertaken using an Exeter analytical CE-440 analyser.
Thermogravimetric analysis was undertaken on a TA instruments Q500 instrument. The temperature was ramped from 20 to 1000 °C at a rate of 10 °C min−1. Typically 8 mg of sample was used for analysis. Experiments were performed in the presence of air (BOC) and argon (BOC 99.999%) atmospheres and the gas flow rate applied was 50 ml min−1.
A schematic of the system used for thermal volatilisation and sub-ambient differential distillation analysis (TVA) is presented in Fig. S1 (ESI†). 100 mg of sample was pumped to vacuum overnight on the TVA system prior to analysis. The purpose was the reduction of background levels of carbon dioxide and water within the system. Initial experiments were undertaken where the samples were heated to 1000 °C at 10 °C min−1 and held isothermally at 1000 °C for twenty minutes with continual cryogenic collection of evolved volatiles. Sub-ambient differential distillation was performed on the collected volatiles by heating the primary cold trap from −195 °C to 30 °C. The volatiles were collected in four secondary cold traps and transferred to IR and GC-MS sample cells. The separated volatiles were analysed via a combination of MS, GC-MS and FTIR spectroscopy.
Results and discussion
The nature of the carbon species deposited on the mordenite samples after 300 min of reaction has been investigated as a function of reaction temperature. XRD analyses (not shown) confirm that the mordenite framework remains intact under all the reaction conditions investigated in this manuscript. Post-reaction C elemental analyses have been performed and the results are shown in Fig. 1. Upon inspection of the figure it can be seen that carbon deposition generally increases as a function of reaction temperature, although a plateau at ca. 16 wt% can be seen in the 350–450 °C temperature interval. The data presented is the mean of duplicate analyses and care has been taken to carefully and thoroughly mix samples prior to analysis to minimise the potential influence of any heterogeneity arising from bed profile effects. Furthermore, care has also been taken in the reactor shutdown procedure in which the methanol feed was stopped prior to cooling the reactor under a flow of argon so as to ensure that further reaction/adsorption of gas-phase methanol did not occur upon cooling. Elemental analyses were also performed within a short interval of time after the samples were generated so as to reduce potential artefacts arising as a result of the loss of volatile species from the materials. Despite these considerations, there was still a large degree of variability apparent in the analyses of the 150 °C and 200 °C generated samples. The deposition of carbon is concomitant with decreasing uptake in the N2 physisorption isotherms. These are presented in Fig. S2 (ESI†). Pre-reaction, the sample exhibits a composite type I—type IV isotherm from the BDDT classification and post-reaction type II isotherms are observed indicating the loss of micro- and meso- porosity from the samples. This is indicative of the fact that some, if not all, of the carbon deposition has occurred in the pore structure of the mordenite samples.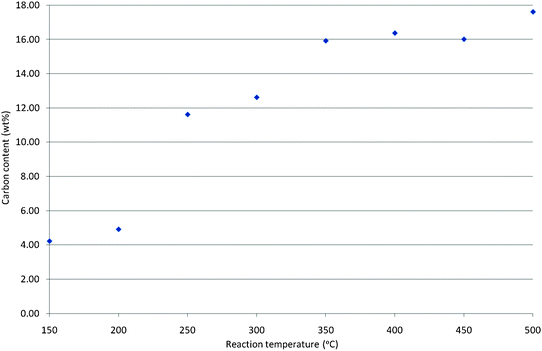 | ||
| Fig. 1 Carbon analyses as function of temperature for mordenite samples coked with methanol for 300 min on stream. | ||
