Design strategies for engineering selectivity in bio-inspired heterogeneous catalysts
David J.
Xuereb
and
Robert
Raja
*
Department of Chemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK. E-mail: R.Raja@soton.ac.uk; Fax: +44 (0)2380 593781; Tel: +44 (0)2380 592144
First published on 11th March 2011
Abstract
In an era where the requirement for greener chemical processes is so exigent, the growing need for developing novel routes to environmentally benign and sustainable catalytic procedures is highly desirable. Heterogenising bio-inspired transition-metal complexes on a diverse range of porous supports provides a viable alternative for achieving some of these goals, particularly in terms of reducing waste and by increasing efficiency and selectivity in industrially significant catalytic processes. Choosing an appropriate spatial-restricting support is vital for facilitating enhancements in rate and stereoselectivity, as this plays a pivotal role in optimising the orientation of desired transition-states through varying confinement effects, by utilising a myriad of pore-window apertures for regulating diffusion of organic molecules. The nature of the active site can also be further attuned by adopting an appropriate encapsulation strategy, which could eventually assist in maximising the hydrophilic/hydrophobic character of the support. The nature of the active site and its involvement in the catalytic process can be characterised by using a wide-range of physico-chemcial spectroscopic techniques, which provide valuable insights for drawing mechanistic relationships, which in turn facilitates structure–property correlations.
 David J. Xuereb | David Xuereb graduated from Cardiff University in 2008. He is studying towards a PhD at the University of Southampton with Dr Robert Raja. His research is primarily focused on the heterogenization of organocatalysts for fine chemical applications, including C–C bond formation and oxidation reactions. Other areas of interest include the utilization of solid oxidants for chemical processes and the catalytic function of metal coordinated amino acid-derivatized frameworks and porous inorganic–organic hybrid materials. |
 Robert Raja | Robert Raja completed his PhD in 1997 and was awarded a Royal Commission for the Exhibition of 1851 Fellowship at the Royal Institution of Great Britain. He worked with Bayer Chemicals, Leverkusen, Germany (2000–2003) as a Research Chemist developing one of the first series of heterogeneous, organometallic chiral catalysts for fine-chemical and pharmaceutical applications. He returned to Cambridge University in 2004 as a Senior Research Associate concentrating on the discovery and design of novel single-site heterogeneous catalysts. In 2006, he was appointed as Reader/Associate Professor in Chemistry at the University of Southampton where he focuses on engineering active sites for industrially significant catalytic transformations, using environmentally benign reagents, energy-renewable feedstocks and sustainable processes. He has won several awards and prizes, the most notable being the Erskine Fellowship (2008) awarded by the University of Canterbury (New Zealand) and the Barrer Award (2005) by the Royal Society of Chemistry. |
Introduction
Incentive for biocatalysts in industry: drawbacks and solutions
Enzymes have been found to catalyse a wide-range of organic transformations with a high-degree of activity and selectivity. The most fundamental transformations from simple oxidations of straight chain alkanes to intricate rearrangements are catalysed with exceptional selectivity. It is this unsurpassed regio-, chemo- and stereoselectivity that has caught the eye of the pharmaceutical industrial giants of the present day. The importance of enantiomerically pure drug syntheses is evident from the growth of the global pharmaceutical market to $808 billion in 2009, at a compound annual growth rate (CAGR) of 9.3% between 1999 and 2009.1 The market is forecast to grow to over $1000 billion in 2014 demonstrating the requirement for highly efficient and selective catalysts to meet the growing demand.Already an increasing number of bio-transformations are carried out on an industrial scale, utilising biocatalytic processes2,3 using primarily fixed-bed immobilised enzyme reactors or batch reactors (Table 1). Many of the products formed are isolated as a particular enantiomer, which reiterates the importance of using enzymes in industry and their notable characteristic for effecting highly stereoselective organic and bio-organic catalytic transformations.
| Product | Enzyme | Production scale (tpy) |
|---|---|---|
| High fructose corn syrup (HFCS) | Glucose isomerase | >1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 000000 |
| Lactose-free milk | Lactase | >100000 |
| Acrylamide | Nitrilase | >10000 |
| Cocoa butter | Lipase | |
| Nicotinamide | Nitrilase | >1000 |
| D-Pantothenic acid | Aldonolactonase | |
| (S)-chloropropionic acid | Lipase | |
| 6-Aminopenillianic acid | Penicillin amidase | |
| 7-Aminocephalosporanic acid | Glutaryl amidase | |
| Aspartame® | Thermolysin | |
| L-Aspartate | Aspatase | |
| D-Phenylglycine | Hydantoinase/carbamoylase | |
| D-p-OH-phenylglycine | Hydantoinase/carbamoylase |
The biggest advantage of enzymes is their supreme selectivity. Enzymes are used to increase chemo- and regio-selectivity; but their best asset is enabling enantiomeric control. In many cases enzymes can achieve enantioselectivies of >99% enantiomeric excess (ee) in an undemanding fashion. Enzymes are also highly active under mild conditions such as temperature, pressure and pH, which makes them easier to work with on a small scale; but are also more appealing on an industrial scale on a cost and efficiency basis. Another benefit is that reactions are preferentially carried out in aqueous media therefore providing a ‘greener’ route. Using water as a solvent meets industrial processing goals such as attaining sustainable development and carrying out manufacturing by more environmentally-benign methods. A further advantage of enzymes is their wide-diversity in terms of the reactions that they are able to catalyse, which means they can be applied to an increasing number of applications on an industrial scale. Progressively, biocatalysts are combined or used in conjunction with chemical catalysts for a network of reactions.4
Over the past few decades, irrespective of the advantages of using biocatalysts, many reservations have been raised focusing on their drawbacks.5 The availability of enzymes for commercial exploitation has been limited due to a lack of in-depth, in situ characterisation as well as proprietary precincts, which leads to a shortfall of desired reactions from specific substrates to targeted products. This restricted substrate specificity is a huge shortcoming because, even though in principle a large breadth of chemical reactions can be catalysed, only certain substrates could be directly employed due to the size-selection preferences of the enzyme. Enzymes also require co-substrates and co-factors which lead to cost considerations, but of greater significance is the enzymes lack of stability in varying conditions and desired media. In most cases, harsher temperatures, pressures and pH are required for maximising efficiencies and yields leading to narrow operation windows within specific reactions. Given the above, the stability of the enzyme is rather limited and often leads to their deactivation, via bimolecular pathways, which is a major cause for concern in downstream processing. Another drawback is development cycles are too long for new industrial biocatalysts. A main reason for this is a deficient knowledge base of biotechnology and, compared to patent lifetimes around the same length, such durations can easily be deemed too long for many industrial applications. Compared with other types of alternatives, for example, homogeneous and heterogeneous catalysts, the main advantage of enzymes as catalysts is their unsurpassed selectivity, which is offset, to a great degree, by their lack of stability (Table 2). Immobilising an entire enzyme can be thought of as an arduous task, but by mimicking the catalytic function of its active site, stable catalysts that are amenable for industrial processing can be developed. Metalloenzyme mimics have long held the fascination of synthetic chemists and both structural and functional enzyme mimics have been rationally developed through the interaction of transition-metal complexes containing suitable amino acid residues. Developing strategies for anchoring these active entities on stable inorganic frameworks leads to the creation of highly active and selective catalysts that can facilitate highly stereoselective transformations for the production of fine-chemicals. Furthermore, through the immobilisation of a functional mimic of the active site in an enzyme, problems associated with substrate specificity may also be overcome. This would also aid the recovery and reusability of the catalyst and make it amenable for continuous processing.
Zeolites and mesoporous silicas are suitable hosts for anchoring and encapsulating single-site bio-derived catalysts because of their large surface areas and robust composition. The different routes used for immobilisation such as in situ encapsulation or post-synthetic modification of the framework can alter the nature of the active catalyst.
In this article, we wish to highlight some of the methodologies and strategies for heterogenising bio-inspired catalysts on different solid supports, consequently providing a measure of how successful each framework type is for catalysis and what applications can be envisaged in the future from these materials. However, it is important to understand the function and composition of the metalloenzyme, in order to devise a suitable strategy for encapsulation or immobilisation of the active site.
Devising active sites analogous to metalloenzymes
A metalloenzyme is a lengthy protein that contains a small metal complex in the active site that is coordinated by amino acids from the protein scaffold. The amino acids stabilise and isolate the metal active-centre providing a specific binding pocket for particular substrates to attach (Fig. 1).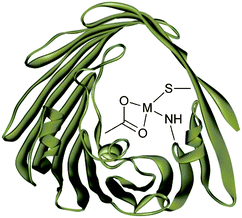 | ||
| Fig. 1 Schematic of an active site within a metalloenzyme. | ||
Proteins coordinate to metal ions by dative bonds formed through nitrogen, oxygen and sulfur atoms that make up the enzyme backbone. Amino, amidato, amido, carbonyl and carboxylate ligands can be located at the C- or N-termini of the peptide chain, within the chain itself (except amino and carboxylate) and in side-chains.6 The peptide chain is made up of small building blocks of amino acids, which are composed of an acidic carboxyl group (–COOH), a basic amino group (–NH2), a hydrogen atom and a characteristic side chain (–R). Amino acids are good metal-complexing agents because of the electron donating effect of the substituents. Forming chelate rings through the amino and carboxylate groups is a common feature of amino acid coordination spheres, due to dissociation of the acidic proton to form a bidentate N, O-donor. Side-chains can also coordinate to the metal centre, e.g. amino group of lysine and arginine, imidazole group of histidine, mercapto group of methionine or carboxylato groups of aspartate and glutamate7 (see Fig. 2).
