Nature of dispersed vanadium oxide: influence of the silica support structure and synthesis methods
Rita
Chlosta †
a,
Genka
Tzolova-Müller
a,
Robert
Schlögl
a and
Christian
Hess
*b
aFritz-Haber-Institut der Max-Planck-Gesellschaft, Abteilung Anorganische Chemie, Faradayweg 4-6, 14195 Berlin, Germany
bEduard-Zintl-Institut für Anorganische und Physikalische Chemie, Technische Universität Darmstadt, Petersenstr. 20, 64287 Darmstadt, Germany. E-mail: hess@pc.chemie.tu-darmstadt.de
First published on 30th June 2011
Abstract
Dispersed vanadium oxide samples were prepared on the basis of two differently structured high surface area silica materials (Aerosil 300, SBA-15). For each support material incipient wetness impregnation and a grafting/ion exchange procedure were applied to prepare catalyst samples with comparable vanadium density. The influence of the silica support material and preparation method on the vanadium oxide structure and dispersion has been studied using diffuse reflectance UV-Vis spectroscopy, visible Raman spectroscopy and X-ray photoelectron spectroscopy (XPS). By independent spectroscopic characterization a fully consistent picture regarding the relation of the structure (UV-Vis, Raman) and dispersion (XPS) is developed. Based on the present structural data and recent findings the dispersed vanadium oxide is proposed to consist of monomers and oligomers with a distorted tetrahedral (VO4) structure containing one short V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. A different degree of hydroxylation of vanadium gives rise to two VO stretching bands at 1027 and 1040 cm−1. The structure and dispersion of vanadium oxide are more strongly influenced by the support material than by the synthesis method. In this regard the Aerosil 300 samples show a higher degree of oligomerization, i.e. less dispersion of the surface vanadium oxide species, than the SBA-15 samples. Likewise incipient wetness impregnation leads to more oligomerized species than grafting/ion-exchange.
O bond. A different degree of hydroxylation of vanadium gives rise to two VO stretching bands at 1027 and 1040 cm−1. The structure and dispersion of vanadium oxide are more strongly influenced by the support material than by the synthesis method. In this regard the Aerosil 300 samples show a higher degree of oligomerization, i.e. less dispersion of the surface vanadium oxide species, than the SBA-15 samples. Likewise incipient wetness impregnation leads to more oligomerized species than grafting/ion-exchange.
Introduction
Supported vanadium oxides exhibit a large potential as heterogeneous catalysts in a variety of oxidation reactions such as the selective oxidation of methane1–6 and methanol7–9 to formaldehyde or the oxidative dehydrogenation of ethane and propane.10–13 Regarding technical applications, catalytic processes based on the oxidative transformation of alkanes to olefins or oxygenates are still scarce. In industry, there has been an increasing demand for propene over the last years mainly due to its use in polypropylene production. The oxidative dehydrogenation (ODH) of propane is considered as an alternative to the existing commercial processes. Compared to (non-oxidative) dehydrogenation ODH is thermodynamically not limited and can be operated at lower temperatures due to the exothermicity of the reaction. However, the reactivity of propene is higher than that of propane. Thus the main challenge is to minimize the amount of COx, which is favored at high levels of propane conversion. The activity and selectivity of supported vanadium oxide catalysts depend, among many other factors, on the properties of the oxide material used for vanadium oxide immobilization.14 Therefore, an optimization of the catalytic performance requires detailed knowledge on the influence of the support material.13,15Based on the literature the support material determines the catalytic performance by structural aspects and the interaction of the surface vanadium oxide species with the underlying oxide material. In our efforts to unravel the relation between the structure and activity we first concentrated on structural aspects of the supported vanadium oxide species. To this end silica systems are investigated which are considered as inert support due to their weak interaction with the deposited vanadium oxide species. On the other hand it is known that caused by the weak bonding between the support and supported species, these systems may lead to thermally activated aggregation of the surface species and ultimately to the formation of crystalline V2O5. However, as shown previously at moderate loadings this problem can be overcome by the use of high-surface area support materials. Previous studies of supported vanadium oxides on different high-surface area silicas have revealed that such materials are active and selective for the oxidative dehydrogenation of propane.16,17 However, a detailed comparison of the structural properties of mesoporous silica and conventional silica based vanadium oxide catalysts has not been reported.