In order to directly probe the nature of the deposited carbon species, 13C MAS NMR spectroscopic studies have been undertaken on samples generated at 150 °C, 250 °C, 300 °C and 500 °C. Spectra which have employed 1H cross-polarisation to compensate for the low abundance of the 13C isotope are presented in Fig. S3 (ESI†). To aid probe tuning, 50wt% talc was added to the 500 °C sample. In the case of coked mordenite samples, resonances in the ranges 10–35 ppm and 120–125 ppm have been attributed to the carbon atoms of alkyl groups and aromatic species respectively.8 No sharp peaks characteristic of adsorbed methanol (reported to occur at 49.2 ppm)8 were observed in any of the samples. Sharp peaks at 16.3 ppm and 132.8 ppm have been interpreted as resulting from hexamethylbenzene.8Hexamethylbenzene deposited without heating on the mordenite used in this study generated resonances at 17.1 ppm and 131.9 ppm. In the case of the spectra presented in Fig. S3, it is apparent that aromatic species are present in all post-reaction materials as evidenced by the broad peaks in the 120–150 ppm range. In both the 150 °C and 500 °C generated samples, these are the only species evident. In the former case, this may be a consequence of the low overall carbon content (ca. 4 wt%), whereas in the latter case it may be more reflective of the presence of graphitic/polyaromatic species. By means of comparison, naphthalene deposited without heating on the mordenite used in this study results in narrow resonances occurring at 125.9 ppm, 128.2 ppm and 133.9 ppm. For samples run under the methanol feedstream at 250 °C and 300 °C, broad features with superimposed sharper resonances are also observed in the 10–35 ppm region implying the presence of alkyl groups. In both cases, features similar to those attributed to hexamethylbenzene are apparent, along with additional signals which can be possibly ascribed to retained hydrocarbons. In relation to the latter species, it is of interest to note that octane deposited without heating on the zeolite used in this study produces corresponding narrow resonances at 13.7 ppm, 22.5 ppm, 28.4 ppm and 38.1 ppm.
To obtain more information from the 13C NMR spectra, samples have been generated by reaction with 25% 13CH3OH enriched feeds at 300 °C and 500 °C respectively. Direct polarisation has been applied and resultant spectra are presented in Fig. 2. As anticipated, the spectra obtained were comparable to those using the 1H cross-polarisation technique. However, particularly in the case of the post-300 °C sample, more features are apparent due to the enhanced signal to noise ratio. It should be noted that the resonance observed at 111 ppm in the spectra of both samples is an artefact which arises from the use of a Teflon rotor cap and which is apparent as a result of the application of direct polarisation rather than 1H cross polarisation. In the case of the 300 °C generated sample, a greater range in the alkyl region is apparent which indicates the presence of various alkylaromatic and/or adsorbed hydrocarbon species. Furthermore, a small peak at ca. 49 ppm, indicative of adsorbed methanol is also present, along with a resonance at 56 ppm which could be attributed to dimethylether and/or methanol adsorbed on silanol species.11,12 The high degree of asymmetry evident in the aromatic feature also suggests that at least two different aromatic species are present. The corresponding spectrum for the sample reacted at 500 °C shows only a broad feature associated with aromatic species. It is therefore clearly evident, as is also apparent from the cross-polarised spectra presented in Fig. S3, that the nature of the carbonaceous residues deposited upon reaction of mordenite with methanol after 300 min reaction time is a strong function of reaction temperature, with aromatic species being evident in all cases and additional hydrocarbon/alkyl species being particularly evident at 250 °C and 300 °C reaction temperature.