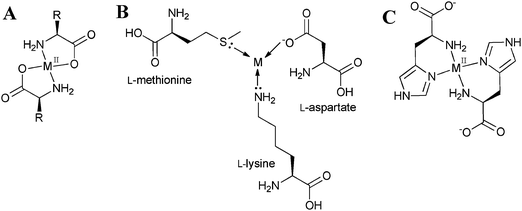 | ||
| Fig. 2 (A) Typical coordination sphere of an amino acid containing transition-metal complex. (B) Examples of amino acids that can also coordinate through the side chains. (C) Possible coordination sphere of a bis-complex of histidine coordinated through the imidazole group. | ||
Iron and copper ions frequently occur as metal centres in biological oxidation systems and act as proficient catalysts, the activity and selectivity of which can be attributed to their inherent electronic properties and accessible redox capabilities. Metalloenzymes containing iron active sites, which can be very diverse due to electronic and geometric differences arising from ligand environments, consist of a substantial group of dioxygen activating enzymes that show significant promise for functionalising a wide range of organic substrates with high efficiency and selectivity.8 Heme-enzymes play critical roles in the biological hydroxylation of saturated carbon–hydrogen bonds, epoxidation of olefins, heteroatom oxidation, oxidation of aromatics and dealkylations.9 Mononuclear non-heme enzymes catalyse oxidative transformations either by FeIII-sites activating substrates for reactions with dioxygen (e.g. intradiol dioxygenases and lipoxygenases) or through FeII-sites activating oxygen by direct binding to O2, resulting in iron–oxygen intermediates that react with the substrate10 (e.g. Rieske dioxygenases and extradiol dioxygenases) (Fig. 3). Binuclear iron enzymes are also useful O2 activation compounds; for example, soluble methane monooxygenase (sMMO), a di-iron containing enzyme, has a carboxylate-bridged dinuclear iron centre11 that is capable of oxidizing aromatics and methane (the most inert hydrocarbon with a C–H bond energy of 104 kcal mol−1).12
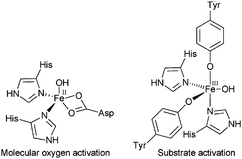 | ||
| Fig. 3 Mono-nuclear enzyme active sites. FeII sites directly bind O2 for substrate insertion, while high spin FeIII sites activate the substrate for direct reaction with O2. | ||
Furthermore, copper metal centres play a major role in biological dioxygen activation systems13 (Fig. 4). Single copper-ion-containing enzymes {e.g. particulate methane monooxygenases (pMMO)} are effective in highly demanding hydroxylation reactions and oxidations of inert hydrocarbons such as methane.14 Mononuclear copper enzymes {e.g. galactose oxidase (GO)} also play an important role in the two-electron reduction of O2 to peroxide coupled with oxidation of organic molecules, such as oxidation of primary alcohols to aldehydes, which in turn can serve as substrates to yield carboxylic acids.15 Dicopper species that contain a binuclear copper centre as the active site (e.g. tyrosinase and catechol oxidase) can selectively oxidize phenols to catechols and further to quinone.16,17
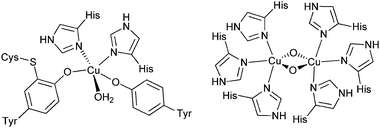 | ||
| Fig. 4 Mono-nuclear and bi-nuclear examples of different copper sites in enzymes. | ||
Rationale for heterogenised bio-inspired catalysts
Understanding the nature and the coordination sphere of the active metal centre in a metalloenzyme provides valuable insights for developing design strategies for heterogenising bio-inspired catalysts for chemical transformations. Structural and functional enzyme mimics have been developed through the assembly of transition-metal complexes with natural amino acids,18 designed organic ligands,19 or by the use of artificial proteins.20 Amino acid–transition-metal complexes can perform as isolated active sites when encapsulated in inorganic host materials such as aluminosilicates or covalently bonded to mesoporous silica frameworks and polymers. These supports act as substitutes to the protein scaffold and create a spatial environment similar to that of the natural enzymes, therefore presenting an alternative to conventional biological and chemical processes. The replica heterogeneous catalysts are more versatile to pH changes, exhibit better thermal stability and display superior solvent and substrate versatility compared to biological metalloenzymes. The catalyst may also be recovered and recycled numerous times due to the heterogeneous nature, which is desirable from an industrial perspective.Catalyst stability, activity and selectivity may be enhanced through chemical modifications of the active site by altering immobilisation procedures or by changing the support type. Somorjai and Park21 have recently highlighted the seven molecular components that influence the selectivity of heterogeneously-catalysed reactions and have elegantly demonstrated how it would be possible to correlate these molecular factors for the development of hybrid systems; which take advantage of and facilitate enzyme selectivity in a heterogeneous medium. One of the most important properties of the support is the degree of the hydrophilicity or hydrophobicity, which can play an important role in determining the catalytic outcome. Corma and co-workers22,23 have shown that the hydrophobic–hydrophilic properties of a heterogeneous catalyst plays a determinant role in enhancing both the activity and selectivity of a reaction, due to different polarities and transition-states that the reactant can adopt. Each framework type has its own inherent degree of water attraction but can also be altered by post-treatments. Choice of support is based upon consideration of experimental conditions of intended reaction, with each methodology of heterogenisation having explicit benefits most notably in selectivity.
Encapsulation strategies encompassing channels and cages in microporous solids
Zeolites are porous crystalline aluminosilicates with three-dimensionally connected channels. The primary building units are single TO4 tetrahedra, where T is an aluminium or silicon atom. Each T atom is tetrahedrally bonded to four oxygen atoms forming a bridge to neighbouring T atoms. Zeolites have the ability to act as catalysts for chemical reactions which take place within their internal cavities. They are thermally stable, chemically robust and easy to separate from reaction mixtures by filtration or centrifugation. Zeolites can incorporate guest molecules, more specifically metal-complexes, within their structure, due to the availability of empty channels and cages, which provide a stereochemicaly demanding void space for catalysis to occur. A metal cation can be readily exchanged into the zeolite to balance the overall net negative charge on the framework.24 The ionic interactions between the support and the metal-ion are strong enough to hold the guest metal in place. Using this rationale, careful combinations of metal complexes immobilised as active sites throughout an inorganic matrix can result in catalysts that can mimic the functional aspects of enzymes.25,26 Aluminosilicates are ideal supports and by astute choice of the metal complex, novel single-site heterogeneous catalysts can be created.27,28 Zeolite encapsulated active centres offer many advantages, such as the organized pores and channels, which can impede or assist diffusion on a size selective basis. In these architectures, zeolites serve as a substitute for the protein scaffold of natural enzymes and provide a controlled steric environment, where the catalysis proceeds.29,30 These heterogeneous catalysts are resistant to harsh reaction conditions, stable at high temperatures, facilitate separation of products from reactants and aid catalyst recyclability. Romanovsky and co-workers31–33 first reported the synthesis of metallophthalocyanines inside the supercages on the zeolite Na–Y in 1977. Due to similarities in the structure and chemical properties between metallophthalocyanines and metalloporphyrins, the latter34,35 have often been used as substitutes, for mimicking the catalytic function of cytochrome P450 enzymes. Researchers from DuPont36–39 were among the early pioneers to exploit the chemistry of natural enzymes, adopt strategies for the encapsulation of transition-metal complexes within porous hosts, and apply them to the rational design of zeolite-based catalysts, both as mimics of natural enzymes and for processes of industrial interest. In an attempt to prepare heterogeneous analogues of haemoglobin and myoglobin,40,41 which reversibly bind to dioxygen, cobalt(II) salen complexes were encapsulated within the supercages of zeolite-Y.36,37 Such encapsulated complexes formed adducts with dioxygen, which were more stable than those formed by the same complexes in free solution.36 The isolation of the active centres within the zeolitic cavities suppresses dimerisation reactions that lead to the formation of the inactive μ-oxo and peroxo-complexes of cobalt.38,39 In a similar attempt to prepare analogues of cyctochrome P-450, Fe(II)–Pd(0) particles were encapsulated in zeolite A and these exhibited remarkable substrate and regioselectivity in the oxidation of unactivated alkanes with molecular oxygen.38,42 Although these systems enabled selectivity control and inhibition of auto-oxidation, they displayed very little, in terms of mechanistic analogy, with the actual enzymatic process. Jacobs et al.43 reported a composite catalytic system that achieves realistic mimicry of cytochrome P-450, by incorporating iron phthalocyanine complexes in crystals of zeolite Y, which were in turn, embedded in a polydimethyl siloxane membrane. The polymer acts as a mimic of the phospholipid membrane where the active centre of cyctochrome P-450 is located and this system oxidises cyclohexane at room temperature with rates comparable to that of the natural enzyme. These workers had established earlier44–46 that a range of metal complexes could be successfully immobilised within the internal cavities of a zeolitic framework and demonstrated the viability of a supramolecular system, where complexes of manganese(II) with bipyridine, when encapsulated in the supercages of zeolites X and Y, have the potential to oxidise alkanes and alkenes under mild conditions.47 Since the early work of the DuPont scientists,36–39 there have been numerous efforts aimed at the activation of molecular oxygen by metal complexes,48,49 and extensive studies have been reported for the selective oxidation of organic compounds using a range of oxygen sources.50–53A number of routes54 are known for preparation of metal complexes inside zeolites (Fig. 5): ion exchange,55 flexible ligand approach,56 zeolite synthesis,57 ligand adsorption58 and ship-in-bottle methods.59
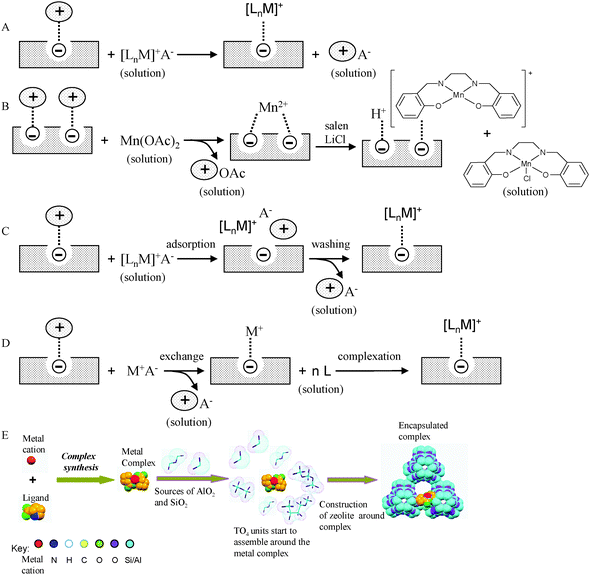 | ||
| Fig. 5 Methods for encapsulating metal complexes within zeolites: Ion exchange (A), flexible ligand (B), ligand adsorption (C), ‘ship in a bottle’ (D) and zeolite synthesis methods (E). | ||
The ship-in-bottle method involves assembling the final active metal complex inside the pores of the host by reacting smaller precursors to afford the larger complex that becomes sterically entrapped inside the cages. This route was employed for encapsulating metal–phthalocyanine or metal–porphyrin complexes within the zeolite.59
The ion-exchange strategy, for example, involves the exposure of a sodium-ion charge balanced zeolite to a solution containing other cations, facilitating an exchange of the sodium ions. This method has been used for encapsulating metal–amino acid complexes inside a zeolite structure—an example of this is entrapped Cu–histidine complex60 (Fig. 6). The strategy initially involves ion-exchanging a sodium-enriched zeolite Y (Na–Y) with a solution of Cu–histidine at pH 7.3. The capability of the copper complex to undergo ion-exchange is dependant on its charge and stability, which are both influenced by the pH of the reaction. At low pH values of around 2, less than 1% of the Cu2+ is coordinated to the histidine ligand. On increasing the pH to 3, Cu2+ forms mono-histidine complexes and within a pH range 6–10, bis complexes are formed. Bis complexes are formed by coordination of four nitrogen atoms from the imidazole and the amine group of each molecule, as well as the carboxylate oxygens that can also coordinate to the metal. In solution, two histidine ligands coordinate to the Cu2+ ion in a square-planar geometry. However, in a confined space in the cages of the zeolite, encapsulated by the ion-exchange method, a framework oxygen atom replaces one of the histidine ligands60 (Fig. 6).