Recently, a new insight has been gained regarding the structure of silica supported vanadium oxide both experimentally and theoretically.18–25 In the following, important aspects of these findings will be briefly discussed. While it is now generally accepted that dehydrated vanadium oxide at low vanadium density (typically at <2 atoms nm−2) adopts a pyramidal VO4 structure with one short vanadyl bond, it is still a subject of debate whether exclusively monomeric species or a mixture of monomeric and non-monomeric surface species are present.24 Besides, for monomeric species different structures have been suggested depending on the number of anchoring V–O–Si bonds.25 As a common structural motif non-monomeric species contain the V–O–V fragment (e.g. in dimers). A recent experimental and theoretical study on alumina and silica supported vanadium oxide model systems has demonstrated that the vibrational features observed between 900 and 950 cm−1 originate from the Al–O–V and Si–O–V out-of-phase interface modes rather than V–O–V vibrations as suggested earlier in the literature.18 According to a more detailed theoretical study of the silica system the V–O–V mode of a dimer should appear around 775 cm−1 partly overlapping with silica-related bands.19 Thus, a clear distinction between monomeric and oligomeric species solely based on vibrational spectroscopy appears to be difficult. However, recent experiments on silica SBA-15 supported vanadium oxide catalysts employing X-ray absorption spectroscopy in the soft (NEXAFS)20 and hard X-ray range (EXAFS)21 as well as IR spectroscopy (using NO as probe molecule)23 have provided clear evidence for the presence of oligomeric species (besides monomeric species). A mixture of monomeric and non-monomeric species even at low loadings had been proposed earlier on the basis of a detailed UV-Vis, Raman and IR spectroscopic study.26
In this contribution we report on the influence of the support material and synthesis method on the structure of silica supported vanadium oxide catalysts with comparable vanadium density. As high surface area silica materials stabilized SBA-15 and Aerosil 300 have been employed. After an introduction of the synthesis procedures, the results of the characterization of the support materials (BET) and the deposited surface vanadium oxide species (UV-Vis, Raman, XPS) will be presented. In the final part, the results will be discussed in light of the literature data. A new structural model for silica supported vanadium oxide is proposed, which is consistent with the present findings and previous spectroscopic results.
Experimental
Synthesis
As silica support materials Aerosil 300 (Evonik Degussa GmbH) and SBA-15 are used. Because of its difficult handling Aerosil 300 was pretreated before use by adding water drop by drop until complete wetting and drying at room temperature for 16 h. SBA-15 was prepared as described in the literature.27Fig. 1 summarizes the preparation routes used. The grafting/ion-exchange route consists essentially of three steps, surface functionalization, ion exchange and calcination as described in detail elsewhere.28 Briefly, 2.5 g of the silica material were suspended in 100 ml toluene under stirring and heated to 65 °C. To this suspension 6.4 ml 3-aminopropyltrimethoxysilane (APTMS, 97%, Aldrich) was added and stirred for 12 h. Subsequently, the solvent was removed and the functionalized silica material was washed with toluene. The resulting product was dried overnight and again suspended in 150 ml 0.3 M HCl under stirring for 12 h. After solvent removal and washing with water the functionalized material was dried at 110 °C. As the next step, 2 g of the functionalized silica material were stirred in 60 ml distilled water, and 146 mg butylammonium decavanadate, ([BuNH3]6V10O28), synthesized according to Roman et al.29) were added. The solution was stirred for 12 h at room temperature. The resulting yellow content was filtered, washed with distilled water, and finally calcined in air at 550 °C for 12 h. The resulting samples are named VxOy/SBA-15 and VxOy/A300, respectively.
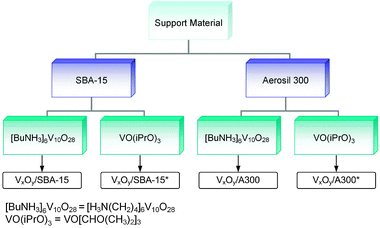
As a second method to incorporate vanadium into silica incipient wetness impregnation was employed using vanadium (V) triisopropoxide oxide (VO(iPrO)3, 95–99%, Alfa Aesar) as a vanadium source. Following the protocol of Gao et al.30 the silica material was dried for 16 h at 110 °C in air. Under an argon atmosphere (glove box), 0.147 ml of VO(iPrO)3 (in the case of SBA-15) were mixed with 5 ml isopropanol. In air, this solution was added dropwise to 1 g of the dry silica material. After drying overnight at room temperature, it was calcined for 12 h at 550 °C in air. In the following, these samples are referred to as VxOy/SBA-15* and VxOy/A300*, respectively.