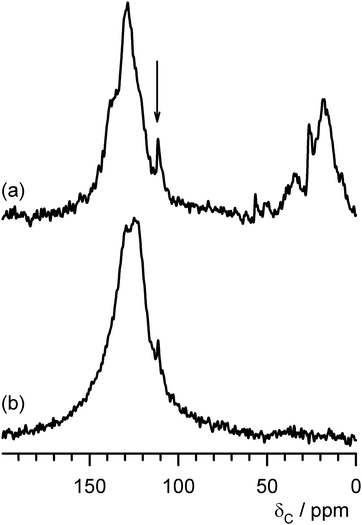 | ||
| Fig. 2 13C DP MAS NMR of mordenite samples coked with 25% 13C enriched methanol for 300 min on stream at (a) 300 °C and (b) 500 °C. The arrow indicates the position of the signal arising from the Teflon rotor caps. | ||
In comparing the 300 °C and 500 °C generated samples, a question concerning the similarity of the aromatic fraction can be raised. For example, the extent to which the 300 °C sample contains the same aromatic fraction as the 500 °C sample, in addition to further alkyl/hydrocarbon fragments, is not clear. However, for the 300 °C sample, the NMR features strongly suggest that at least some of the aromatic fraction is present in the form of alkylaromatic species which is plainly not the case in the 500 °C sample. The presence of an alkylaromatic fraction is consistent with the formation of a hydrocarbon pool which is argued to be necessary for methanol to hydrocarbon reactions on zeolites. In order to further distinguish the 300 °C and 500 °C generated carbonaceous species, thermogravimetric analysis was undertaken in an air atmosphere. The first derivative mass change profiles are presented in Fig. 3 (those for all the samples are given in Fig. S4 in the ESI†). Significant differences between the two samples are apparent. The main mass loss feature occurs over a wider range and at a lower temperature in the case of the 300 °C sample and this is consistent with the presence of a broader spectrum of more reactive species. In the case of both samples, lower temperature loss features are observed (very broad <300 °C for the 500 °C sample and at ca. 400 °C for the 300 °C generated sample.) It is probable that these features relate to simple desorption processes rather than oxidation. In order to probe this effect, TGA under an Ar atmosphere employing the same temperature ramp rate was run and the first derivative profiles, which confirm the occurrence of simple desorption processes, are shown in Fig. 4 (note the difference in scale with respect to Fig. 3.)
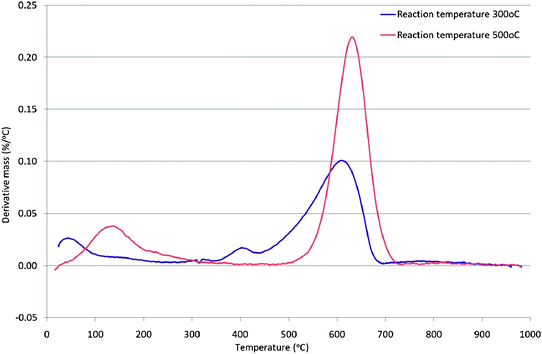 | ||
| Fig. 3 First derivative TGA profile under air of mordenite samples coked with methanol for 300 min on stream at 300 °C and 500 °C. | ||
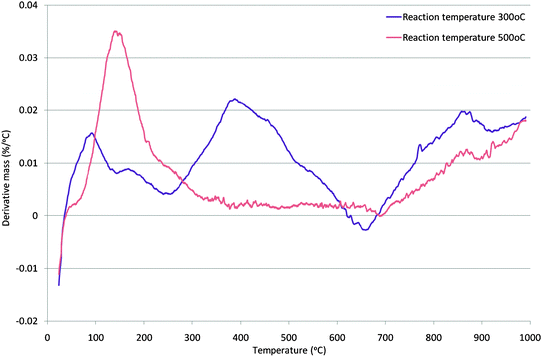 | ||
| Fig. 4 First derivative TGA profile under argon of mordenite samples coked with methanol for 300 min on stream at 300 °C and 500 °C. | ||
To determine in detail the species evolved on heating the coked samples, thermal volatilisation analysis (TVA) has been undertaken on the 300 °C and 500 °C reacted samples. TVA is a technique which is generally applied to polymer degradation studies13–15 and to the best of our knowledge this is the first time it has been applied to the study of coked zeolites. Details of the technique can be found elsewhere13–15—in summary, it involves heating a sample to high temperature under vacuum and performing detailed product analyses on the evolved species which occur in various fractions. The data for the 300 °C treated sample is presented in Fig. 5. In this sample, there is a significant quantity of evolved volatiles and products which are not condensed in a liquid nitrogen trap. Using on-line mass spectrometry, the products evolved have been determined to be methane in the temperature range 300–500 °C, methane and hydrogen from 750–900 °C and hydrogen between 900 and 1000 °C. This data is indicative of the possible occurrence of three different cracking processes, each of which consumes hydrogen generating hydrogen deficient