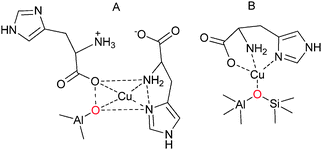 | ||
| Fig. 6 A combination of XRF, UV/Vis/NIR, ESR, EXAFS and XANES, IR and Raman spectroscopy is used to elucidate the composition of Cu–histidine complex encapsulated within zeolite Y.60 Based on one form of a proposed structure by Baute et al.154 (A) a refined complex is determined as one histidine ligand coordinating in a tridentate facial-like manner and the fourth position occupied by a framework oxygen atom from a Brønsted acid site (in red) (B). | ||
The zeolite synthesis method follows a kind of templated synthesis approach. Preformed transition-metal complexes are included in the zeolite synthesis mixture and the zeolite is formed around the metal complex, using it as a sort of template, and thereby encapsulating it.57 Metal phthalocyanines, porphyrins and amino acids provide examples of such entrapped catalysts (Fig. 7). The flexible-ligand method involves the synthesis of metal complexes in the zeolite cavity by coordination of the ligand with the exchanged metal cations. Complexes with previously exchanged metal ions are able to diffuse freely through the zeolite pores. This approach is well suited for the encapsulation of metal–salen complexes, as the salen ligand offers the flexibility to move throughout the channels.56 However, such intrazeolite complexes are physically-trapped (size-inclusion) inside the zeolitic pores and are not necessarily, anchored to the surface. Furthermore, this method poses the added complications of not having a well-defined active centre, as contamination by uncomplexed or partially-complexed metal ions as well as the presence of free ligands cannot be ruled out. Balkus Jr., Bedioui and their colleagues devised a method61 for synthesising zeolite Na–X around cobalt(II) and copper(II) hexadecafluorophthalocyanines using the zeolite synthesis method shown in Fig. 5. They introduced electron-withdrawing halogen substituents in the phthalocyanine ligand, with a view to enhancing the oxidative stability of the metal complex, which in turn, dramatically improved the catalytic potential of the encapsulated catalyst.61–63
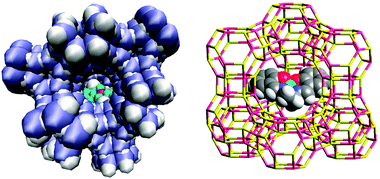 | ||
| Fig. 7 Visual representation of amino acid coordinated transition metal complex (Fe3+–proline) encapsulated within zeolite X (left) and copper-phthalocyanine entrapped within a zeolite supercage (right). | ||
Transition-metal complexes containing amino acids immobilized within zeolitic supports generate isolated active centres that function as effective selective oxidation catalysts using benign oxidants such as air and display high turnovers and selectivity in oxidation reactions.55,59,60,64,65 Weckhuysen et al.64 pioneered the immobilisation of Cu(histidine) complexes within the cages of zeolite Y using an ion-exchange strategy. Using a combination of spectroscopic tools, they demonstrated that, in the case of [Cu(His)2]+, the two histidine ligands are coordinated to the Cu2+ in the coordination plane by the amido N and the imidazole N, while the carboxylate group coordinates in an apical plane. However, an equilibrium exists between the NNNN (histamine-like coordination) and NNNO (one histidine is bound histamine-like and the other glycine-like) bonding in the coordination plane of the bis(histidine) complexes, as the carboxylate oxygen of one of the histidine ligands can partially replace one of the imidazole nitrogens.55 The type of bonding though, between an amino acid and Cu2+ is similar for all α-amino acids with the exception of histidine.66 The general coordination for a Cu complex with two α-amino acids is the binding of both the amino acid by an amino nitrogen and a carboxyl oxygen; i.e. a NNOO coordination or a glycine-like bonding. Recently, Weckhuysen's group have elegantly demonstrated67,68 the isolation and stabilisation of a mononuclear copper(II) complex with a 3,3-bis(1-methylimidazol-2-yl)propionate (abbreviated: MIm2Pr) ligand (see Fig. 8), using the zeolite-encapsulation strategy that has been described earlier,64 for the room temperature oxidation of benzyl alcohol to benzaldehyde. On immobilisation, the authors observe a neutral bis-ligand species (Fig. 8, right), which is identical to the homogeneous analogue, but can easily be washed out from the zeolite structure. This further facilities access of the substrate molecule (benzyl alcohol) to the immobilised, mononuclear, monoligand [Cu(MIm2Pr)] charged species (Fig. 8, left), which displays a higher catalytic potential than its homogeneous analogue, Cu(MIm2Pr)2.
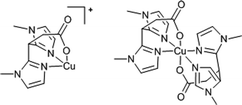 | ||
| Fig. 8 Charged 1∶1 Cu-complex which can coordinate to the zeolite framework oxygen atoms (left) and the neutral homogeneous 1∶2 complex coordinated through two NNO-ligands (right).68 | ||
This approach has been rationally extended to the adsorption and intercalation of metal-substituted amino acid complexes in clays69–78 and further to the immobilisation amino-acid-derived ligands and related complexes in polymer matrices.87,88,77–84 Cu(II)–lysine and Cu–(histidine) complexes intercalated between layers of saponite clays by cation exchange display promising catalytic potential in several oxidation reactions using tert-butylhydroperoxide as the oxidant.69 Hydrotalcite-functionalised bio-inspired Mo(II) complexes have been proven to be more stable than their homogeneous counterparts in olefin epoxidation70 whilst intercalation of chiral catalytic centres (such as L-proline) in anionic clays has resulted high enantiomeric excess (94%) in the asymmetric aldol reaction of benzaldehyde and acetone.71 The clay structure and composition influence the activation of the reactant and its suspension stability provides ease of access of the substrate to the imbedded catalytic-active sites. The catalytic efficiency of these materials has been attributed to the peptide bond formation which derives from the above structure-composition effect.72 Spectroscopy and computational modelling have helped elucidate the nature of the active site, when bonding to a range of bio-monomers (purine, adenine, guanine, hypoxanthine, etc.) and its coordination geometry, which have helped gain a deeper insight into the interplay of bonds, conformations and configurations between the active complexes and their hosts.73–76
Oxovanadium(IV) and copper(II) complexes of amino acid derived tridentate ligands (L-alanine and L-isoleucine) when anchored to polymer matrices (e.g. chloromethylated polystyrene) display higher activities and turnovers, compared to the non-polymer-bound analogues, in the oxidation p-chlorotoluene and epoxidation of olefins.79 Chiral copper(II) coordination polymers have proved effective in the enantioselective oxidative coupling of 2-naphthols, Baeyer–Villiger oxidations, Kharasch–Sosnovsky reactions and in the kinetic resolution of secondary alcohols by asymmetric acylation with ee's reaching up to 90%.80,81 Anchoring Cu(II)– and Mn(II)–valine onto cross-linked styrene–divinylbenzene results in stable, recyclable catalysts that are highly active for a range of industrially-significant reactions, including the epoxidation of olefins (styrene, cis-cyclooctene, norbornylene and cyclohexene) and oxidation of alcohols (cyclohexanol and benzyl alcohol).82,83 In particular, the use of in situ vibrational spectroscopic techniques showed that amino acid complexes that were covalently anchored to polymeric supports displayed a greater stability, with retention of higher activities, for electron transfer reactions.85,86 The merits of immobilising single-site homogeneous catalysts has paved the way for exploring the interface, as well as combining the advantages of homogeneous as well as heterogeneous catalysts, for potential industrial exploitation.89–91
Heterogeneous Fe3+–proline complexes, prepared by the zeolite-synthesis and flexible ligand methods (described earlier) have shown65 promising potential in the oxidation of cyclohexane and benzyl alcohol (Scheme 1). The oxidation of cyclohexane to cyclohexanone is an important chemical reaction, because cyclohexanone is a vital precursor in the manufacture of ε-caprolactam (for nylon-6) and adipic acid (for nylon 6,6).
 | ||
| Scheme 1 Cyclohexane oxidation to cyclohexanol and cyclohexanone (top). Benzyl alcohol oxidation to benzaldehyde (bottom). | ||
Interesting changes in activity and selectivity were observed by simply changing the oxidant (Fig. 9). The encapsulated Fe3+–proline selectivity oxidises the hydrocarbon to cyclohexanone (99.5 mol%) with O2, whilst using tert-butylhydroperoxide (TBHP) as the oxidant produced preferentially cyclohexanol. In both cases far more conversion was achieved with the heterogeneous analogues due to the generation of isolated active sites in the encapsulation procedure.57
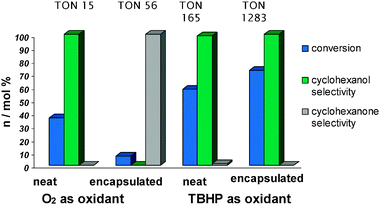 | ||
| Fig. 9 Catalytic results for the cyclohexane oxidation with zeolite encapsulated Fe3+–proline complex. | ||
In order to ascertain whether the Fe3+–proline complex was encapsulated within the supercages of the zeolite, gas adsorption studies and surface area measurements65 were carried out (Fig. 10). The distinct decrease in the BET surface area and micropore volume shows the complex resides within the cages of the aluminosilicate, as opposed to being adsorbed on the external surface. Powder XRD studies revealed the structural integrity of the zeolite was intact after the encapsulation process and further confirmed that there are no phase impurities or defect structures in the framework and that no additional peaks due to neat complex on the outer surface were detected.65
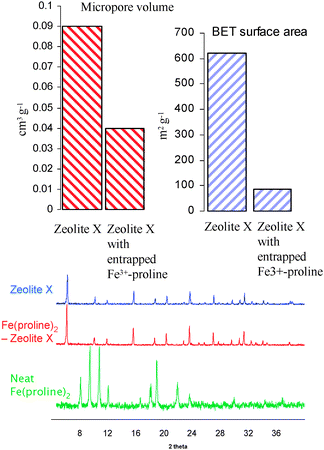 | ||
| Fig. 10 N2 physisorption isotherms and pXRD patterns for zeolite with and without encapsulated complex. | ||
Spectroscopic data of the encapsulated catalysts offer more information about the nature of the active site such as the coordination sphere, complex structure and metal oxidation state. This is not limited to just zeolite models, this can be taken in general terms whereby these techniques are used in characterizing the active site on other systems too. Although in this instance, using the example of an iron–proline complex entrapped within zeolite X, the spectroscopic information can provide detailed information on the catalytic sites that in future can be used to elucidate a plausible mechanistic pathway. In this case the FTIR spectra (Table 3) of neat and encapsulated Fe3+–proline confirm that the carboxyl group of the ligand is deprotonated due to the strong band at 1662 cm−1 resulting in an antisymmetric (νas COO−) and a symmetric stretching mode (νs COO−), around 1650 and 1400 cm−1, respectively. The absence of a signal around 1750 cm−1 (which is typical of the COOH species) further confirms that the carboxyl group is involved in binding to the metal ion. The absence of the δNH2+ vibration at 1550 cm−1 is evident, which substantiates that the amino group is not protonated and most likely coordinates to the iron centre. The νN–H bands of neutral N–H groups are observed at 3285 cm−1 for zeolite entrapped Fe3+–proline and at 3210 cm−1 for neat Fe3+–proline, although in Fe3+–proline–X, the bands are readily superimposed into a broad signal between 3700 and 2800 cm−1, which can correspond to hydrogen bonding between proline and the zeolite inner surface.
DR UV-Vis spectroscopy reveals the nature of Fe species confined within zeolite-X.65 The heterogeneous Fe3+–proline-based catalyst produces bands at 37600 cm−1 and 45400 cm−1 which are due to ligand to metal charge transitions (LMCT) involving iron in tetrahedral or octahedral geometries. The band at 45400 cm−1 can be assigned to highly distributed and segregated Fe3+ sites92 therefore providing evidence for the complex occupying isolated single-sites throughout the inorganic motif.