Physical characterization
Surface areas of the prepared samples were determined by nitrogen adsorption/desorption isotherms and standard multipoint BET analysis methods. The pore volume was determined from the adsorption branch of the N2 isotherm curve at the P/P0 = 0.95 signal point. The pore-size distributions were calculated from the isotherms using the NLDFT method.Visible Raman spectroscopy
Raman spectra were recorded with a fiber probe using 514 nm laser excitation (3 mW) of an Argon ion laser (Melles Griot) at 5 cm−1 spectral resolution (Kaiser Optical, HL5R with CCD). Dehydrated samples were prepared by in situ treatment in 20% O2/N2 (50 ml min−1) at 350 °C for 30 min and subsequent cooling to room temperature.Diffuse reflectance UV-Vis spectroscopy
Diffuse reflectance UV-Vis spectra were measured using a Perkin Elmer Lambda 950 spectrometer equipped with a Harrick diffuse reflectance attachment and a reaction chamber. Spectra were recorded at room temperature. Prior samples were dehydrated in situ in 20% O2/N2 (20 ml min−1) at 300 °C for 30 min. The Kubelka–Munk function F(R∞) was used to convert diffuse reflectance data into absorption spectra using BaSO4 as white standard.XPS
The experiments were performed on a modified LHS/SPECS EA200 MCD system equipped with a Mg Kα source (1253.6 eV, 168 W). A detailed description of the setup as well as the experimental procedures has been given elsewhere.31 Briefly, the binding energy scale of the XPS system was calibrated using Au4f7/2 = 84.0 eV and Cu2p3/2 = 932.67 eV from foil samples. To account for charging of the powder samples the peak of the Si2p signal was set to 103.6 eV. Further data reduction included satellite deconvolution and subtraction of a Shirley background. Quantitative data analysis was performed on the basis of peak areas by fitting with 30/70 Gauss–Lorentz product functions. Atomic ratios were calculated using empirical cross sections.32 Samples were pretreated in 20% O2/N2 at 350 °C for 30 min in an attached flow cell before being transferred to the analysis chamber without exposure to air. Spectra were recorded at room temperature.Results
Physical characterization
Fig. 2 shows the nitrogen adsorption–desorption isotherms for the SBA-15 (left panel) and Aerosil (right panel) supported vanadium oxide samples together with those of the bare silica support materials as reference. The results of the physisorption characterization are summarized in Table 1. Regarding the SBA-15 based samples X-ray diffraction and TEM reveal that the hexagonal structure of SBA-15 is preserved upon deposition of vanadium oxide. As shown in the left panel of Fig. 2 the isotherms show a type IV hysteresis typical for mesoporous materials. Compared to bare SBA-15 the specific surface area decreases to 75% for the incipient wetness impregnated sample (VxOy/SBA-15*) and to 65% for the grafting ion-exchange sample (VxOy/SBA-15). The large drop in surface area in the case of VxOy/SBA-15 can be attributed to a modification of the SBA-15 surface during synthesis as discussed previously.33 With deposition of vanadium oxide the pore size distribution and pore volume slightly shifted to smaller values. This behavior suggests that the vanadium oxide species are located inside the SBA-15 pores. | ||
| Fig. 2 Left panel: N2 isotherms and pore size distributions of (a) blank SBA-15, (b) VxOy/SBA-15* and (c) VxOy/SBA-15. Right panel: N2 isotherms and pore size distributions of (a) blank Aerosil 300, (b) VxOy/A300 and (c) VxOy/A300*. | ||
| V loading/ wt% V | V density/ atoms nm−2 | S BET/ m2 g−1 | d p/ nm | Vp/ ml g−1 | |
|---|---|---|---|---|---|
| SBA-15 | 0 | 0 | 858 | 7.3 | 1.06 |
| VxOy/SBA-15 | 3.0 | 0.5 | 557 | 7.0 | 0.71 |
| VxOy/SBA-15* | 3.1 | 0.59 | 646 | 7.0 | 0.83 |
| Aerosil 300 | 0 | 0 | 300 | — | — |
| VxOy/A300 | 1.37 | 0.54 | 265 | — | — |
| VxOy/A300* | 1.3 | 0.5 | 265 | — | — |
Aerosil 300 is a non-porous silica support. As shown in the right panel of Fig. 2 no significant amount of porosity is created in the Aerosil samples upon vanadium oxide deposition. For VxOy/A300 a hysteresis appears in the isotherm at a relative pressure of 0.8–0.9, which corresponds to pore diameters bigger than 70 nm. As the average particle size of the Aerosil 300 sample is about 7 nm this can be attributed to agglomeration of silica particles. The isotherm of the incipient wetness impregnated sample shows a high amount of adsorbed volume for relative pressures above 0.9 indicating unrestricted monolayer–multilayer adsorption.