co-products. Methane generation is most likely related to the cracking of methyl groups from the various methylated aromatics and other hydrocarbons. It is also seen to occur over a temperature range which corresponds to the loss of deposited alkyl/hydrocarbon species upon reaction in the presence of methanol. During the TVA temperature ramp, condensable products are trapped in a liquid nitrogen trap and post-analysis is performed by measuring the evolution of products, as determined by mass spectrometry, as a function of increasing trap temperature. A limitation with this is that whilst the spectrum of condensed products is determined, the temperature at which they are evolved is not, since the analysis is undertaken at the end of the TVA experiment. Significant products observed in this fraction are identified in Fig. 5(b). Ethene, carbon dioxide, propene, water, traces of aromatics and butene (not labelled) are observed. The CO2 generated most likely originates from the decomposition of oxygenated products. It should also be borne in mind that it is possible for secondary reactions to occur during the TVA process so, for example, reforming processes could be in operation. A third product fraction analysed was the liquid fraction which comprises those species which condense at ambient temperature. This was analysed by GCMS and the composition is shown in Fig. 5(c). It is apparent that there is a multitude of aromatic products which span toluene to dimethyl naphthalene. Inspection of this fraction shows the possible complexity of the aromatic phase which is not readily apparent from the 13C MAS NMR and TGA studies. Whilst, as stated above, it must be considered that secondary reactions may be occurring, the study of this fraction indicates the significant potential of TVA for investigation of coked catalysts. Caution must be exercised in equating these species to those deposited, the speciation of which would involve dissolution/chemical extraction procedures. It is also apparent that the TVA procedure only removes a fraction of the total carbon present. Around 30% removal of the retained carbon occurs on 300 °C generated samples, as determined by post-TVA carbon analysis.
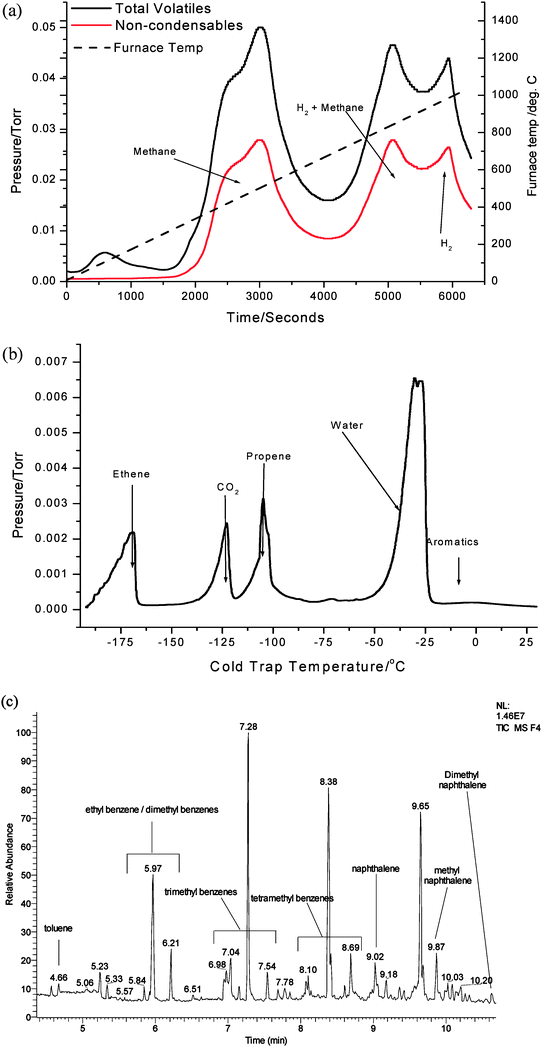 | ||
| Fig. 5 TVA analysis of mordenite sample coked with methanol for 300 min on stream at 300 °C (a) total volatiles and components not condensed in the liquid nitrogen trap, (b) sub-ambient thermal volatilisation analysis of products evolved by warming the liquid nitrogen trap and (c) GCMS analysis of the collected liquid fraction. | ||
The TVA fractions of the 500 °C treated sample contain far fewer components, in accordance with the expectation from the 13C MAS NMR spectroscopic studies. The results are shown in Fig. 6. The volatiles and liquid nitrogen-condensable fraction analysis is shown in Fig. 6(a). In addition to water evolution at lower temperatures, carbon monoxide and methane are observed products between 500 °C and 700 °C and hydrogen is produced between 700 °C and 1000 °C. Compared to the 300 °C generated sample, the cold trap fraction (Fig. 6(b)) is much simpler with only CO2 and H2O observed, which result from background species. An ambient temperature condensable liquid fraction was not formed, which is in stark contrast to the 300 °C treatment and further emphasises the significant difference in the carbonaceous species present upon the materials. Post-TVA carbon analysis demonstrated that ca. 9% removal of carbon retained occurred as a result of the TVA procedure.