Electron Paramagnetic Resonance (EPR) spectrum65 of the neat iron complex consists of a broad resonance at g = 2, which is a feature of strongly interacting Fe3+ ions. The zeolite encapsulated complex also reveals this signal, which is highly likely due to interacting Fe3+–proline centres located near the extremities of the cages of zeolite X. More importantly this spectrum produces another signal at g = 4.26 which is indicative of isolated iron(III) ions and further validates the notion of the complexes being well-dispersed throughout the material. The above characterisation evidence further substantiates that the complexes are present in the mono-nuclear form, well-separated from others by the cavities and channels of the zeolitic host, giving rise to isolated single-site heterogeneous catalysts, that can be utilized in industrially significant reactions.54,57,65 The impressive selectivities, activity and reusability achieved through this encapsulation method augur well for future sustainable applications.
Covalent anchoring strategies using mesoporous silica
Porous inorganic solids can be used for anchoring well-separated catalytic centres in order to replicate the behaviour of homogeneous and enzymatic catalysts. This strategy can be specifically adopted for larger channel architectures, such as hydrophilic mesoporous silicas,93 so as to accommodate a broader scope of substrates. A major drawback of zeolites as supports is the microporous nature of the pore architecture, which regulates the diffusion of reactant molecules to, and product molecules from, the active site that is contained within a small pore or channel. This is not the case with mesoporous silica frameworks, because the pore diameters can be in excess of 500 Å. In fact, with mesoporous silicas, a range of pore diameters are potentially available (from 25–250 Å), hence judicious choice of pore aperture imparts desired spatial constraints or freedom with respect to the mechanistic pathway. A notable example is MCM-41 (M41S) which has ordered pores of hexagonally packed one-dimensional channels comprising of a very large surface area.94 The silanol groups at the surface of the channels can be organofunctionalised and then used to host organometallic and transition-metal-containing catalysts by coordinating to metal centres.95 Introduction of functional groups via silylation is the most reliable method to prepare inorganic–organic hybrid materials. The hydrophilicity of the silica may also be altered by decreasing the amount of silanol groups on the surface by grafting less polar organic compounds therefore enhancing surface hydrophobicity, which in turn can influence catalytic activity.96 RSi(OEt)3 and RSiCl3 are the most popular silylating agents,97 which can be facilely used to functional the surface of the mesoporous silica.As with most solid supports, practical advantages include high mechanical and thermal stability and straightforward separation of the catalyst from the reaction mixture, in order to be re-used without degrading. In addition, silica frameworks also do not swell on exposure to solvent or reactants and are readily available in a range of pore diameters.
It was originally hoped that deliberate restrictions of space inside the pore, particularly in the vicinity of a tethered active site, would induce chirality in a target molecule (Fig. 11). The approaching prochiral compound would experience interactions with the chiral ligands and the metal centre but also more significantly with the inner wall. The sum of these interactions would be approximately the same order of the two transition-states that determine enantiomeric excess; therefore with the additional interaction on the pore wall potentially affecting the stereoselective outcome.
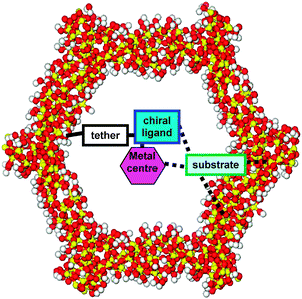 | ||
| Fig. 11 Substrate interactions that govern selectivity when a transition-metal complex is covalently anchored within a mesopore. | ||
This has been suitably demonstrated in the allylic amination of the Trost-Tsuji reaction (Scheme 2) by making a direct comparison with 3 different catalysts (1 homogeneous and 2 heterogeneous, Fig. 12); containing the same (S)-1-[(R)-1,2′-bis(diphenylphosphino)ferrocenyl]ethyl-N,N′-dimethylethylenediamine palladium dichloride active centre.98,99 The homogeneous catalyst comprised of the active metal complex attached to a soluble silsesquioxane100 moiety. One heterogeneous analogue was covalently attached to nonporous silica, and the other to mesoporous silica with pores diameters in the range of ca. 30 Å. Results show (Table 4) that the homogeneous catalyst forms the straight chain product almost exclusively and this is mirrored by a very similar outcome in the case of the unconfined heterogeneous catalyst. On the other hand, the catalyst tethered in the mesopore gives an equal amount of the regioisomers, but more significantly exhibits an ee of 95% of the branched product, which is far superior to the ee of 43% of the diminutive yield of the same product from the unconfined solid catalyst. These results indicate that there are no spatial restrictions in the uninhibited catalysts that would preferentially produce branched isomers and consequently give impressive ee's.
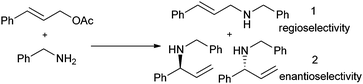 | ||
| Scheme 2 Allylic amination of cinnamyl acetate with benzylamine (Trost-Tsuji reaction). | ||
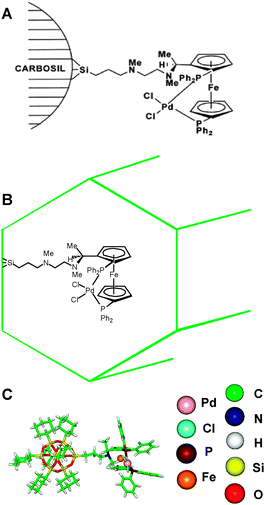 | ||
| Fig. 12 Depiction of the dppf–diamine palladium dichloride catalyst attached to nonporous silica (A), spatially confined mesoporous silica, MCM-41 (B) and to a soluble silsesquioxane moiety (C).95 | ||
| Form of Pd catalyst | Conv. (%) | 1 (%) | 2 (%) | ee (%) |
|---|---|---|---|---|
| Homogeneous | 76 | 99+ | — | — |
| Tethered-non-porous | 98 | 98 | 2 | 43 |
| Tethered-mesoporous | 99+ | 50 | 50 | 95 |
Further proof of this concept was observed in the one step conversion of ethyl nicotinate to ethyl nipecotinate using the same catalysts.101 Once again the confined form gave conversions far superior to that of the non-confined analogue with an order of magnitude improvement in ee. The soluble catalyst attached to the silsesquioxane produced a racemate, thereby proving the chiral advantage of creating a hindered active site that is immobilised within the channels of the framework.
This rationale can promote the cost effectiveness of using anchored inexpensive chiral ligands rather than more extravagant auxiliaries as in homogeneous catalysts. Greater enantioselectivities can be attained using the restricted conditions in the concave pores of a mesoporous silica because the limiting dimensions hinder the possible orientations of bulky reactants so that interactions with the inner walls dictate stereospecificity. This theory is established further in the asymmetric hydrogenation of E-α-phenylcinnimic acid and methylbenzoylformate102,103 (Scheme 3). RhI(COD) metal centres (where COD: 1,5-cyclooctadiene) are covalently tethered to porous silica with inexpensive ligands, such as (S)-(−)-2-aminomethyl-1-ethylpyrrolidine (AEP). The chiral complex attached to concave surfaces produces better ee's than that of the convex or homogeneous catalysts (Fig. 13).
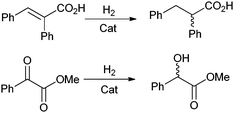 | ||
| Scheme 3 Asymmetric hydrogenation of E-α-phenyl cinnamic acid and methyl benzoylformate. | ||
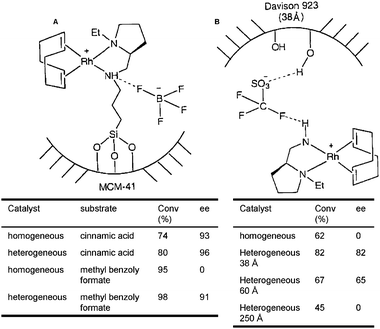 | ||
| Fig. 13 Rhodium diamine complexes covalently anchored to mesoporous silica (A) and results of asymmetric hydrogenations. The same Rh catalyst can be anchored via ionic interactions with the pore wall (B) and employed in the hydrogenation of methyl benzoylformate.95 | ||
A differing methodology for immobilising metal complexes to mesoporous silica is through ionic attachment—an approach that was adopted by de Rege et al.104 Through this approach, a negatively charged counter-ion could be used to attract a metal-cation-containing complex, through dipole charges in the ligands and anchoring the compound through hydrogen bonding with the surface silanol functionalities (Fig. 13). The Rh1(COD)AEP catalyst was immobilized in this way and used again in the industrially-significant hydrogenation of methylbenzoylformate to methyl mandelate.105,106 As expected, the confined, non-covalently anchored catalyst gives the same correlation in results to that of the covalently anchored series, proving that chiral restriction does indeed boost enantioselectivity. Non-covalently tethered rhodium catalysts were immobilised in the same way onto mesoporous silica supports that contained a range of different pore apertures. Results show95 that, with more room available inside the nanospace around the active site, less enantioselectivity was observed (Fig. 14). It reflects that as the declining influence of the spatial constraints from the inner wall decreases, so does the preference for a particular stereoisomer. This methodology can be further applied to the immobilisation of bio-inspired complexes for selective catalytic transformations.
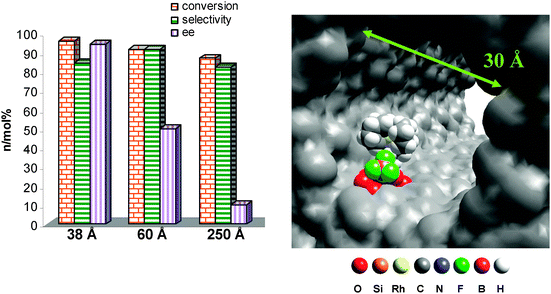 | ||
| Fig. 14 Effect of surface curvature on the enantioselectivity of the reaction with a range of pore apertures (left) and a graphical representation (right) to show how the constrained environment imposed by the pore walls facilitates stereoselectivity. | ||
Using covalent methods similar to those applied in previous immobilisation approaches,107,108 solitary amino acids or peptides can be grafted onto amino-functionalised mesoporous silica by solid phase peptide synthesis (SPPS) methodology109 (Fig. 15). First, the silica surface must be derivatized with an amino group and this is most commonly achieved by the co-condensation of 3-aminopropyl-trimethoxysilane.110 The hydrophobic character inside the pores can be adjusted by co-functionalization with other species e.g. methyl groups111 and this also influences the stability of the amino groups. The material with just the 3-aminopropyl moieties shows a poor hydrothermal stability because the basicity of the functional groups gives rise to the fast hydrolysis of the silica. Grafting a mixture of 3-aminopropyl and methyl groups to the surface improves hydrothermal stability due to a decreasing basic character inside the pores.111
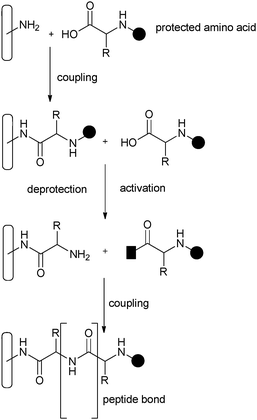 | ||
| Fig. 15 SPPS method of covalently attaching amino acids to silica. | ||
Amino acids (such as Fe3+–proline that were previously encapsulated within the cages of zeolites using the “zeolite synthesis method” described earlier) can be covalently anchored onto a mesoporous support by the formation of very stable bonds, with surface bound amide groups, in a coupling reaction through the formation of a peptide bond. This technique was a major breakthrough allowing the synthesis of peptides and small proteins, which could be attached to an insoluble support.109 This solid phase method is beneficial because the material can be easily recovered by filtration and any unreacted reagents left at the end of the synthetic coupling step can be removed facilely by a washing procedure. SPPS comprises of repeated cycles of coupling and deprotection of successive amino acids. The free N-terminal amine of an already attached peptide chain is coupled to a single N-protected amino acid. This unit is then deprotected, providing a new N-terminal amine to which a further amino acid can be attached.