Spectroscopic characterization
For characterization of the structure and dispersion of the supported vanadium oxide species UV-Vis spectroscopy, visible Raman spectroscopy and XPS were used. Fig. 3 depicts diffuse reflectance UV-Vis spectra of the catalyst samples in their dehydrated state. For comparison of the shape of the bands the absorption was normalized to the peak maximum. A clear shift of the peak maxima to higher wavelengths can be seen for the Aerosil samples (285 nm) as compared to the SBA-15 samples (260 nm/270 nm). Furthermore, the peaks for the Aerosil samples are considerably broader than those of the SBA-15 samples. Among the SBA-15 samples a peak shift from 260 nm (VxOy/SBA-15, grafting/ion-exchange) to 270 nm (VxOy/SBA-15*, incipient wetness impregnation) is observed but the shape of the curves do not show any significant differences. Both Aerosil samples show a peak maximum at 285 nm. In the case of sample VxOy/A300* an additional shoulder is observed at around 425 nm. Based on previous studies on supported vanadium oxide catalysts, an intensity shift to higher wavelengths is associated with an increase of the degree of oligomerization of the surface vanadium oxide species.26,34,35 Thus for the samples studied here the degree of oligomerization increases in the order: VxOy/SBA-15 < VxOy/SBA-15* < VxOy/A300 < VxOy/A300*. In summary, the use of Aerosil and (to a smaller extent) incipient wetness impregnation leads to larger vanadium oxide aggregates as compared to SBA-15 and grafting/ion-exchange, respectively. | ||
| Fig. 3 Diffuse reflectance UV-Vis spectra of (a) VxOy/SBA-15, (b) VxOy/SBA-15*, (c) VxOy/A300 and (d) VxOy/A300*. | ||
Visible Raman spectra of the silica supported vanadium oxide samples as well as the bare silica supports are shown in Fig. 4. The silica support materials are characterized by bands at around 490, 606, 810 and 980 cm−1, which are attributed to 4-membered siloxane rings (D1 defect mode), 3-membered siloxane rings (D2 defect mode), the symmetrical Si–O–Si stretching mode and the Si–OH stretching mode of free surface silanol groups, respectively.36–39 Upon vanadium oxide deposition a new band appears at 1039 cm−1, which is assigned to the VO (vanadyl) stretch vibration of tetrahedrally coordinated vanadium in OV(−O)3.26,30,40,41 No band around 995 cm−1 is observed for any of the samples. Thus, visible Raman analysis gives no indication for the presence of crystalline V2O5.41
 | ||
| Fig. 4 Raman spectra of (a) VxOy/SBA-15, (b) VxOy/SBA-15*, (c) VxOy/A300, (d) VxOy/A300*, (e) bare SBA-15 and (f) bare Aerosil 300. Spectra are offset for clarity. | ||
Fig. 5 shows the region of the vanadyl vibration in more detail. Obviously, the band around 1040 cm−1 consists of several contributions. For comparison, the spectra were normalized to the intensity maximum of the vanadyl band and fitted within the range 860–1210 cm−1 using a minimum number of five Gaussian functions centered at 915 ± 3 cm−1, 982 ± 3 cm−1, 1027 ± 2 cm−1, 1040 ± 1 cm−1 and 1060 ± 3 cm−1. According to Magg et al.,18 the bands at 915 cm−1 and 1060 cm−1 are readily assigned to out-of phase and in-phase Si–O–V interface modes, respectively. The band at 982 cm−1 is attributed to the Si–OH stretch vibration (see above). The vanadyl band at 1040 cm−1 possesses a shoulder towards lower wavenumbers (1027 cm−1). In general, the observed differences in the Raman spectra are small. Interestingly, the relative intensity ratio of the vanadyl-related bands (I1027/I1040) is higher for the SBA-15 samples (0.79) than for the Aerosil samples (0.47) as will be discussed later in more detail.
 | ||
| Fig. 5 Raman spectra of the vanadyl band of (a) VxOy/SBA-15, (b) VxOy/SBA-15*, (c) VxOy/A300 and (d) VxOy/A300*. Spectra are offset for clarity. | ||
For further characterization of the dispersion of the catalyst samples XPS was used. Fig. 6 shows the X-ray photoelectron spectra of SBA-15 and Aerosil supported vanadium oxide samples. All spectra were normalized to the Si2p peak. The left panel depicts the O1sV2p region dominated by the O1s contribution of the silica support. The right panel gives an enlarged view of the V2p3/2 region together with a least-square fit to the data. For quantitative analysis the V2p3/2 region was deconvoluted into three peaks with binding energies at 519.0 eV, 517.6 eV and 515.8 eV. As discussed previously, these binding energies are attributed to highly dispersed vanadium oxide, bulk-like V5+ and V3+, respectively.41 The results from the peak-fitting analysis are summarized in Table 2. The amount of V3+ is comparable for all samples due to partial reduction of the samples during the XPS analysis. In contrast, significant differences are observed for the ratio of the highly dispersed vanadium oxide to the bulk-like V5+ species e.g. larger vanadium oxide aggregates (see Table 2). The latter ratio represents a measure of the dispersion of the vanadium oxide species and decreases in the order: VxOy/SBA-15 > VxOy/SBA-15* > VxOy/A300 > VxOy/A300*. The dispersion is influenced by both the support material and the synthesis method. The support material with the higher specific surface area, i.e.SBA-15, allows for higher vanadium oxide dispersion. In addition, for each of the support materials samples prepared by grafting/ion-exchange possess a higher dispersion as those prepared by incipient wetness impregnation.