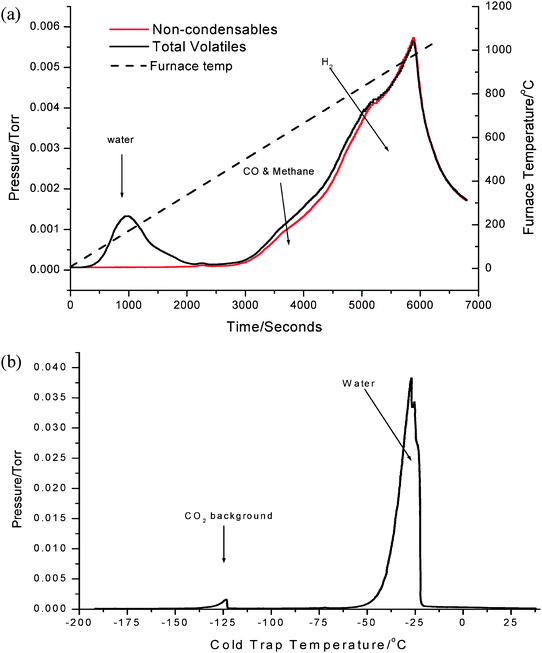 | ||
| Fig. 6 TVA analysis of mordenite sample coked with methanol for 300 min on stream at 500 °C (a) total volatiles and components not condensed in liquid nitrogen trap and (b) sub-ambient thermal volatilisation analysis of products evolved by warming the liquid nitrogen trap. | ||
In addition to temperature, time on stream is a reaction variable which would be expected to significantly influence the nature of the carbonaceous species deposited. On the basis of the results presented above, 300 °C was selected as the reaction temperature. The studies involving shorter times on stream, which would be expected to result in lower levels of carbon deposition, were undertaken with 25% 13CH3OH enriched feeds to facilitate 13C MAS NMR spectroscopic analysis. The post-reaction C elemental analyses, as a function of time on stream, are presented in Fig. 7. Due account was taken of the influence of reactor dead volume and the times quoted correspond directly to the exposure of the catalyst to the methanol feedstream. The precautions outlined previously in relation to Fig. 1 were undertaken. Upon inspection of Fig. 7 it is apparent that, as expected, there is a general increase in the amount of carbon deposited as a function of reaction time. However, this increase is not continuous and there is a possible plateau at ca. 12.0–12.5 wt% between 120 and 420 min. Caution must be exercised in view of the potential limitations of the measurement, but it does imply that the catalyst has reached some form of a constant level of carbonaceous material in this interval before a further increase in carbon deposition is observed. It could be hypothesised that this is associated with the formation, lifetime and changing chemistry of the adsorbed hydrocarbon pool. As anticipated, the N2 physisorption isotherms show a progressive decrease in uptake with time on stream and these are shown in Fig. S5 (ESI†). 13C MAS NMR spectroscopy has been undertaken with the aim of characterising the changing nature of the carbonaceous species present and the resultant spectra are shown in Fig. 8. Studies involving 2 min and 5 min on stream employed 13C enriched feeds and direct polarisation spectra have been recorded, whereas those at 60 min, 180 min, 420 min and 600 min used normal feeds and cross-polarisation from 1H was employed. Inspection of the spectra shows the dramatic changes which occur as a result of time on stream. At the early times on stream, species with narrow line widths, indicating high degrees of mobility are apparent. At 2 min on stream, adsorbed methanol (50 ppm) and a range of alkyl/hydrocarbon 12.9 ppm, 15.8 ppm, 24.4 ppm) species are evident. At least some of these species are associated with alkyl groups attached to aromatic species (the development of an aromatic feature at ca. 140 ppm is apparent—the feature at 111 ppm is the rotor cap, as previously described.) The species at ca. 13 ppm is close to the position for CH3 groups present in the deposited octane mentioned previously (these occur at 13.7 ppm in that case), the species at ca. 16 ppm has been discussed previously in relation to methyl species attached to aromatics, and the species evident at ca. 24 ppm is consistent with