Two major termini are used in solid phase peptide synthesis; Fmoc and t-Boc protecting groups. Each method involves different protection, deprotection and cleavage steps. To remove Fmoc from a peptide chain, basic conditions are used such as piperidine in DMF. To remove a t-Boc protecting group acidic conditions are used, for example trifluoroacetic acid. It is a common conception that Fmoc chemistry has many advantages over t-Boc chemistry, as it generates peptides of higher quality and in better yield. Activating agents are used when coupling a carboxyl group of an amino acid to make the reaction proceed at a much faster rate. There are two main types of activating groups: triazolols (HOBt, HOAt) and carbodiimides (DCC, DIC) (Fig. 16).
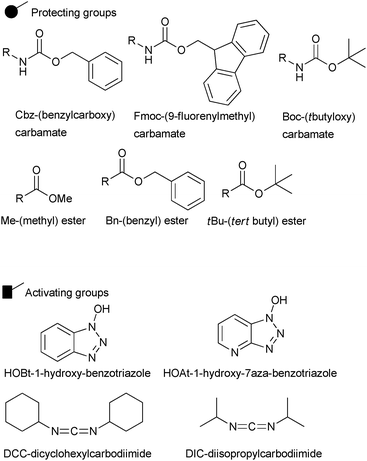 | ||
| Fig. 16 Common activating and protecting groups used in SPPS. | ||
A number of amino acids (Fig. 17) and short peptide complexes with iron and copper have been immobilised on mesoporous silica via SPPS and these have shown promising trends in the oxidation of hydrocarbons.112 This elegant method was implemented by Pirngruber et al.112 in order to mimic the active core of the enzyme methane mono-oxygenase (MMO). Instead of immobilising single amino acids on porous silica, a short peptide motif, found in the active site of the enzyme of His-x-x-Glu, was covalently anchored and coordinated to transition-metal centres via an auto-assembly route, in order to mimic the structure of the metalloprotein more closely. The main difficulty is preventing the formation of incomplete peptide chains but this can be overcome by capping unreacted surface amino groups and maximising the yield of the coupling steps. Immobilising a short peptide chain rather than single amino acids made it possible to incorporate more coordination sites, therefore significantly increasing the metal loading. Additional benefits of improvement in catalytic properties were observed by complexation of the metal-ions to the peptide chain when compared to that of the grafted single amino acids. One problem associated with self-assembly is that mixtures of complexes with varying coordination modes arise, which makes spectroscopic characterisation difficult. However, the use of EXAFS along with geometrical constraints allowed for credible structural models of the complexes to be developed.112 The immobilised complexes have also been tested in the oxidation of cyclohexane using hydrogen peroxide as the oxidant to produce cyclohexanol and cyclohexanone.113 The complexes of iron and copper with the covalently anchored HisGlyGlyGlu peptide sequence gave superior activities when compared to iron and copper immobilized on a non-peptide-modified support or one single amino acid derivatized silica.113 Studies proved no metal leaching from the bio-derivatized support, because the metal content stayed constant before and after reaction, as can be detected through ICP analysis.
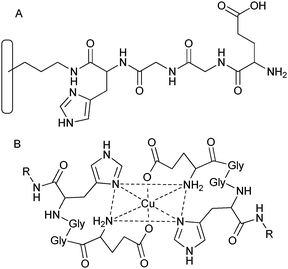 | ||
| Fig. 17 Synthesis of the HisGlyGlyGlu peptide chain on functionalised silica by SPPS113 (A) and how the peptide link may coordinate to a copper centre to form a metal complex (B). | ||
A similar route to immobilization of organic moieties to silica is by using ‘click’ chemistry114 protocols. In this instance the tether is generated quickly and reliably by joining smaller units together. First the silica is derivatized, usually with amino functionalities, tethered by the most popular reaction within click chemistry; azide alkyne Huisgen cycloaddition using a Cu catalyst.115 This procedure gives good yield and, in fact, has been used to immobilize proline on hydrophobic supports.116 This route for immobilisation on hydrophilic silica provides amino acid derivatised frameworks that can coordinate to transition metal centres (Fig. 18), giving rise to highly effective heterogeneous catalysts.
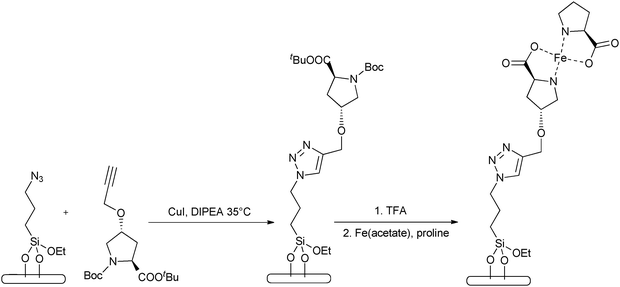 | ||
| Fig. 18 Immobilisation of amino acids on silica by the ‘click chemistry’ method, followed by coordination to transition-metal centres. | ||
More recently chiral copper proline-derived complexes have been immobilised on mesoporous silica and employed as heterogeneous catalysts in the asymmetric epoxidation of α-β-unsaturated ketones, using H2O2 and triethyl amine as an additive117 (Fig. 19). Triethyl amine was added to the mixture because α-β-unsaturated ketones require basic conditions in order to achieve impressive conversion and selectivity.118 The covalently anchored copper complex produces conversions in excess of 85% with greater than 82% ee for a range of substrates (see Table 5) and is also recycled effectively for up to 5 times with marginal loss in yield and no drop in enantioselectivity. This example further illustrates the proof of concept of covalently attaching bio-inspired metal complexes and highlights the impressive selectivities that can be achieved with this novel approach.
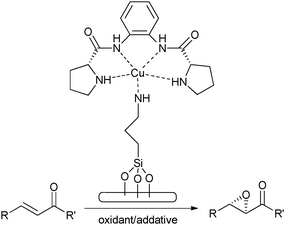 | ||
| Fig. 19 Epoxidation of unsaturated ketones using a heterogenised prolinamide copper complex. | ||
| Substrate | Conv. (%) | ee (%) |
|---|---|---|
| Chalcone | 88 | 83 |
| 4-phenyl-3-buten-2-one | 89 | 82 |
| Cyclohex-2-enone | 92 | 83 |
| Naphthalene-1,4-dione | 89 | 82 |
Making a direct comparison between the catalytic results of the same transition-metal complex (Fe3+–proline) encapsulated within a zeolite framework (using the zeolite synthesis approach that has been outlined earlier) to the one that has been and covalently anchored to the silica in the preceding section, provides crucial information in identifying the merits of the respective immobilisation strategies. Using the iron–proline complex as a case study in the selective oxidation of benzyl alcohol to benzaldehyde, both methods of immobilisation (zeolite encapsulation versus covalent anchoring to mesoporous silica) increase the activity of the catalyst as observed by the improvement of turnover numbers (TON's) in comparison to the corresponding homogeneous analogue (Fig. 20). The improvement is far more apparent in the case of the covalent anchoring of the metal complex to mesoporous silica, as TON's are significantly enhanced compared to the zeolite-encapsulated analogue, by as much as 80% when TBHP is used as the oxidant. This can be accredited to the more robust and stable immobilization procedure that the covalent attachment strategy to the silica surface imparts. With the catalyst chemically bound to the mesoporous silica surface via covalent bonds, the active centres are less likely to aggregate, dimerise and therefore deactivate.
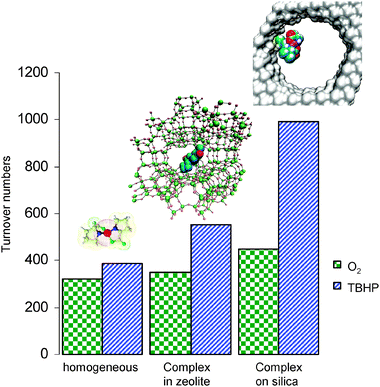 | ||
| Fig. 20 Improvements in TON's for benzyl alcohol oxidation on immobilisation of Fe3+–proline complex in zeolite X and further enhancements on covalent attachment to mesoporous silica. | ||
The catalyst anchored on mesoporous silica also exists as a more isolated single-site entity, because of the chemical bonds that restrain the immobilized complex to a definite space, via the pendant silanol groups of the mesoporous silica. Within the zeolite architecture, more than one complex could be potentially be encapsulated in each cage, furnishing a less well-defined active site which in turn would produce a lower activity. Diffusion rates of substrate molecules to the active centre and products on entering and leaving the porous support also play a decisive role in determining the activity of the catalyst. The pore apertures of the microporous zeolite encapsulated iron–proline complex are far smaller than the mesopores of silica, therefore reactants and products will be impeded to a greater extent on gaining access to and departing from the active site. The structure–property differences of the supports additionally affect the catalytic turnover, as the degree of hydrophilicity and hydrophobicity is significantly altered. Oxidation reactions, which rely on the use of aqueous oxidants such as hydrogen peroxide or acetyl peroxyborate (APB),119,120 depend upon a fine balance of hydrophilic/hydrophobic interactions.