 | ||
| Fig. 6 Left panel: X-ray photoelectron O1sV2p spectra of (a) VxOy/SBA-15, (b) VxOy/SBA-15*, (c) VxOy/A300 and (d) VxOy/A300*. Right panel: V2p3/2 detailed spectra together with the results from the least-square fit. Spectra are offset for clarity. | ||
| Peak area 519.0/eV | Peak area 517.6/eV | Peak area 515.8/eV | Σ | Area ratio 519.0 eV/517.6 eV | |
|---|---|---|---|---|---|
| SBA-15 | 0.1084 | 0.0341 | 0.0258 | 0.1683 | 3.18 |
| SBA-15* | 0.0913 | 0.0357 | 0.0248 | 0.1518 | 2.56 |
| A300 | 0.0336 | 0.0144 | 0.0211 | 0.0691 | 2.33 |
| A300* | 0.0296 | 0.024 | 0.0253 | 0.0789 | 1.22 |
Discussion
The structure of silica supported vanadium oxide has been the subject of numerous studies.18–26,28,30,40 In the following we will focus on catalysts with low vanadium density (0.5–0.7 atoms nm−2), i.e. densities at which no crystalline V2O5 is present, and catalysts in their dehydrated state. Recently, new findings have revived the debate on the vanadium oxide structure of such low-loaded systems mainly towards two aspects: the presence of non-monomeric species as deduced from IR data23 and detailed analysis of X-ray absorption spectra (NEXAFS, EXAFS)21,22 and the (re)assignment of the vanadium oxide-related vibrational bands.18,19 Combining these recent experimental and theoretical results with the present UV-Vis and Raman data as well as previous spectroscopic findings the following information on the surface vanadium oxide species is currently available:- NEXAFS data and IR data (using NO as probe) on SBA-15 supported vanadium oxide (0.7 atoms nm−2) reveal the presence of non-monomeric besides monomeric species.21,23 According to the IR results V–O–V containing species dominate over monomeric species.
- EXAFS data for SBA-15 supported vanadium oxide starting at 0.7 atoms nm−2 up to densities, at which crystalline V2O5 is observed in Raman, can only be fully described if V–V distances are taken into account.22 The data are consistent with a model based on an ordered arrangement of vanadium oxide dimers. However, due to averaging nature of the latter technique the presence of other (minority) species may not be ruled out.
- Regarding vibrational band assignments recent detailed theoretical work on silica supported monomeric vanadium oxide model systems19 has demonstrated that vibrational features observed between 873 and 962 cm−1 originate from a Si–O–V out-of-phase interface mode. The Si–O–V in-phase mode is predicted to be present between 1020 and 1080 cm−1. The vanadyl modes are to be found between 1013 and 1047 cm−1. Oligomers (e.g. dimers) are predicted to show similar frequencies as monomers. Additional V–O–V modes are expected in the range 774–761 cm−1 partly overlapping with silica bands and are thus of limited analytical use.
- Previously, UV-Vis absorption bands of supported vanadium oxide catalysts have been assigned by comparison with those of vanadium oxide reference compounds.26,28,30UV-Vis absorption bands at around 250/290 nm and 280 nm were attributed to tetrahedrally coordinated mono and dimeric vanadates, respectively. Bands at 288/363 nm (NH4VO3) and at 281/353 nm (NaVO3) were assigned to tetrahedrally coordinated polyvanadate, respectively. Thus, the observed UV-Vis spectra for the SBA-15 samples with their maxima at around 260 and 270 nm point to the presence of a mixture of monomeric and dimeric species in both samples with a larger amount of dimeric species in the case of sample VxOy/SBA-15*. The UV-Vis spectra of the Aerosil samples show maxima at around 285 nm and a broad shoulder at around 350 nm strongly suggesting the presence of tetrahedrally coordinated oligomers. Sample VxOy/A300* exhibits an additional shoulder at around 425 nm, which may be attributed to some ribbon-like vanadium oxide with vanadium in square-pyramidal coordination.