alkyl groups attached to aromatic species such as ethylbenzene and xyleneetc. The peak at ca. 60 ppm, which is also evident in the spectrum of the sample has previously been tentatively assigned to dimethylether or methanol bound to silanol groups. After a further three minutes on stream, the apparent changes are dramatic. The aromatic fraction has grown significantly and can be seen to relate to more than one component. The other features observed at 2 min on stream are also still apparent, but in the alkyl region a resonance at ca. 18 ppm, which can be attributed to methyl groups attached to aromatic species, is evident. For the other spectra, the features are less easy to discern, as a result of the 1H cross-polarisation technique being employed. However, it can be seen that the general format of the spectra is similar comprising a distribution of aromatic and alkyl species. Taken together, the time on stream studies indicate that the working carbonaceous state of the catalyst is rapidly established (within 5 min) at 300 °C with the feedstream composition applied in this study.
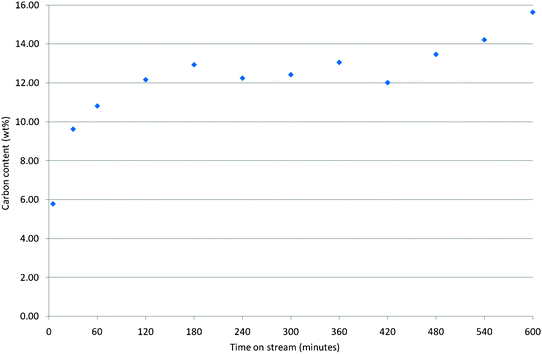 | ||
| Fig. 7 Carbon analyses as function of time on stream for mordenite samples coked with methanol at 300 °C. | ||
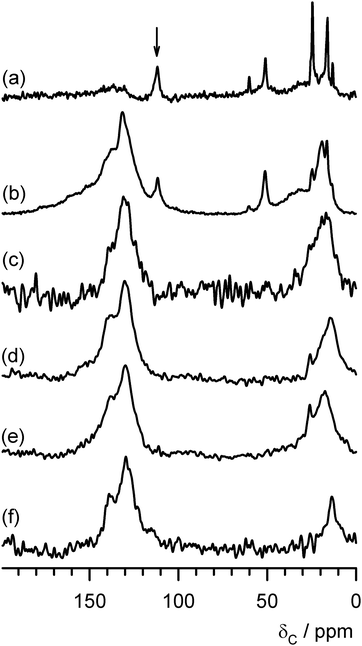 | ||
| Fig. 8 13C MAS NMR spectra of mordenite coked at 300 °C with methanol as a function of time on stream (a) direct polarisation spectrum after 2 min on stream with a 25% 13C enriched feed, (b) direct polarisation spectrum after 5 min on stream with a 25% 13C enriched feed, (c) cross-polarisation (1H) after 60 min on stream, (d) cross-polarisation (1H) after 180 min on stream, (e) cross-polarisation (1H) after 420 min on stream, (f) cross-polarisation (1H) after 600 min on stream. The arrow indicates the position of the signal arising from the Teflon rotor caps. | ||
Conclusions
The nature of the carbonaceous residues deposited upon mordenite by interaction with methanol has been found to be a strong function of temperature and time on stream. At reaction temperatures as low as 150 °C aromatic species are formed as evidenced by 13C MAS NMR spectroscopic studies and at 250 °C and 300 °C, there is a mixture of alkyl and aromatic species present which is consistent with the development of a hydrocarbon pool. At 500 °C, only aromatic species are evident. Consistent with this, TVA analysis demonstrates that a rich mixture of species are removed from 300 °C treated samples as compared to 500 °C upon ramping samples in vacuo. Furthermore, comparisons of the reactivity with air of the carbonaceous species formed after reaction at 300 °C and 500 °C have demonstrated that the former material contains a wider distribution of species which are susceptible to oxidation at lower temperature.Upon investigation of the nature of the carbonaceous species formed at 300 °C as a function of time on stream, the formation of aromatic species is evident after only 2 min reaction time and the 13C MAS NMR spectrum indicates the presence of some alkylaromatic species. After 5 min reaction time, alkylaromatic species appear to be well established. This suggests that the active hydrocarbon pool is established within 5 min in stream under these reaction conditions.