New directions and scope in heterogeneous organocatalysis
Among the many applications and varying substrates that can be oxidised or transformed by these bio-derived catalysts immobilised on silica, the heterogenised amino acids (on their own), which are not coordinated to the transition-metal centres, can also individually act as organocatalysts, displaying high activities and selectivities in some of the most demanding and fundamental organic transformations.Organocatalysis has become a prominent facet in some of the most indispensable fundamental transformations in organic chemistry, which can be attributable to the exceptional stereoselectivity that can be achieved. In recent years amino acids, especially that of L-proline, have been used to catalyse essential transformations used in the fine chemical and pharmaceutical industries, such as the direct asymmetric aldol reaction,121,122 Mannich reactions,123 Michael Reactions124 and α-functionalisation of carbonyl compounds.125
Using a rationale, similar to that described earlier for the immobilisation of transition-metal amino acid complexes; amino acids with functional side chains that can react with tethering silanes such as lysine, arginine, glutamic acid, cysteine or amino acid derived organocatalysts could be covalently anchored to the inner walls of mesoporous silica. These materials have shown promise as heterogeneous organocatalysts for industrial significant reactions, such as the Hajos–Parrish–Eder–Sauer–Wiechert reaction126,127 and intramolecular [4+2] cycloadditions.128 This approach has the potential to improve recoverability and recyclability, use far less mol% of catalyst and potentially give rise to significant improvements in activity and selectivity. Organocatalysts can be covalently anchored to mesoporous silica (Fig. 21) by building on current methodologies that are reported in the literature.129 Tethering is carried out through the side-chain or a functionalized side-chain of a derivatised amino acid and not through a SPPS technique (described earlier), because the mechanism of catalysis relies on the amino and carboxylate groups to be free for substrate binding.130
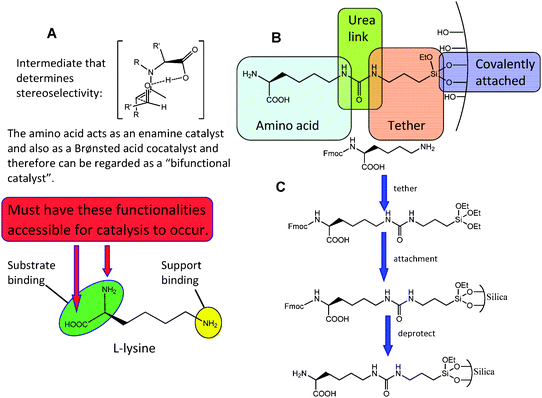 | ||
| Fig. 21 Rationale (A) and generalised strategy (B) for derivatizing (tethering) an amino acid through the side chain or functionalised side chain. Specific approach employing L-lysine (C) as an exemplar amino acid. | ||
Through spatial constraints, nature of the active site and a precise balance of substrate–catalyst–pore wall interactions, the stereoselectivity of the catalytic reaction could also be suitably enhanced. Judiciously controlling the orientation of the channels and the pore-diameter of the mesoporous support could facilitate enhancements in the desired stereochemical outcome, as observed earlier with anchored transition-metal amino acid complexes. The range of substrates and scope of reactions that can be catalysed by heterogenised amino acids and their derivatives (Fig. 22) is ever expanding131 and immobilisation of these organic moieties could result in the design and development of more stable, highly active, selective single-site heterogeneous catalysts. The potential for expanding the scope and industrial applicability of heterogenised organocatalysts demands a greater understanding of the synergistic effects, between the support and the amino acid at a molecular level, which could provide valuable insights on structure–property relationships and its mechanistic significance. A potential hurdle associated with acquiring this knowledge-base is the lack of spectroscopic handles an organic moiety has when attached on to an inorganic support. Since there is no metal present and the bulk of material is the support, this reduces the number of spectroscopic techniques that are available at one's disposal for in-depth characterisation with a view to elucidating the nature of the active site. In addition, several techniques are surface sensitive and are therefore ineffective if the organocatalyst is encapsulated within the host. Nevertheless, solid-state methods of characterisation such as MAS NMR and FT-IR are able to provide some meaningful insights into the nature of the active-site.
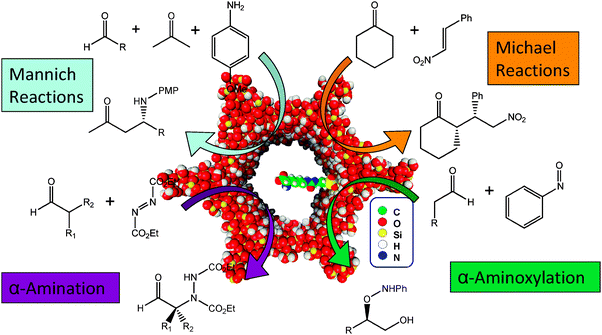 | ||
| Fig. 22 Examples of applications with immobilised amino acids such as lysine that act as enamine catalysts.125 | ||
In light of the numerous advantages highlighted over the course of this article, there are evidentially a number of limitations associated with such catalysts and immobilisation procedures. We have emphasised earlier the importance and benefits of the synthesis methodologies, in order to highlight the pioneering routes to heterogenisation, as well as revealing of the breadth and scope of what can be achieved in the near future. But, in reality, in-depth, in situ characterisation techniques and a more detailed and fundamental understanding of the nature of the active centre and its mechanistic significance in the catalytic process are less well-understood that impede the implementation of bio-inspired heterogeneous catalysts in industry. These catalysts have shown promising results in laboratory-based environments; yet questions still linger on their stability, with only a small minority of them being subject to the stringent tests that are required for development at an industrial scale. It must be noted that immobilisation of a homogeneous catalyst on to a support has a number of constraints at present. The reasons for inhibiting catalysis are unclear; but it would be sensible to assume that some of these factors are due to unfavourable effects of the support; which ironically are the very same factors that are responsible for boosting catalytic properties. Ultimately, an explicit understanding of why heterogenisation improves catalysis in some cases and not in others has still not fully been explored. The influence of support type, hydrophilicity/hydrophobicity, framework topology, diffusion and adsorption all play important roles, but currently the extent of each factor has not been quantified and, as long as this remains the case, the possibility of being able to design better catalysts with a view to predicting structure–property relationships may be a distant realisation.
However, preliminary investigations into structure–activity correlations have been acquired by recent advances in spectroscopy, space-resolved profiling and combining high-throughout experimentation with data modelling. Being able to recognize the relationship between the support and the active site can lead to optimisation strategies for catalytic processes. An example of this is the epoxidation of bulky olefins and methyl oleate by porous titanosilicates.132 Through in-depth characterisation, high-throughput experimentation and molecular modelling, information on the roles of different catalyst variables were uncovered. Associations of the properties of the support silicate, dispersion and measure of active-sites, presence of surface modifiers, substrate selection, activity and selectivity were established. Using these connections made it viable to maximise the activity of the Ti-catalyst, by providing a particular catalyst environment and controlling the hydrophilicity of the support.22,23
Characterising the nature of the active site is imperative in being able to optimise catalysis as well as predict catalytic outcomes. By building on results obtained with IR, UV-Vis, EPR, SEM/TEM, BET, NMR and X-ray absorption spectroscopy (EXAFS and XANES), the nature of active-sites and the coordination geometry in their immediate vicinity can be quantified to elucidate the catalytic significance of the results, from a more fundamental perspective. Moreover, in situ measurements using appropriate probe molecules need to be suitably deployed, for studying changes in oxidation states, conformational flexibility of the ligands, local coordination environment and emergence of transition-states, which provides valuable insights for a better understanding of the rate and energetic interactions associated with these catalytic processes. Through spatially-resolved methods of detection of the chemical structure and temperature133 and using a combination of complementary techniques such as UV-Vis/Raman and FT-IR/ED-EXAFS, under in situ conditions, in intricately designed reaction cells, further improves the quality of information obtained and affords a greater knowledge about the investigated system.134 Multiple technique operando set-ups render the measurement of spectra in a time-resolved, spatially resolved fashion,135 which provides detailed insights into the working principles of the heterogeneous catalytic system. Another technique currently being pioneered, which in the near future could elucidate reaction mechanisms on catalytic surfaces, is 4D electron microscopy.136,137 The powerful focusing capabilities of electrons allow structures to be resolved down to the atomic scale and the technique has been used to directly determine the structure and dynamics of substrates on hydrophilic supports.138,139 Being able to establish structures at interfaces with atomic scale resolution may one day be applied and advanced to more complex structures, such as to observe catalyst environments and conformations on analogous surfaces. Zewail140 has described 4D electron microscopy as having the potential to decipher the fundamental forces of complex structures, such as the catalytic properties of heterogeneous nanoparticles or in phase transitions.
The activity of an anchored catalyst in a porous host often depends on diffusion of solvent and substrate molecules. There is a lacuna in the field with respect to modelling kinetic and thermodynamic data for predicting diffusion limitations and reaction pathways, with a view to understanding the overall mechanism of the reaction. This can be achieved by performing a range of kinetic studies, with a view to identifying the rate-determining step and the overall limitations within a reaction. With a greater understanding of the thermodynamic parameters, kinetic modelling studies can be undertaken with a view to studying the effects of bifunctional and multifunctional active sites. Density Functional Theory (DFT) can elucidate the properties of a support and therefore postulate the potential implications it can have on a catalytic reaction. There is scope for quantifying the support characteristics such as acidity and reactivity,141 which further demonstrates the valuable role theoretical chemical tools offer in the studies of inorganic frameworks such as zeolites. DFT orientated approaches that are tailored to the design of new immobilised, encapsulated or bio-inspired catalysts need to explicitly identify the individual steps in a catalytic process at a molecular level, by calculating energy barriers and the stability of the intermediates,142,143 thereby presenting information that can be rationally applied to the development of new strategies for designing new catalysts or optimising existing ones.144 Theoretical ab initio calculations could also be performed to model the active site, providing further insights into the interactions between the transition-metal and other substituents that make up the framework. This also facilitates the calculation of binding energies and transition states, for building a complete reaction profile, which could potentially reveal the key steps that may require improvement from a design perspective. By combining recent advances in spectroscopy and operando techniques along with theoretical calculations and computational modelling, the nature of the active site, its interactions with the support or framework and its ability to facilitate synergistic enhancements in catalytic potential can be more quantitatively rationalised.145–150 By using the above advances concurrently, it should be possible to develop structure–activity correlations relating to bio-inspired systems, in single-site heterogeneous catalysts, by establishing the exact nature of different types of active sites and in what ways the geometry of the metal or ligand affects the ensuing catalysis.151
All of these techniques outlined above can provide valuable insights for correlating experimental observations with theoretical predictions. The key to understanding the influence of the support on catalysis, how it can be altered and what repercussions this would have if suitably modified, will play an important role, from a spectroscopic and computational viewpoint, in elucidating the nature of the active-site and its direct influence on the catalytic outcome, in terms of selectivity and activity.
Conclusions
Bio-derivatised frameworks such as amino acids immobilised within inorganic host matrices are starting to attract considerable attention for applications in catalysis for the synthesis of commodity and fine-chemicals.65,94,152–154 Well-characterised and well-defined transition-metal complexes, which mimic the catalytic function of the active centres in metalloenzymes, can be anchored in a site-isolated fashion on to inorganic supports, giving rise to highly active and selective heterogeneous catalysts. By changing the properties of the support, i.e. support type, framework topology, pore aperture or hydrophilicity/hydrophobicity and by understanding how this affects the catalytic outcome, facilitates structure–property correlations to be established. This provides further avenues for not only optimising reaction conditions but to specifically tailor the support for desired outcomes in activity and selectivity. Although several advances have been made from a design perspective, there is still considerable scope for a greater understanding of the function of the host and the influence it can have mechanistically on facilitating the stereo-chemical outcome for predicting product distribution and enantiomeric excesses. This would create a novel approach towards the rational design and optimisation in heterogeneous catalysis, where support tuning and modification would take primary precedence, outweighing direct approaches to altering the catalyst composition, in order to change or modify the nature of the active site. Further, by taking advantage of spatial constraints within a mesopore, the nature of the active site can be suitably attuned so as to facilitate a precise balance between substrate–catalyst–pore wall interactions, thereby enhancing the stereoselectivity in catalytic reactions and its overall turnover compared to analogous homogeneous systems. In situ measurements using appropriate probe molecules could be suitably deployed, for studying changes in oxidation states, conformational flexibility of the ligands, local coordination environment and emergence of transition-states, which should provide valuable insights for a better understanding of the rate and energetic interactions associated with these catalytic processes. In the near future, this could become a reality, as advances in in situ characterisation, operando techniques and computational modelling could facilitate structure–activity relationships to be established for probing the mechanism and ultimately designing novel active catalysts.Acknowledgements
The authors wish to thank EPSRC (UK) and the British Italian Partnership Program for financial support. We also wish to acknowledge our colleagues at the University of Turin (Chemistry) and Joanna Dzierzak for helpful discussions.References
- Pharmaceutical Market Trends 2010–2014, Key market forecasts & growth opportunities (4th edn), URCH Publishing, 2010.