- All Raman spectra presented here show a vanadyl stretching band with a shoulder towards lower wavenumbers pointing to (at least) two contributions located at 1027 and 1040 cm−1. The observation of two different VO stretching bands suggests the presence of two types of surface vanadium oxide species. Interestingly, their relative intensity ratio (I1027/I1040) is higher for the SBA-15 samples (0.79) than for the Aerosil samples (0.47). As the Raman spectra were obtained under normal Raman conditions (at most weak resonance enhancements) the ratios resemble the distribution of these two species in the SBA-15 and Aerosil samples assuming the same VO Raman cross section for both species. While visible in a large number of published Raman spectra of silica supported vanadium oxide only few authors have mentioned the occurrence of a shoulder towards lower wavenumber at around 1030 cm−1.25,42 Previously, Raman bands with comparable frequencies have been observed for (partially) hydrated vanadium oxides.43 Under UV excitation (244 nm, 325 nm) the latter feature dominates the vanadyl stretching region. Recently, it has been assigned to a partially hydroxylated vanadium oxide species by comparison with the VO stretching band position of (partially) hydrated vanadium oxide.25
- Previous IR experiments on dehydrated silica supported vanadium oxide have demonstrated the presence of vanadium oxide-related bands around 3660 cm−1, which have been attributed to V–OH stretch vibrations.26,35,45 However, quantification is not possible as the molar extinction coefficient of V–OH is unknown.
As a structural model consistent with all spectroscopic results we propose that the surface vanadium oxide consists of different species with a distorted tetrahedral (VO4) structure containing one short VO bond but a varying degree of hydroxylation and oligomerization (see Scheme 1). Hydroxylated vanadium species, i.e. species with hydroxy ligands V–OH (monomers, oligomers), are characterized by a visible Raman vanadyl band at 1027 cm−1, whereas vanadium species without V–OH show a visible Raman vanadyl band at 1040 cm−1. The smaller degree of oligomerization of the SBA-15 supported vanadium oxides as compared to the corresponding Aerosil systems (see UV-Vis results) is fully consistent with a higher fraction of hydroxylated vanadium atoms V–OH in smaller oligomers (see Raman results). Scheme 1 shows possible structural motifs for silica supported vanadium oxide species. For SBA-15 the presence of smaller oligomers (e.g. n = 1, 2, 3) dominates, whereas the Aerosil samples contain larger oligomers. The UV-Vis data of VxOy/A300* strongly indicate further condensation leading to the formation of ribbon-like elements. Although not explicitly mentioned in the above discussion the presence of monomeric species, circular structures (e.g. tetramers) as well as linkages of vanadium centres via two V–O–V bridges is not excluded.
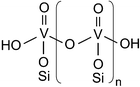 | ||
| Scheme 1 Possible structural motifs for silica supported VxOy species. | ||
In a previous study, the dispersion of the surface vanadium oxide species was investigated using XPS by determining the vanadium to silicon intensity ratio.41 The V/Si intensity ratio was evaluated by using empirically derived atomic sensitivity factors.32 Comparison of the present work with results from the literature46,47 reveals that the observed V2p3/2 binding energies of 517.6 eV and 515.8 eV can be attributed to V5+ and V3+ in the bulk-like environment, respectively. Previously, high binding energy position of the 519.0 eV band had been explained by a final state effect resulting from the Coulomb interaction between the photoelectron and positively charged small vanadium oxide entities, which are too small to screen the charge.41 The ratio of this highly dispersed vanadium oxide species to the bulk-like V5+ species, e.g.vanadium oxide aggregates, defines the dispersion of the surface vanadium oxide species. Previous XPS experiments on SBA-15 supported vanadium oxide have shown that the effect of sample reduction can be efficiently removed by time-dependent analysis without significantly changing the ratio of the highly dispersed to the aggregated species.41 Thus, no influence of the partial reduction of vanadium oxide during XPS experiments on the determination of the dispersion is expected.
Summarising, based on XPS analysis the dispersion of the surface vanadium oxide species decreases in the following order: VxOy/SBA-15 > VxOy/SBA-15* > VxOy/A300 > VxOy/A300*. The higher dispersion in the case of SBA-15 as compared to Aerosil can be explained by the higher surface area of the mesoporous material facilitating the distribution of vanadium oxide over the surface. With respect to the synthesis method, for both support materials, the grafting/ion-exchange method gave a higher dispersion than incipient wetness impregnation. This behavior may be explained by considering the distribution of the vanadium precursor over the silica surface prior to calcination. During the ion-exchange step (driven by electrostatic interactions) the decavanadate ions are first homogeneously distributed over the silica surface and subsequently fragmented into smaller vanadium oxide entities during the calcination step. In contrast, in the case of incipient wetness impregnation less control over the surface distribution of the precursor species is attained leading to increased aggregation during calcination and overall lower dispersions.
In comparison to the above order of dispersion, the degree of oligomerization of the vanadium oxide species, as deduced from the UV-Vis data, increases in the reverse order. While such a behavior may have been expected it should be mentioned that information on the structure and dispersion have been deduced from independent experiments thus underlining the consistency of the results.
Summary
Two high surface area silica materials (Aerosil 300, SBA-15) and two synthesis methods (grafting/ion-exchange, incipient wetness impregnation) have been compared regarding their influence on the structure and dispersion of silica supported vanadium oxide. At comparable vanadium density, the support material has a stronger effect on the structure and dispersion as the synthesis method. The use of the higher surface area support (SBA-15) leads to a lower degree of oligomerization of the surface vanadium oxide species, i.e. a higher dispersion, as compared to Aerosil 300. Regarding the synthesis procedure a smaller but distinct influence on the vanadium oxide structure/dispersion has been observed. For both support materials incipient wetness impregnation leads to more oligomerized vanadium oxide species as compared to grafting/ion-exchange. Thus, within limits, the structure/dispersion of the deposited surface vanadium oxide species can be controlled by both the choice of the support and the synthesis method.Acknowledgements
The authors would like to thank Gisela Lorenz for conducting the N2 adsorption–desorption experiments. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) via an Emmy Noether grant (C.H.) and the collaborative research center SFB 546.References
- A. Parmaliana, F. Fursteri, A. Mezzapica, M. S. Scurrell and N. Giordano, J. Chem. Soc., Chem. Commun., 1993, 751–753 RSC.
- R. G. Herman, Q. Sun, C. Shi, K. Klier, C. B. Wang, H. Hu, I. E. Wachs and M. M. Bhasin, Catal. Today, 1997, 37, 1–14 CrossRef CAS.
- M. A. Banares, J. L. Alemany, M. L. Granados, M. Faraldos and J. L. G. Fierro, Catal. Today, 1997, 33, 73–83 CrossRef CAS.
- H. Berndt, A. Martin, A. Brückner, E. Schreier, D. Müller, H. Kosslick, G. U. Wolf and B. Lücke, J. Catal., 2000, 191, 384–400 CrossRef CAS.
- V. Fornes, C. Lopez, H. H. Lopez and A. Martinez, Appl. Catal., A, 2003, 249, 345–354 CrossRef CAS.
- D. A. Ruddy, N. L. Ohler, A. T. Bell and T. D. Tilley, J. Catal., 2006, 228, 277–285 CrossRef.
- P. Forzatti, E. Tronconi, A. S. Elmi and G. Busca, Appl. Catal., A, 1997, 157, 387–408 CrossRef CAS.
- J. M. Tatibouet, Appl. Catal., A, 1997, 148, 213–252 CrossRef.
- L. J. Burcham, G. T. Gao, X. T. Gao and I. E. Wachs, Top. Catal., 2000, 11, 85–100 CrossRef.
- T. Blasco and J. M. L. Nieto, Appl. Catal., A, 1997, 157, 117–142 CrossRef CAS.
- O. V. Buyevskaya, A. Brückner, E. V. Kondratenko, D. Wolf and M. Baerns, Catal. Today, 2001, 67, 369–378 CrossRef CAS.
- J. M. L. Nieto, Top. Catal., 2006, 41, 3–15 CrossRef.
- A. Dinse, B. Frank, C. Hess, D. Habel and R. Schomäcker, J. Mol. Catal. A: Chem., 2008, 289, 28–37 CrossRef CAS.
- B. M. Weckhuysen and D. N. Keller, Top. Catal., 2003, 78, 25–46 CAS.
- P. Gruene, T. Wolfram, K. Pelzer, R. Schlögl and A. Trunschke, Catal. Today, 2010, 157, 137–142 CrossRef CAS.
- Y.-M. Liu, Y. Cao, N. Li, W.-L. Feng, W.-L. Dai, S.-R. Yan, H.-Y. He and K.-N. Fan, J. Catal., 2005, 224, 417–428 CrossRef.
- I. E. Kondratenko, M. Cherian, M. Baerns, D. Su, R. Schlögl, X. Wang and I. E. Wachs, J. Catal., 2005, 234, 131–142 CrossRef.
- N. Magg, B. Immaraporn, J. B. Giorgi, T. Schroeder, M. Bäumer, J. Döbler, Z. Wu, E. Kondratenko, M. Cherian, M. Baerns, P. C. Stair, J. Sauer and H.-J. Freund, J. Catal., 2004, 226, 88–100 CrossRef CAS.
- J. Döbler, M. Pritzsche and J. Sauer, J. Phys. Chem. C, 2009, 113, 12454–12464 Search PubMed.
- M. Cavalleri, K. Hermann, A. Knop-Gericke, M. Hävecker, R. Herbert, C. Hess, A. Oesterreich, J. Döbler and R. Schlögl, J. Catal., 2009, 262, 215–223 CrossRef CAS.
- M. Hävecker, M. Cavalleri, R. Herbert, R. Follath, A. Knop-Gericke, C. Hess, K. Hermann and R. Schlögl, Phys. Status Solidi B, 2009, 264, 1459–1469 CrossRef.
- A. Walter, C. Hess, R. Herbert and T. Ressler, Chem. Cent. J, 2010, 4 Search PubMed.
- T. V. Venkov, C. Hess and F. C. Jentoft, Langmuir, 2007, 23, 1768–1777 CrossRef CAS.
- C. Hess, ChemPhysChem, 2009, 10, 319–326 CrossRef CAS.
- Z. Wu, S. Dai and S. H. Overbury, J. Phys. Chem. C, 2010, 114, 412–422 CAS.
- M. Schraml-Marth, A. Wokaun, M. Pohl and H. L. Krauss, J. Chem. Soc., Faraday Trans., 1991, 87, 2635 RSC.
- D. Y. Zhao, Q. S. Huo, J. L. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- C. Hess, J. D. Hoefelmeyer and T. D. Tilley, J. Phys. Chem. B, 2004, 108, 9703–9709 CrossRef CAS.
- P. Roman, A. Aranzabe, A. Luque and J. M. Gutierrez-Zorilla, Mater. Res. Bull., 1991, 26, 731–740 CrossRef CAS.
- X. Gao, S. R. Bare, B. Weckhuysen and I. E. Wachs, J. Phys. Chem. B, 1998, 102, 10842–10852 CrossRef CAS.
- C. Hess, U. Wild and R. Schlögl, Microporous Mesoporous Mater., 2006, 95, 339–349 CrossRef CAS.
- D. Briggs and M. P. Seah, Practical surface analysis, Wiley, Chichester, 1990 Search PubMed.
- R. Herbert, D. Wang, R. Schomäcker, R. Schlögl and C. Hess, ChemPhysChem, 2009, 10, 2230–2233 CrossRef CAS.
- F. Arena, F. Frusteri, G. Martra, S. Cosuccia and A. Parmaliana, J. Chem. Soc., Faraday Trans., 1997, 93, 3849–3854 RSC.
- M. Baltes, K. Cassiers, P. V. D. Voort, B. M. Weckhuysen, R. Schoonheydt and E. F. Vansant, J. Catal., 2001, 197, 160–171 CrossRef CAS.
- C. J. Brinker, R. Kirkpatrick, D. R. Tallant, B. C. Bunker and B. Montez, J. Non-Cryst. Solids, 1988, 99, 418–428 CrossRef CAS.
- D. R. Tallant, B. C. Bunker, C. J. Brinker and C. A. Balfe, Mater. Res. Soc. Symp. Proc., 1986, 73, 261–267 CrossRef CAS.
- R. H. Stolen and G. E. Walrafen, J. Chem. Phys., 1976, 64, 2623–2631 CrossRef CAS.
- B. C. Brinker, D. R. Tallant, E. P. Roth and C. S. Ashley, Mater. Res. Soc. Symp. Proc., 1986, 61, 387 CrossRef.
- G. T. Went, S. T. Oyama and A. T. Bell, J. Phys. Chem., 1990, 94, 4240–4246 CrossRef CAS.
- C. Hess, G. Tzolova-Müller and R. Herbert, J. Phys. Chem. C, 2007, 111, 9471–9479 CAS.
- H. Miyata, S. Tokuda and T. Yoshida, Appl. Spectrosc., 1989, 43, 522–526 CrossRef CAS.
- L. Abello, E. Husson, Y. Repelin and G. Lucazeau, J. Solid State Chem., 1985, 56, 379 CrossRef CAS . The Raman spectra in this study were recorded at 647 nm excitation. Spectra of some of our samples taken at 632 nm excitation44 are in excellent agreement with those of Abello et al. and the 514 nm spectra presented here.
- C. Hess, J. Catal., 2007, 248, 120–123 CrossRef CAS.
- L. D. Nguyen, S. Loridant, H. Launay, A. Pigamo, J. L. Dubois and J. M. M. Millet, J. Catal., 2006, 237, 38–48 CrossRef CAS.
- G. A. Sawatzky and D. Post, Phys. Rev. B: Condens. Matter, 1979, 20, 1546–1555 CrossRef CAS.
- M. A. Eberhardt, A. Proctor, M. Houalla and D. M. Hercules, J. Catal., 1996, 160, 27–34 CrossRef CAS and references therein.
Footnote |
| † Present address: Uhde GmbH, Friedrich-Uhde-Strasse 15, 44141 Dortmund, Germany. |
| This journal is © The Royal Society of Chemistry 2011 |