Acknowledgements
The authors are grateful to Mrs Kim Wilson, University of Glasgow, for kindly carrying out the CH analyses. JSJH and LJF would like to acknowledge the generosity of BP Chemicals in the provision of an EPSRC Industrial CASE award. The NMR spectra were recorded at the EPSRC service in Durham.References
- M. Stocker, Microporous Mesoporous Mater., 1999, 29, 3–48 CrossRef CAS.
- J. F. Haw, W. Song, D. M. Marcus and J. B. Nicholas, Acc. Chem. Res., 2003, 36, 317–326 CrossRef CAS.
- T. Mokrani and M. Scurrell, Catal. Rev.–Sci. Eng., 2009, 51, 1–145 CAS.
- I. M. Dahl and S. Kolboe, J. Catal., 1994, 149, 458–464 CrossRef CAS.
- D. M. McCann, D. Lesthaeghe, P. W. Kletnieks, D. R. Guenther, M. J. Hayman, V. van Speybroek. M. Waroquier and J. F. Haw, Angew. Chem., Int. Ed., 2008, 47, 5179–5182 CrossRef CAS.
- M. Bjorgen, S. Akyalcin, U. Olsbye, S. Benard, S. Kolboe and S. Svelle, J. Catal., 2010, 275, 170–180 CrossRef CAS.
- J. F. Haw and D. M. Marcus, Top. Catal., 2005, 34, 41–48 CrossRef CAS.
- J. W. Park, S. J. Kim, M. Seo. S. Y. Kim, Y. Sugi and G. Seo, Appl. Catal., A, 2008, 349, 76–85 CrossRef CAS.
- P. Dejaifve, A. Auroux, P. C. Gravelle, J. C. Vedrine, Z. Gabelica and E. G. Derouane, J. Catal., 1981, 70, 123–136 CrossRef CAS.
- J. M. Fougerit, N. S. Gnep and M. Guisnet, Microporous Mesoporous Mater., 1999, 29, 79–89 CrossRef CAS.
- Y. Jiang, M. Hunger and W. Wang, J. Am. Chem. Soc., 2006, 128, 11679–11692 CrossRef CAS.
- L. J. Carlson, P. K. Isbester and E. J. Munson, Solid State Nucl. Magn. Reson., 2000, 16, 93–102 CrossRef CAS.
- I. C. McNeill, J. Pol. Sci. A1, 1966, 4, 479–2485 Search PubMed.
- I. C. McNeill, L. Ackerman, S. N. Gupta, M. Zulfiqar and S. Zulfiqar, J. Polym. Sci., Polym. Chem. Ed., 1977, 15, 2381–2392 CrossRef CAS.
- J. P. Lewicki, K. Pierichowski, P. T. De la Croix, B. Janowski, D. Todd and J. J. Liggat, Polym. Degrad. Stab., 2010, 95, 1099–1105 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cy00103e |
| This journal is © The Royal Society of Chemistry 2011 |