- A. Liese, K. Seelbach and C. Wandrey, Industrial biotransformations: A collection of processes, VCH, Weinheim, 2000 Search PubMed.
- A. J. J. Straathof, S. Panke and A. Schmid, Curr. Opin. Biotechnol., 2002, 13, 548 CrossRef CAS.
- A. S. Bommarius and B. R. Riebel, Biocatalysis: fundamental and applications, Wiley-VCH, 2004 Search PubMed.
- J. P. Rasor and E. Voss, Appl. Catal., A, 2001, 9, 738.
- R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239 CrossRef CAS.
- O. Yamauchi, A. Odani and M. Takani, J. Chem. Soc., Dalton Trans., 2002, 3411 RSC.
- X. Shan and L. Que, J. Inorg. Biochem., 2006, 100, 421 CrossRef CAS.
- J. W. Whittaker, Chem. Rev., 2003, 103, 2347 CrossRef CAS.
- M. L. Neidig and E. I. Solomon, Chem. Commun., 2005, 5843 RSC.
- B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947 CrossRef CAS.
- J. Zhang, H. Zheng, S. L. Groce and J. D. Lipscomb, J. Mol. Catal. A: Chem., 2006, 251, 54 CrossRef CAS.
- E. I. Solomon, P. Chen, M. Metz, S. K. Lee and A. E. Palmer, Angew. Chem., Int. Ed., 2001, 40, 4570 CrossRef CAS.
- R. L. Lieberman and A. C. Rosenzweig, Dalton Trans., 2005, 3390 RSC.
- J. P. Klinman, Chem. Rev., 1996, 96, 2541 CrossRef CAS.
- Y. Matoba, T. Kumagai, A. Yamamoto, H. Yoshitsu and M. Sugiyama, J. Biol. Chem., 2006, 281, 8981 CrossRef CAS.
- C. Eicken, B. Krebs and J. C. Sacchettini, Curr. Opin. Struct. Biol., 1999, 9, 677 CrossRef CAS.
- M. Luechinger, A. Kienhöfer and G. D. Pirngruber, Chem. Mater., 2006, 18, 1330 CrossRef CAS.
- K. Suzuki, P. D. Oldenburg and L. Que Jr., Angew. Chem., Int. Ed., 2008, 47, 1887 CrossRef CAS.
- K. Kerman and H. B. Kraatz, Angew. Chem., Int. Ed., 2008, 47, 6522 CrossRef CAS.
- G. A. Somorjai and J. Y. Park, Angew. Chem., Int. Ed., 2008, 47, 9212 CrossRef CAS.
- M. A. Camblor, A. Corma, P. Esteve, A. Martinez and S. Valencia, Chem. Commun., 1997, 795 RSC.
- M. J. Climent, A. Corma, A. Velty and M. Susarte, J. Catal., 2000, 196, 345 CrossRef CAS.
- E. G. Derouane, CATTECH, 2002, 6, 11 CrossRef.
- R. Raja and J. M. Thomas, J. Mol. Catal. A: Chem., 2002, 181, 3 CrossRef CAS.
- R. Raja and P. Ratnasamy, J. Catal., 1997, 170, 244 CrossRef CAS.
- R. Raja and P. Ratnasamy, Catal. Lett., 1997, 48, 1 CrossRef CAS.
- D. E. De Vos, B. F. Sels and P. A. Jacobs, CATTECH, 2002, 6, 14 CrossRef CAS.
- R. Raja and P. Ratnasamy, Appl. Catal., A, 1996, 143, 145 CrossRef.
- R. Raja and P. Ratnasamy, Appl. Catal., A., 1996, 158, L7.
- V. Y. Zakharov, O. M. Zakharov, B. V. Romanovsky and R. E. Mardaleishvili, React. Kinet. Catal. Lett., 1977, 6, 133 CAS.
- A. N. Zakharov and B. V. Romanovsky, J. Inclusion Phenom., 1985, 4, 30.
- B. V. Romanovsky and A. G. Gabrielov, J. Mol. Catal., 1992, 74, 293 CrossRef.
- J. E. Lyons, P. E. Ellis Jr. and H. K. Myers Jr., J. Catal., 1995, 155, 59 CrossRef CAS.
- P. E. Ellis Jr. and J. E. Lyons, Coord. Chem. Rev., 1990, 105, 181 CrossRef CAS.
- N. Herron and C. A. Tolman, J. Am. Chem. Soc., 1987, 109, 2837 CrossRef CAS.
- N. Herron, Inorg. Chem., 1986, 25, 4714 CrossRef CAS.
- D. R. Corbin and N. Herron, J. Mol. Catal., 1994, 86, 343 CrossRef CAS.
- N. Herron, J. Coord. Chem., 1988, 19, 25 CAS.
- J. P. Collman, R. R. Gagne, T. R. Halbert, J. C. Marchon and C. A. Reed, J. Am. Chem. Soc., 1973, 95, 7868 CrossRef CAS.
- R. D. Jones, D. A. Sumerville and F. Basolo, Chem. Rev., 1979, 79, 139 CrossRef CAS.
- N. Herron, New J. Chem., 1989, 13, 761 Search PubMed.
- R. F. Parton, I. F. J. Vankelecom, M. J. A. Casselman, C. P. Bezoukhanova, J. B. Uytterhoeven and P. A. Jacobs, Nature, 1994, 370, 541 CrossRef CAS.
- R. F. Parton, D. De Vos and P. A. Jacobs, in Zeolite Mincroporous Solids: Synthesis, Structure and Reactivity, ed. E. G. Derouane, F. Lemos, C. Naccache and R. F. Ribeiro, Kluwer Academic Publishers, London, 1991, p. 555 Search PubMed.
- D. R. C. Huybrechts, R. F. Parton and P. A. Jacobs, Stud. Surf. Sci. Catal., 60, 225 Search PubMed.
- R. F. Parton, J. B. Uytterhoeven and P. A. Jacobs, Stud. Surf. Sci. Catal., 59, 395 Search PubMed.
- P. P. Knops-Gerrits, D. De Vos, F. Thibault-Starzyk and P. A. Jacobs, Nature, 1994, 369, 543 CrossRef CAS.
- B. R. Cook, T. J. Reinert and K. S. Suslick, J. Am. Chem. Soc., 1986, 108, 7281 CrossRef CAS.
- S. Ernst, Y. Traa and U. Deeg, Stud. Surf. Sci. Catal., 84, 925 Search PubMed.
- A. Zsigmond, F. Notheisz, M. Bartok and J. E. Backvall, Stud. Surf. Sci. Catal., 78, 417 Search PubMed.
- R. J. Taylor, R. S. Drago and J. P. Hage, Inorg. Chem., 1992, 31, 253 CrossRef CAS.
- E. R. Birnbaum, M. W. Grinstaff, J. A. Labinger, J. E. Bercaw and H. B. Gray, J. Mol. Catal. A: Chem., 1995, 104, L119 CrossRef.
- H. Diegruber, P. J. Plath, E. G. Schultz-Elkoff and M. Mohl, J. Mol. Catal., 1984, 24, 115 CrossRef CAS.
- D. Xuereb, J. Dzierzak and R. Raja, Catalysis by Metal Complexes: Heterogenized Homogeneous Catalysts for Fine Chemical Production, ed. C. Bianchini and D. J. Cole-Hamilton, P. W. N. M. van Leeuwen, Springer, Dordrecht, 2010, chap. 2 (see also chapter 3: J. M. Fraile, J. I. Garcia, J. A. Mayoral, and E. Pires) Search PubMed.
- B. M. Weckhuysen, A. A. Verberckmoes, L. Fu and R. A. Schoonheydt, J. Phys. Chem., 1996, 100, 9456 CrossRef CAS.
- B. Fan, H. Li, W. Fan, C. Jin and R. Li, Appl. Catal., A, 2008, 340, 67 CrossRef CAS.
- J. Dzierzak, M. Lefenfeld and R. Raja, Top. Catal., 2009, 52, 1669 CrossRef CAS.
- K. J. Balkus Jr., M. Eissa and R. Levado, J. Am. Chem. Soc., 1995, 117, 10753 CrossRef.
- R. Grommen, P. Manikandan, Y. Gao, T. Shane, J. J. Shane, R. A. Schoonheydt, B. M. Weckhuysen and D. Goldfarb, J. Am. Chem. Soc., 2000, 122, 11488 CrossRef CAS.
- J. G. Mesu, T. Visser, A. M. Beale, F. Soulimani and B. M. Weckhuysen, Chem.–Eur. J., 2006, 12, 7167 CrossRef CAS.
- K. J. Balkus Jr., A. G. Gabrielov, S. L. Bull, F. Bedioui, L. Roué and J. Devynck, Inorg. Chem., 1994, 33, 67 CrossRef.
- A. G. Gabrielov, K. J. Balkus Jr., F. Bedioui and J. Devynck, Microporous Mesoporous Mater., 1994, 2, 119 CAS.
- K. J. Balkus Jr., A. A. Welch and B. E. Gnade, J. Inclusion Phenom. Mol. Recognit. Chem., 1991, 10, 141 CrossRef.
- B. M. Weckhuysen, A. A. Verberckmoes, I. P. Vannijvel, J. A. Pelgrims, P. L. Buskens, P. A. Jacobs and R. A. Schoonheydt, Angew. Chem., Int. Ed. Engl., 1995, 34, 2652 CAS.
- J. Dzierzak, E. Bottinelli, G. Berlier, E. Gianotti, E. Stulz, R. M. Kowalczyk and R. Raja, Chem. Commun., 2010, 46, 2805 RSC.
- H. Sigel and D. B. McCormick, J. Am. Chem. Soc., 1971, 93, 2041 CrossRef CAS.
- P. C. A. Bruijnincx, M. Lutz, A. L. Spek, E. E. Van Faassen, B. M. Weckhuysen, G. Van Koten and R. J. M. Klein Gebbink, Eur. J. Inorg. Chem., 2005, 779 CrossRef CAS.
- K. Kervinen, P. C. A. Bruijnincx, A. M. Beale, J. G. Mesu, G. Van Koten, R. J. M. Klein Gebbink and B. M. Weckhuysen, J. Am. Chem. Soc., 2006, 128, 3208 CrossRef CAS.
- L. Fu, B. M. Weckhuysen, A. A. Verberckmoes and R. A. Schoonheydt, Clay Miner., 1996, 31, 491 CrossRef CAS.
- C. I. Fernandes, N. U. Silva, P. D. Vaz, T. G. Nunes and C. D. Nunes, Appl. Catal., A, 2010, 384, 84 CrossRef CAS.
- Z. An, W. Zhang, H. Shi and J. He, J. Catal., 2006, 241, 319 CrossRef CAS.
- J. Bujdak and B. M. Rode, J. Mol. Catal. A: Chem., 1999, 144, 129 CrossRef CAS.
- A. M. Davis, G. Joanis and L. Tribe, J. Comput. Chem., 2008, 29, 983 CrossRef CAS.
- B. M. Weckhuysen, H. Leeman and R. A. Schoonheydt, Phys. Chem. Chem. Phys., 1999, 1, 2875 RSC.
- Z. Csendes, I. Lajter, V. Burgis, L. J. T. Kiss and I. Palinko, Vib. Spectrosc., 2010, 53, 132 CrossRef CAS.
- A. Aranyi, Z. Csendes, J. T. Kiss and I. Palinko, J. Mol. Struct., 2009, 924–926, 166 CrossRef CAS.
- J. E. Krohn and M. Psapatsis, Langmuir, 2005, 21, 8743 CrossRef CAS.
- S. Munsch, M. Hartmann and S. Ernst, Chem. Commun., 2001, 1978 RSC.
- M. R. Maurya, M. Kumar, A. Kumar and J. C. Pessoa, Dalton Trans., 32, 4220 Search PubMed.
- S. Jammi, L. Rout, P. Saha, V. K. Akkilagunta, S. Sanyasi and T. Punniyamurthy, Inorg. Chem., 2008, 47, 5093 CrossRef CAS.
- T. Punniyamurthy and L. Rout, Coord. Chem. Rev., 2008, 252, 134 CAS.
- V. B. Valodkar, G. L. Tembe, A. Ravindranathan, R. N. Ram and H. S. Rama, J. Mol. Catal. A: Chem., 2004, 208, 21 CrossRef CAS.
- V. B. Valodkar, G. L. Tembe, A. Ravindranathan, R. N. Ram and H. S. Rama, J. Macromol. Sci., Part A: Pure Appl. Chem., 2004, 41, 839 Search PubMed.
- Y. R. De Miguel, J. Chem. Soc., Perkin Trans. 1, 2000, 4213 RSC.
- Z. Csendes, V. Burgis, L. Lacko, I. Labadi, J. T. Kiss and I. Palinko, Anal. Bioanal. Chem., 2010, 397, 549 CrossRef CAS.
- B. Korbelly, J. T. Kiss, K. Hernadi and I. Palinko, J. Mol. Struct., 2007, 834–836, 345 CrossRef CAS.
- N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217 CrossRef CAS.
- B. Meunier, Chem. Rev., 1992, 92, 1411 CrossRef CAS.
- L. Canali and D. C. Sherrington, Chem. Soc. Rev., 1999, 28, 85 RSC.
- J. M. Thomas and R. Raja, Top. Catal., 2010, 53, 848 CrossRef CAS.
- J. M. Thomas, J. C. Hernandez-Garrido, R. Raja and R. G. Bell, Phys. Chem. Chem. Phys., 2009, 11, 2799 RSC.
- S. Bordiga, R. Buzzoni, F. Geobaldo, C. Lamberti, E. Giamello, A. Zecchina, G. Leofanti, G. Petrini, G. Tozzola and G. Vlaic, J. Catal., 1996, 158, 486 CrossRef CAS.
- J. M. Thomas, Nature, 1994, 368, 289 CrossRef.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710 CrossRef CAS.
- J. M. Thomas and R. Raja, Acc. Chem. Res., 2008, 41, 708 CrossRef CAS.
- A. P. Wight and M. E. Davis, Chem. Rev., 2002, 102, 3589 CrossRef CAS.
- H. Yang, G. Zhang, X. Hong and Y. Zhu, Microporous Mesoporous Mater., 2004, 68, 119 CrossRef CAS.
- J. M. Thomas, Angew. Chem., Int. Ed., 1999, 38, 3588 CrossRef.
- B. F. G. Johnson, S. A. Raynor, D. S. Shephard, T. Maschmeyer, J. M. Thomas, G. Sankar, S. T. Bromley, R. D. Oldroyd, L. G. Gladden and M. D. Mantle, Chem. Commun., 1999, 1167 RSC.
- F. J. Feher, D. A. Newman and J. F. Walzer, J. Am. Chem. Soc., 1989, 111, 1741 CrossRef CAS.
- S. A. Raynor, J. M. Thomas, R. Raja, B. F. G. Johnson, R. G. Bell and M. D. Mantle, Chem. Commun., 2000, 1925 RSC.
- M. D. Jones, R. Raja, J. M. Thomas, B. F. G. Johnson, D. W. Lewis, J. Rouzard and K. D. M. Harris, Angew. Chem., Int. Ed., 2003, 42, 4326 CrossRef CAS.
- J. Rouzard, M. D. Jones, R. Raja, B. F. G. Johnson, J. M. Thomas and M. Duer, Helv. Chim. Acta, 2003, 86, 1753 CrossRef CAS.
- F. M. de Rege, D. K. Morita, K. C. Ott, W. Tumas and R. D. Broene, Chem. Commun., 2000, 1797 RSC.
- J. M. Thomas, B. F. G. Johnson, R. Raja and M. D. Jones, European Patent Filing No., EP1469005 B1, 2004 Search PubMed.
- J. M. Thomas, B. F. G. Johnson, R. Raja and M. D. Jones, US Patent Filing No., US2004220347, 2004 Search PubMed.
- J. M. Thomas, Faraday Discuss., 1995, 100, C9 RSC.
- J. M. Thomas, T. Maschmeyer, B. F. G. Johnson and D. S. Shephard, J. Mol. Catal. A: Chem., 1999, 141, 139 CrossRef CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149 CrossRef CAS.
- A. Walcarius, M. Etienne and B. Lebeau, Chem. Mater., 2003, 15, 2174 CrossRef CAS.
- M. Luechinger, R. Prins and G. D. Pirngruber, Microporous Mesoporous Mater., 2005, 85, 111 CrossRef CAS.
- G. D. Pirngruber, L. Frunz and M. Lüchinger, Phys. Chem. Chem. Phys., 2009, 11, 2928 RSC.
- L. Frunz, R. Prins and G. D. Pirngruber, Chem. Mater., 2007, 19, 4357 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- J. E. Hein, J. C. Tripp, L. B. Krasnova, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2009, 48, 8018 CrossRef CAS.
- D. Font, C. Jimeno and M. A. Pericàs, Org. Lett., 2006, 8, 4653 CrossRef CAS.
- E. A. Prasetyanto, N. H. Khan, H. Seo and S. Park, Top. Catal., 2010, 53, 1381 CrossRef CAS.
- K. Jakka, J. Liu and C. G. Zhao, Tetrahedron Lett., 2007, 48, 1395 CrossRef CAS.
- R. Raja, J. M. Thomas, M. Greenhill-Hooper and V. Doukova, Chem. Commun., 2007, 1924 RSC.
- M. Greenhill-Hooper, R. Raja and J. M. Thomas, WO Patent. Appl., 2006043075 A1, 2006 Search PubMed.
- B. List, P. Pojarliev and C. Castello, Org. Lett., 2001, 3, 573–575 CrossRef CAS.
- B. List, Synlett, 2001, 1675–1686 CrossRef CAS.
- B. List, J. Am. Chem. Soc., 2000, 122, 9336 CrossRef CAS.
- B. List, P. Pojarliev and H. J. Martin, Org. Lett., 2001, 3, 2423 CrossRef CAS.
- S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS.
- Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615 CrossRef CAS.
- U. Eder, G. Sauer and R. Weichert, Angew. Chem., Int. Ed. Engl., 1971, 10, 496 CrossRef.
- R. M. Wilson, J. Am. Chem. Soc., 2005, 127, 11616 CrossRef CAS.
- F. Calderón, R. Fernandez, F. Sanchez and A. Fernandez-Mayoralas, Adv. Synth. Catal., 2005, 347, 1395 CrossRef CAS.
- B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef CAS.
- M. J. Gaunt, C. C. C. Johansson, A. McNally and N. T. Vo, Drug Discovery Today, 2007, 12, 8 CrossRef CAS.
- P. Serna, L. A. Baumes, M. Moliner and A. Corma, J. Catal., 2008, 258, 25 CrossRef CAS.
- A. Urakawa and A. Baiker, Top. Catal., 2009, 52, 1312 CrossRef CAS.
- F. C. Meunier, Chem. Soc. Rev., 2010, 39, 4602 RSC.
- S. T. Tinnemans, J. G. Mesu, K. Kervinen, T. Visser, T. A. Nijhuis, A. M. Beale, D. E. Keller, A. M. J. van der Eerden and B. M. Weckhuysen, Catal. Today, 2006, 113, 3 CrossRef CAS.
- B. Barwick, H. S. Park, O.-H. Kwon, J. S. Baskin and A. H. Zewail, Science, 2008, 322, 1227 CrossRef CAS.
- F. Carbone, O.-H. Kwon and A. H. Zewail, Science, 2009, 325, 181 CrossRef CAS.
- S. Chen, M. T. Seidel and A. H. Zewail, Angew. Chem., Int. Ed., 2006, 45, 5154 CrossRef CAS.
- C.-Y. Ruan, V. A. Lobsatov, F. Vigliotti, S. Chen and A. H. Zewail, Science, 2004, 304, 80 CrossRef CAS.
- A. H. Zewail, Science, 2010, 328, 187 CrossRef CAS.
- J. B. Nicholas, Top. Catal., 1997, 4, 157 CrossRef CAS.
- C. R. A Catlow, S. A. French, A. A. Sokol and J. M. Thomas, Philos. Trans. R. Soc. London, Ser. A, 2005, 363, 913 CrossRef CAS.
- B. Hammer and J. K. Norskov, Adv. Catal., 45, 71 Search PubMed.
- J. M. Thomas, J. Chem. Phys., 2008, 128, 182502 CrossRef.
- J. Paterson, M. Potter, E. Gianotti and R. Raja, Chem. Commun., 2011, 47, 517 RSC.
- E. Gianotti, V. N. Shetti, M. Manzoli, J. A. L. Blaine, W. C. Pearl Jr., R. D. Adams, S. Coluccia and R. Raja, Chem.–Eur. J., 2010, 16, 8202 CAS.
- M. Vishnuvarthan, A. J. Paterson, R. Raja, A. Piovano, F. Bonino, E. Gianotti and G. Berlier, Microporous Mesoporous Mater., 2011, 138, 167 CrossRef CAS.
- S. T. Bromley, G. Sankar, C. R. A. Catlow, T. Maschmeyer, B. F. G. Johnson and J. M. Thomas, Chem. Phys. Lett., 2001, 340, 524 CrossRef CAS.
- S. A. French, A. A. Sokol, S. T. Bromley, C. R. A. Catlow and P. Sherwood, Top. Catal., 2003, 24, 161 CrossRef CAS.
- S. T. Bromley, S. A. French, A. A. Sokol, C. R. A. Catlow and P. Sherwood, J. Phys. Chem. B, 2003, 107, 7045 CrossRef CAS.
- M. Boronat, P. Concepcion, A. Corma, M. T. Navarro, M. Renz and S. Valencia, Phys. Chem. Chem. Phys., 2009, 11, 2876 RSC.
- R. A. Schoonheydt and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2009, 11, 2794 RSC.
- C. H. Lee, H. C. Lin, S. H. Cheng, T. S. Lin and C. Y. Mou, J. Phys. Chem., 2009, 113, 16058 Search PubMed.
- D. Baute, D. Arieli, F. Neesse, H. Zimmermann, B. M. Weckhuysen and D. Goldfarb, J. Am. Chem. Soc., 2004, 126, 11733 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |