Gold catalysis at the gas–solid interface: role of the support in determining activity and selectivity in the hydrogenation of m-dinitrobenzene
Received
11th February 2011
, Accepted 3rd April 2011
First published on 5th May 2011
Abstract
The catalytic gas phase hydrogenation of m-dinitrobenzene (1 atm, 423 K) over laboratory synthesised (1 mol%) Au supported on Al2O3, TiO2, Fe2O3 and CeO2 and a reference Au/TiO2 (World Gold Council) has been investigated. The catalysts were prepared by deposition-precipitation (DP) and impregnation (IMP), where the former route generated smaller (surface area weighted) mean Au particle sizes (1.5–2.8 nm) compared with the IMP synthesis (3.5–9.0 nm). The catalysts have been characterized in terms of temperature programmed reduction (TPR), H2 chemisorption/temperature programmed desorption (TPD), BET area, powder X-ray diffraction (XRD) and high resolution transmission electron microscopy (HRTEM) analyses. Hydrogen consumption over the T range 371–457 K during activation of Au on TiO2, Al2O3 and CeO2 can be associated with Au3+ → Au0 reduction. XRD analysis demonstrated the presence of metallic gold in Au/Fe2O3pre-TPR. A partial and complete reduction of the hematite support to magnetite (Fe2O3 → Fe3O4) was observed post-TPR to 423 and 673 K, respectively; H2-TPD results suggest the participation of spillover hydrogen in this step. Exclusive –NO2group reduction and time invariant conversions were observed for all the catalysts considered in this study. An increase in the specific hydrogenation rate with a decrease in the mean Au size (from 9 to 3 nm) was observed. m-Nitroaniline was generated as the sole product over Au/TiO2 and Au/Fe2O3 whereas Au/CeO2 promoted the exclusive formation of m-phenylenediamine and a mixture of both products was obtained over Au/Al2O3. Our findings establish a basis for the development of a sustainable (clean and continuous) process for the hydrogenation of m-dinitrobenzene where product composition can be controlled through the choice of the oxide support.
1. Introduction
Aromatic amines are important intermediates in the manufacture of fine chemicals, herbicides, pesticides, dyes and pigments.1 The conventional route to amino-arenes is the batch hydrogenation of the corresponding nitro-compound either in dilute acid media using Fe (Béchamp process)2 or over Ni,3Pt4 and Pd5catalysts. Both approaches suffer major sustainability drawbacks in terms of low product yields and multi-stage waste (toxic iron oxide sludge and azo/azoxy-derivates) treatment/disposal.6 Reaction selectivity is particularly challenging in the hydrogenation of m-dinitrobenzene (m-DNB) in terms of the exclusive formation of either m-nitroaniline (m-NAN, i.e. partial –NO2reduction) or m-phenylenediamine (m-PDM, i.e. complete –NO2reduction).7,8 The catalytic activity of Au in hydrogen mediated reactions is appreciably lower than that of Group VIII metals.9–11 The higher rates delivered by Ni-, Pd-, or Pt-based catalysts have been associated with the greater facility of these metals (relative to Au) to dissociatively adsorb H2.12 However, supported Au at the nano-scale (≤10 nm) exhibits unique selectivities in a number of hydrogenation reactions.13,14 To date, the hydrogenation of aromatic nitro compounds over Au catalysts has focused on batch reaction in the liquid phase10,15–20 conducted at high H2 pressures (3–4 MPa).18–20
The catalytic response in hydrogenation reactions over supported Au systems can be governed by contributions due to the dispersion, morphology and electronic properties of the Au phase which, in turn, can be influenced by interactions with the support.21,22Oxides have been the most widely used Au carriers where the support redox and/or acid–base properties can impact on catalytic performance.23 The redox character has a direct role to play where a partial reduction of the support generates Au–oxide interactions that affect Au morphology,24,25 resulting in a preponderance of a particular crystallographic plane with distinct catalytic properties.26 Support acid–base properties27,28 can result in electron transfer24,29 from/to the metal phase giving rise to positively (electron-deficient) or negatively charged Au nano-particles.26 The available literature that addresses support effects in hydrogenation reactions over Au is limited but Milone and co-workers30 have shown that the use of a reducible support (notably Fe2O3) strongly influenced activity/selectivity in benzalacetone and cinnamaldehyde hydrogenation, where the formation of electron rich Au species, via support–metal electron transfer, enhanced C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O activation. Lopez-Sanchez and Lennon,22 using Au/TiO2 and Au/Fe2O3 to promote the hydrogenation of propyne, reported differences in activity–selectivity, which they ascribed to different metal–support interactions in both systems. Campo et al.,31 in the gas phase hydrogenation of crotonaldehyde over Au/CeO2 and Au/Nb2O5, recorded a distinct catalytic response in that Au/CeO2 was highly selective to crotyl alcohol but Au/Nb2O5 was non-selective and they attributed this to the formation of unselective Au particles on Nb2O5 with a different morphology. Bailie and Hutchings32 obtained differences in selectivity for the hydrogenation of crotonaldehyde over Au supported on ZnO and ZrO2 and they established that treatment with thiophene affected selectivity (increasing rate of formation of crotyl alcohol), proposing that sites at the Au–support interface were responsible for carbonyl activation.
O activation. Lopez-Sanchez and Lennon,22 using Au/TiO2 and Au/Fe2O3 to promote the hydrogenation of propyne, reported differences in activity–selectivity, which they ascribed to different metal–support interactions in both systems. Campo et al.,31 in the gas phase hydrogenation of crotonaldehyde over Au/CeO2 and Au/Nb2O5, recorded a distinct catalytic response in that Au/CeO2 was highly selective to crotyl alcohol but Au/Nb2O5 was non-selective and they attributed this to the formation of unselective Au particles on Nb2O5 with a different morphology. Bailie and Hutchings32 obtained differences in selectivity for the hydrogenation of crotonaldehyde over Au supported on ZnO and ZrO2 and they established that treatment with thiophene affected selectivity (increasing rate of formation of crotyl alcohol), proposing that sites at the Au–support interface were responsible for carbonyl activation.
We have shown previously33–37 that oxide supported Au is 100% selective in the gas phase continuous hydrogenation of a range of substituted mono-nitroarenes to the target aromatic amine. This study represents an extension to that work where we examine the hydrogenation of m-DNB over Au supported on a group of reducible (CeO2, TiO2 and α-Fe2O3) and non-reducible (γ-Al2O3) oxides. We demonstrate that the product composition can be adjusted through the choice of support.
2. Result and discussion
2.1
Catalyst characterization
2.1.1 BET/TPR/XRD analyses.
BET surface areas of the activated (1 mol%) Au catalysts considered in this study are recorded in Table 1. The TPR activation profiles associated with the supports (γ-Al2O3 (A), TiO2 (B) and CeO2 (C)) are given in Fig. 1 (profiles labelled as I) and those generated for supported Au prepared by DP and IMP appear as profiles II and III, respectively. The temperatures associated with maximum H2 consumption (Tmax) during TPR of the supported Au catalysts are provided in Table 1. The profile obtained for the non-reducible γ-Al2O3 support is featureless, with no evidence of H2 uptake or release, a response that was expected and is consistent with the literature.38 In contrast, over the same temperature range (up to 1073 K), the reducible supports exhibited a positive signal (H2 consumption). Titania presented a broad peak with a Tmax at ca. 850 K, which can be linked to H2 consumption at T ≥ 773 K reported previously39 for TiO2 reduction. The ceria support also exhibited a broad positive signal (Tmax at ca. 710 K), which can be attributed to surface reduction.40 TPR profiles generated for the catalysts prepared by DP (profiles II) are characterised by a H2 consumption peak over the range 371–450 K. A single TPR peak (in the range 373–633 K) has been reported in the literature41,42 and ascribed to the reduction of Au3+ species to Au0. The different Tmax values registered for the DP samples can be associated with differences in metal–support interactions for these Au–oxide systems. Indeed, Liu and Yang43 have reported the reduction of gold species at 378 K and 450 K for Au/TiO2 and Au/Al2O3, respectively. Delannoy and co-workers44 have considered the role of the support in modifying Au reducibility for DP systems and reported a decrease in Au(III) reducibility in the sequence TiO2 > CeO2 > Al2O3. The profiles for the IMP samples present a principal peak at 434 K that is in agreement with the literature.45,46 In addition, a secondary peak at 457 K was observed in the case of Au/TiO2–IMP(1) (Fig. 1BIII). Yim and Nam47 reported a similar response for CrOx supported on Al2O3 and TiO2, where the TPR profiles were characterized by one (593–663 K) and two (553–593 K and 693–713 K) reduction peaks, respectively. Idakiev et al.48 associated a TPR peak at 373 K to a Ti4+ → Ti3+ transformation at the metal/support interface for Au/TiO2. The H2 consumption values recorded for all the Au catalysts presented in Fig. 1 (DP and IMP) are close to that required for the reduction of the supported precursor (see Table 1), suggesting that the positive peak(s) recorded during TPR are due to reduction of the precursor to metallic Au.
Table 1
Gold particle size obtained by TEM (dTEM) analysis, hydrogen consumed during TPR (to 423 or 673 K) with associated Tmax, H2 chemisorption values and BET surface area for a series of oxide-supported Au catalysts prepared by deposition-precipitation (DP) and impregnation (IMP)
|
Catalyst
|
d
TEM
/nm |
TPR Tmax/K |
Theoretical TPR H2 consumptiona/μmol g−1 |
Measured TPR H2 consumption/μmol g−1 |
H2 uptake/μmol g−1Au |
BET area/m2 g−1 |
|
Theoretical amount of H2 necessary to reduce the Au precursor.
TPR to 673 K (see Fig. 2, solid line in profiles II and III).
TPR to 423 K (see Fig. 2, dashed line in profiles II and III).
No hydrogen consumption detected during TPR.
|
|
Au/Al2O3-DP(1) |
2.8 |
450 |
67 |
62 |
33 |
142 |
|
Au/CeO2-DP(1) |
1.5 |
423 |
69 |
63 |
63 |
195 |
|
Au/Fe2O3-DP(1) |
2.6 |
366b,c, 512b |
62 |
2093b |
21 |
57 |
| 750c |
|
Au/TiO2-DP(1) |
2.4 |
371 |
78 |
74 |
28 |
48 |
|
Au/TiO2-Ref |
3.3 |
—d |
79 |
—d |
46 |
50 |
|
Au/Al2O3-IMP(1) |
9.0 |
434 |
152 |
146 |
22 |
161 |
|
Au/Fe2O3-IMP(1) |
3.5 |
370b,c, 508b |
62 |
2050b |
27 |
56 |
| 700c |
|
Au/TiO2-IMP(1) |
6.0 |
434, 457 |
193 |
185 |
16 |
47 |
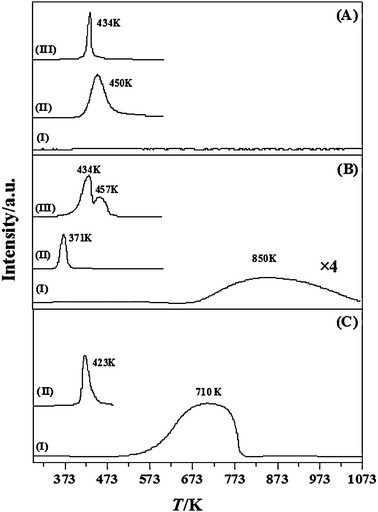 |
| | Fig. 1 TPR profiles generated for: (A) Al2O3 support (I), Au/Al2O3-DP(1) (II) and Au/Al2O3-IMP(1) (III); (B) TiO2 support (I), Au/TiO2-DP(1) (II) and Au/TiO2-IMP(1) (III); (C) CeO2 support (I) and Au/CeO2-DP(1) (II). | |
 |
| | Fig. 2 TPR profiles for (I) α-Fe2O3, (II) Au/Fe2O3-DP(1) and (III) Au/Fe2O3-IMP(1) activated to 423 K (dashed line) or 673 K (solid line). Common XRD patterns for Fe2O3, Au/Fe2O3-DP(1) and Au/Fe2O3-IMP(1): (A) starting (unreduced) samples; (B) samples activated at 673 K. Note: peak assignments based on JCPDS-ICDD reference data: (■) α-Fe2O3 (33-0664); (●) Fe3O4 (19-0629). | |
The X-ray diffractogram patterns for the as prepared Fe2O3, Au/Fe2O3-DP(1) and Au/Fe2O3-IMP(1) are identical and the common profile is presented as an inset (A) in Fig. 2. The three systems present signals at 2θ = 24.1°, 33.2°, 35.6°, 40.9°, 49.5°, 54.1°, 57.6°, 62.4° and 64.0° corresponding to the (012), (104), (110), (113), (024), (116), (018), (214) and (300) planes of α-phase Fe2O3 (hematite, JCPDS-ICDD 33-0664). The XRD patterns of the three samples post-TPR (to 673 K) (see inset (B), Fig. 2) were also identical with reflections at 2θ = 30.1°, 35.4°, 43.1°, 53.4°, 56.9°, 62.5°, 70.9°, 73.9° and 89.6° associated with the (220), (311), (400), (422), (511), (440), (620), (533) and (731) planes of Fe3O4 (magnetite, JCPDS-ICDD 19-0629). The TPR profile for the starting hematite support (see Fig. 2(I)) exhibits one main H2 consumption peak at 647 K that can be associated with the formation of Fe3O4.49 Two peaks at 368 ± 2 K and 510 ± 2 K characterize the TPR profiles (up to 673 K) obtained for Au/Fe2O3-DP(1) and Au/Fe2O3-IMP(1) (Fig. 2, solid lines in profiles II and III, respectively). The total hydrogen consumption during TPR matched (to within ±5%) that required for the reduction of Fe2O3 → Fe3O4 in the three systems. A comparison of the TPR response of Au/Fe2O3 with that of Fe2O3 suggests that Au served to lower the temperature requirements for the reduction of hematite to magnetite. Indeed, a decrease (by up to 200 K) in the reduction temperature of Fe2O3 to Fe3O4 has been reported post-Au incorporation.50,51 The TPR profiles generated for sample activation up to 423 K (reaction temperature) are identified in Fig. 2 by dashed lines. This activation temperature was chosen based on the results presented in Fig. 1, which show that the Au3+ → Au0 reduction step occurred at T ≤ 457 K. The quantity of hydrogen associated with the single peak (368 ± 2 K) recorded in Table 1 exceeded (by up to a factor of 12) the amount required to reduce the Au precursor to the metallic form but was significantly lower (by a factor of 3) than that required to fully transform Fe2O3 to Fe3O4. These findings suggest that the presence of Au results in a “partial” reduction of the iron oxide support at 423 K. Indeed, Liet al.52 reported a reduction peak at ca. 373 K for Au/Fe2O3 prepared by DP and associated this with a partial reduction of Fe2O3 to Fe3O4. In order to confirm this effect, XRD analysis was conducted and the results are presented in Fig. 3. The XRD patterns of the activated (to 423 K) Au/Fe2O3-DP(1) (I) and Au/Fe2O3-IMP(1) (II) show XRD reflections that are characteristic of α-Fe2O3 (hematite) (see profile V). Nevertheless, it should be noted that the relative intensity of the two main peaks at 2θ = 33.2° and 35.6° ((104)/(110)) differs from that which characterises the reference hematite (see dashed square “a” in Fig. 3): α-Fe2O3 = 4/3 (V); Au/Fe2O3-DP(1) = 3/4 (I); Au/Fe2O3-IMP(1) = 1/1 (II). As the XRD peak for the (110) plane of hematite ((V), 2θ = 35.6°) coincides with the main characteristic peak for magnetite ((VI); 2θ = 35.4°, plane (311)), the variation in relative intensities suggests a partial transformation of hematite to magnetite during the reduction of Au/Fe2O3. Moreover, this step appears to be promoted to a greater extent for Au/Fe2O3-DP(1) when compared with Au/Fe2O3-IMP(1). Wang et al.53 suggested that the presence of Auδ+ modifies the polarity of the Fe–O bond, resulting in a more facile reduction of Fe2O3 to Fe3O4. A similar trend is observed for the XRD signals at 2θ = 62.4° and 64.0° (planes (214) and (300) of α-Fe2O3) and the (440) plane (at 2θ = 62.5°) of Fe3O4 (see dashed square “b” in Fig. 3), i.e. modified relative intensities for the characteristic signals of hematite with simultaneous appearance of one of the main peaks of magnetite, a result that is consistent with a partial reduction of the support. Moreover, the formation of magnetitepost-activation (at 423 K) is confirmed by the presence of a new peak at 2θ = 56.9°, associated with the (511) plane of Fe3O4. Thus, the “overconsumption” of H2 during TPR to 423 K (see Table 1) can be attributed to Au induced partial reduction of α-Fe2O3.
 |
| | Fig. 3
XRD patterns associated with passivated/reduced (at 423 K) (I) Au/Fe2O3-DP(1), (II) Au/Fe2O3-IMP(1), (III) Au/Fe2O3-IMP(10) and (IV) as prepared Au/Fe2O3-IMP(10); JCPDS-ICDD reference diffractograms for (V) α-Fe2O3 (33-0664), (VI) Fe3O4 (19-0629) and (VII) Au (04-0784). | |
It should be noted that there were no detectable signals for metallic Au (VII, JCPDS-ICDD 04-0784) in the XRD profiles for Au/Fe2O3-DP(1) (I) or Au/Fe2O3-IMP(1) (II), which suggests a highly dispersed Au phase. It has been established54,55 that one of the main factors determining the Au particle size is metal loading in that a higher content favours agglomeration and the formation of larger Au particles. In order to confirm Au precursor reduction to Au0post-TPR, a higher Au loading (10 mol%) on Fe2O3 was prepared (Au/Fe2O3-IMP(10)) and activated under the same conditions, where the expected formation of larger Au particles should result in the appearance of XRD signals due to Au. The XRD diffractograms for the activated and as prepared Au/Fe2O3-IMP(10) (III and IV, respectively, in Fig. 3) exhibit, in addition to the reflections due to the support, peaks for Au metal (VII, JCPDS-ICDD 04-0784), i.e. 2θ = 38.1°, 44.4°, 64.7° and 77.5° corresponding to (111), (200), (220) and (311) planes, respectively. The activated sample presents stronger Au metal XRD reflections, a response consistent with an increase in the Au particle size upon thermal treatment,55,56i.e. from 7 nm to 12 nm (based on standard line broadening analysis),34 in the as prepared and reduced samples, respectively. The relative intensity of the (104)/(110) hematite peaks in the activated Au/Fe2O3-IMP(10) (4/3) (Fig. 3III) is close to that recorded for the support (4/3, Fig. 3V), indicating a limited degree of α-Fe2O3 reduction in the higher Au loaded sample when compared with Au/Fe2O3-IMP(1) (1/1, Fig. 3II). Moreover, the appearance of an Au XRD peak in the as prepared Au/Fe2O3-IMP(10) (Fig. 3IV) suggests that a reduction of the Fe2O3 supported Au precursor can occur pre-TPR, i.e. during the preparation and drying (at 383 K) steps. In previous work,57 we demonstrated that a supported gold ethanediamine precursor can undergo decomposition to metallic Au during drying (at 373 K). Hodge et al.58 have employed Mössbauer spectroscopy to demonstrate the presence of a mainly metallic Au (up to 86% content) component after subjecting Au/Fe2O3, prepared by co-precipitation, to a drying step (up to 393 K). Milone and co-workers30 reported a single reduction peak in the TPR of Au/Fe2O3-DP that is common to the Fe2O3 support and which they took as proof of Au reductionpre-TPR. There is also evidence in the literature suggesting the presence of unreduced gold species pre-TPR with H2 consumption peaks reported at 468 K,59 489 K38 and 425 K60 for Au/Fe2O3 prepared by DP, IMP and co-precipitation. Our TPR and XRD measurements are consistent with the presence of Au0pre-activation where the presence of small Au particles promotes the transformation (during TPR) of Fe2O3 to Fe3O4.
2.1.2
TEM analysis.
TEM images of three representative samples, i.e.Au/Fe2O3-DP(1) (A), Au/Fe2O3-IMP(1) (B) and Au/Fe2O3-IMP(10) (C), are given in Fig. 4. It is immediately evident that Au is present as discrete particles with a quasi-spherical morphology. The diffractogram patterns for a single gold particle in the case of Au/Fe2O3-DP(1) (AII) and Au/Fe2O3-IMP(1) (BII) are shown in images AIII and BIII, respectively. The d-spacings (0.20/023) are consistent with the (111) and (200) planes of metallic gold. EDX analysis confirmed the presence of Au0 in all the areas mapped. The absence of a signal due to metallic Au in the XRD diffractograms for Au/Fe2O3-DP(1) and Au/Fe2O3-IMP(1) can be attributed to the presence of small Au crystallites (<5 nm) in both catalysts. Au/Fe2O3-IMP(1) exhibited larger particles than Au/Fe2O3-DP(1) with surface area weighted mean particle sizes of 3.5 and 2.6 nm, respectively. This response extends to Au supported on Al2O3 and TiO2 (see Table 1) and agrees with the general consensus which emerges from the literature that catalysts prepared by DP consistently generate small Au particles (<5 nm).54,61Catalyst preparation by IMP, even at low Au loadings (1–2% w/w), typically delivers low Au dispersions (10–35 nm).23 Okumura and co-workers,62 taking Au/Al2O3 (1% w/w) prepared by DP and IMP, quoted mean Au particle sizes of 2.5 nm and 37 nm, respectively. Bailie and Hutchings32 reported that Au/ZnO (5% w/w) prepared by DP and IMP exhibited Au particles of 2–4 nm and >150 nm, respectively. It should be noted that, regardless of the preparation method (DP or IMP), larger Au particles are associated with the non-reducible carrier (Al2O3). This suggests a correlation between the reducibility of the support and Au dispersion, an observation that is in line with the work of Min et al.25 who demonstrated the formation of smaller Au clusters on reducible oxides due to the greater number of nucleation sites. The sequence of decreasing Au particle size obtained from TEM analysis for the Fe2O3 supported samples (Au/Fe2O3-IMP(10) (4.6 nm) > Au/Fe2O3-IMP(1) (3.5 nm) > Au/Fe2O3-DP(1) (2.6 nm)) matches the increasing deviation in the relative intensity of the two principal hematite XRD peaks ((104)/(110)) when compared with the as prepared support, i.e.α-Fe2O3 (4/3) ≈ Au/Fe2O3-IMP(10) (4/3) > Au/Fe2O3-IMP(1) (1/1) > Au/Fe2O3-DPU(1) (3/4); see Fig. 3. This result suggests that the partial reduction of hematite is promoted to a greater extent over smaller Au particles (<5 nm), which can be explained on the basis of the increased Au–support interface associated with higher Au dispersion that must facilitate reduction of the support during TPR.
 |
| | Fig. 4 Representative TEM images of reduced/passivated (at 423 K) (A) Au/Fe2O3-DP(1), (B) Au/Fe2O3-IMP(1) and (C) Au/Fe2O3-IMP(10): (I) medium/high magnification images; (II) images of a single Au particle (with associated diffractogram pattern (III)). | |
2.1.3 H2 chemisorption/TPD analyses.
Hydrogen uptake (see Table 1) on all the supported Au catalysts was low (≤63 μmol g−1Au). We could not find any other report in the literature that provides a measure of room temperature H2 chemisorption on comparable supported Au systems. Our recorded H2 uptake values are significantly lower than those obtained for conventional supported transition metal catalysts with a similar metal content (and dispersion), e.g.Pd/Al2O3 (1870 μmol g−1Pd)63 and Pt/SiO2 (1461 μmol g−1Pt).64 The dynamics of H2 adsorption on Au are still far from being resolved but recent studies suggest that the chemisorption temperature65 and Au coordination number13 are key factors. Bus et al.64 demonstrated an increase in the amount of chemisorbed H2 (by up to a factor of 3) with an increase in the temperature (298 K → 373 K) used during their titration measurements. In terms of Au particle size dependence, there is some consensus that there is an increase in H2 uptake on small clusters11,62 (with higher number of defects) when compared with bulk Au.66Hydrogen temperature programmed desorption (TPD) is a practical approach that can serve to indicate differences in metal/support interactions and electronic properties of supported metals.67 The TPD profiles (up to 873 K), following H2 chemisorption, generated for Au/Fe2O3-DP(1) (I), Au/Fe2O3-IMP(1) (II) and Au/Fe2O3-IMP(10) (III) are shown in Fig. 5. Each profile exhibits a region of desorption over the T range 500–873 K with a maximum at ca. T = 700 K in the case of the lower Au loading: TPD from Au/Fe2O3-IMP(10) generated a broad ill-defined profile. In each case, the total specific (per m2Au) volume of H2 desorbed was appreciably greater (by two orders of magnitude) than that taken up in the chemisorption step. This response suggests that the majority of the hydrogen released from each sample was generated during the TPR step as spillover species.68 This assertion is in line with recent studies that have demonstrated (by FTIR) hydrogen spillover formation associated with Au supported on TiO269 and CeO2–ZrO2.70 In terms of energy requirements, the desorption of spillover hydrogen has been shown to require temperatures in excess of 503 K71,72 while desorption directly from the metal occurs at lower temperatures (T < 473 K).73 Taking the three catalysts presented in Fig. 5, the specific volume of H2 desorbed decreased in the order: Au/Fe2O3-DP(1) (42 μmol m−2) > Au/Fe2O3-IMP(1) (14 μmol m−2) > Au/Fe2O3-IMP(10) (4 μmol m−2). This sequence matches that of increasing Au particle size and there is evidence in the literature that the H2 spillover phenomenon is structure sensitive.67,70 The higher level of surface hydrogen associated with Au/Fe2O3-DP(1) can account for the greater degree of support reduction (transformation of hematite to magnetite) observed for this sample. Indeed, Boccuzzi et al.74 have suggested that spillover hydrogen on Au/Fe2O3 can participate in a superficial reduction of the support.
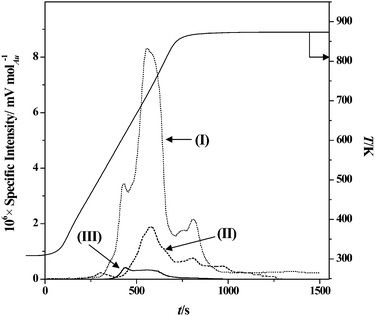 |
| | Fig. 5 H2TPD (to 873 K) for: (I) Au/Fe2O3-DP(1) (dotted line); (II) Au/Fe2O3-IMP(1) (dashed line); (III) Au/Fe2O3-IMP(10) (solid line). | |
2.2
Catalyst activity/selectivity
The gas phase hydrogenation of m-DNB over all the Au catalysts delivered a time-invariant conversion (for up to 5 h on-stream, not shown) and 100% selectivity in terms of –NO2group reduction, i.e. no evidence of hydrodenitrogenation and/or aromatic ring reduction. This combination of stability and selectivity represents a significant development in terms of the application of Au to promote –NO2group hydrogenation. A temporal loss of hydrogenation activity has been observed for reaction over Au supported on Al2O3,75,76CeO2,28Fe2O3,77SiO2,75TiO278,79 and ZrO276 and linked to metal sintering76,78 but has mainly been attributed to coke formation.28,75,78,79 Moreover, catalyst deactivation in the gas phase hydrogenation of nitroarenes over Pd/Al2O3,80–82Pd/hydrotalcite,83Cu/SiO284 and Cu/Kieselghur85 has been reported and ascribed to the deleterious effect of H2O as by-product,83 metal leaching82 and coking.80,81,84,85Nitro-group reduction can be quantified by applying pseudo-first order kinetics86,87| |  | (1) |
where F–NO2 is the total inlet molar –NO2 flow rate and nAu is the number of moles of Au in the catalyst bed; (nAu/F–NO2) has the physical meaning of contact time. The extracted pseudo-first order rate constants (k) are given in Table 2. A direct comparison of rate constants for catalysts bearing different Au particle sizes is only meaningful in terms of specific activities, i.e. per m2 of exposed metal. The Au metal surface area (SAu) was estimated from| |  | (2) |
where ρAu = 18.88 g cm−3 and dTEM is the surface area weighted mean Au particle size, as measured by TEM (see Table 1). The specific pseudo-first order rate constant (k′) was then calculated from| | 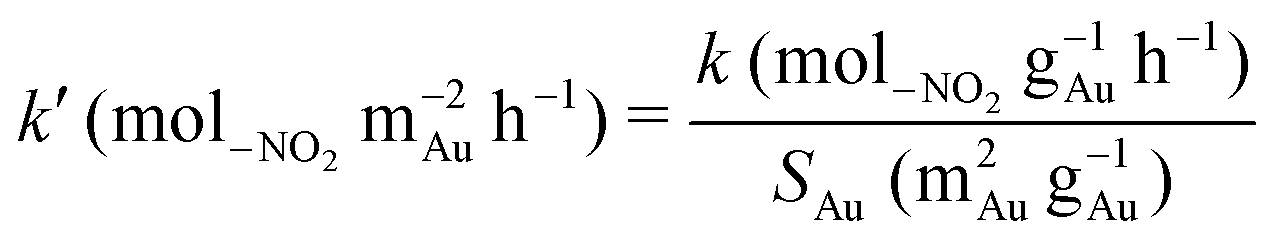 | (3) |
The relationship between the specific activity and mean Au particle size for all the systems considered in this study is presented in Fig. 6. The tendency of increasing activity (k′) with decreasing Au size is consistent with a structure sensitive reaction where an enhanced intrinsic –NO2hydrogenation efficiency is associated with smaller Au particles (9 → 3 nm). The results indicate that the nature of the support does not impact significantly on the hydrogenation rate, which is governed by Au size. Indeed, a similar specific –NO2group reduction rate was obtained over systems with a similar Au particle size on different carriers, as illustrated by points 5 (Au/TiO2-Ref, dTEM = 3.3 nm) and 7 (Au/Fe2O3-IMP(1), dTEM = 3.5 nm). Taking a common support (Al2O3, see points 1 and 6 in Fig. 6), the catalyst with the smaller mean Au diameter (Au/Al2O3-DP(1), dTEM = 2.8 nm) delivered a specific rate (9.4 × 10−4 mol–NO2 m−2Au h−1) that was over two orders of magnitude greater than that (1.4 × 10−6 mol–NO2 m−2Au h−1) obtained over the catalyst bearing larger Au particles (Au/Al2O3-IMP(1), dTEM = 9.0 nm), representing the upper and lower rates recorded in this study. Our findings are in agreement with published studies suggesting that the presence of nanoscale Au particles (≤10 nm) is essential for significant hydrogenation activity.13,88 It should be noted that Au/CeO2-DP(1) with the smallest mean Au size (dTEM = 1.5 nm) deviates somewhat from the general trend, a response that we can attribute to quantum size effects. Differences in the catalytic behaviour of Au particles ≤2 nm have been associated with variations in the electronic character of the active site.89 For Au particles in the 1–3 nm size range, it has been reported that the electronic properties vary considerably with a switch from metal to semiconductor characteristics.90–92 Claus and co-workers,93 studying the gas phase hydrogenation of acrolein over Au/TiO2, linked an order of magnitude decrease in activity (400 → 41 mmol g−1Aus−1) with decreasing Au particle size (down to 1.4 nm) to a transition from metallic to non-metallic character for small Au particles.
Table 2 Pseudo-first order rate constants for nitro-group reduction with product selectivities at a common fractional m-DNB conversion (x0 ≈ 0.1) over a series of oxide supported Au prepared by DP and IMP
|
Catalyst
|
Rate constant, k/h−1 |
Product(s) (selectivity %) |
|
Not possible to achieve x0 = 0.1 due to low catalyst activity under the stated reaction conditions.
|
|
Au/Al2O3-DP(1) |
20 |
m-NAN (30)/m-PDM (70) |
|
Au/CeO2-DP(1) |
24 |
m-PDM (100) |
|
Au/Fe2O3-DP(1) |
18 |
m-NAN (100) |
|
Au/TiO2-DP(1) |
13 |
m-NAN (100) |
|
Au/TiO2-Ref |
7 |
m-NAN (100) |
|
Au/Al2O3-IMP(1) |
<1 |
—a |
|
Au/Fe2O3-IMP(1) |
9 |
m-NAN (100) |
|
Au/TiO2-IMP(1) |
<1 |
—a |
|
Au/Fe2O3-IMP(10) |
1 |
m-NAN (100) |
 |
| | Fig. 6 Relationship between specific rate constant (k′) and Au particle size (dTEM) for (1) Au/Al2O3-DP(1), (2) Au/CeO2-DP(1), (3) Au/Fe2O3-DP(1), (4) Au/TiO2-DP(1), (5) Au/TiO2-Ref, (6) Au/Al2O3-IMP(1), (7) Au/Fe2O3-IMP(1), (8) Au/TiO2-IMP(1) and (9) Au/Fe2O3-IMP(10): DP catalysts represented by open squares; IMP catalysts represented by solid squares. | |
A series of experiments were conducted to study the effect(s) of the nature of the support on reaction selectivity at the same degree of initial conversion. The results are presented in Table 2 where clear differences in the product distribution are in evidence. Exclusive formation of m-NAN (partial –NO2group reduction) was recorded over Fe2O3 and TiO2 supported Au catalysts. In complete contrast, m-PDM (full –NO2group reduction) was the only product generated over Au/CeO2-DP(1) while a mixture of both products was obtained over Au/Al2O3-DP(1). The sole formation of either m-NAN or m-PDM in continuous operation under mild reaction conditions represents an important advancement in the selective hydrogenation of m-DNB as the exclusive formation of either product has been identified as a critical processing challenge. Zhao et al.94 recorded 97% selectivity to m-NAN over 5% w/w Pt/C for reaction under supercritical CO2 (T = 323 K, P = 16 MPa). In liquid phase operation, conditions of high temperatures and/or pressures have been deemed essential to achieve high selectivities to m-NAN or m-PDM. Khilnani and Chandalia95 reported up to 84% selectivity to m-NAN over 5% w/w Pd/C (T = 304 K and P = 34 atm) whereas Liu et al.96 obtained up to 98% selectivity to m-PDM over 20% w/w Ni/SiO2 (T = 373 K and P = 2.6 MPa). The hydrogenation of nitroarenes has been proposed to proceed via a nucleophilic mechanism, where a weak nucleophilic agent (hydrogen) attacks the activated –NO2group with the formation of a negatively charged intermediate.97 We can associate the distinct selectivity response that we observe to the differences in acid–base Lewis character of the supports, which can influence the electronic character of the Au particles and m-DNB adsorption/activation. The presence of Lewis basic sites in CeO298 is known to induce support → metal electron transfer26 that can give rise to Auδ− particles. It follows that the Lewis acid sites (cation vacancies) in Fe2O3 and TiO298 can promote the formation of Auδ+ particles as a result of Au → support electron transfer. Indeed, charge transfer from Au particles to TiO2 has been demonstrated by a positive shift (to higher binding energy) of XPS Au 4f peaks for small particles (up to ca. 4 nm) when compared with metallic Au (Au (111) single crystal).99 The formation of both Auδ+ and Auδ− species is possible in the case of Au/Al2O3 due to the occurrence of both Lewis basic and acid sites on Al2O3.38m-DNB adsorption on Au/CeO2 must result in a repulsion between the π-delocalized electrons and the partially (negatively) charged gold leading to resonance structures with two positive localized charges on the ring where both –NO2groups are activated, ultimately resulting in the formation of m-PDMvia nucleophilic attack. Adsorption on Au/Fe2O3 and Au/TiO2 can generate a resonance form with a single delocalized positive charge on the ring where only one of the –NO2groups is activated, giving rise to the formation of m-NAN. The presence of negatively charged and electron-deficient Au particles in Au/Al2O3 can account for the production of both m-NAN and m-PDM. Our results demonstrate that smaller supported Au particles exhibit a higher intrinsic hydrogenation where selectivity can be tuned or controlled as a function of the support. Moreover, continuous operation under the mild reaction conditions employed represents a significant and novel advance in the selective hydrogenation of m-DNB.
3. Experimental
The CeO2 (HSA5, Rhodia), TiO2 (Degussa, P-25) and γ-Al2O3 (Puralox, Condea Vista Co.) supports were used as received. Hematite (α-Fe2O3) was prepared by precipitation in basic media according to100,101| | | 2[Fe(NO3)3·9H2O] + 3Na2CO3 → 2Fe(OH)3 + 6NaNO3 + 3CO2 + 15H2O | (4) |
100 cm3 aqueous Na2CO3 (1 M) were placed in a three-necked round-bottom flask and heated in a water bath to 358 ± 5 K under constant agitation (300 rpm), provided by a vertically mounted three blade glass impeller. An aqueous solution of Fe(NO3)3·9H2O (300 cm3, 1 M) was then added dropwise (300 cm3 h−1) by means of a microprocessor-controlled infusion pump (100 kd Scientific). Basic conditions (pH > 7.3) were maintained during precipitation by adding Na2CO3 (five additions of 10 g). The solid hydroxide was subsequently aged for 2 h to enhance the BET surface area,102 washed with warm distilled water until the washwater exhibited neutral pH and dried for 3 days at 353 K (2 K min−1) in an atmosphere of ultra pure He (60 cm3 min−1) to produce hematite:| | | 2Fe(OH)3 → Fe2O3 + 3H2O | (5) |
A series of 1 mol% oxide supported Au were prepared by deposition-precipitation (DP) and impregnation (IMP). In the case of the DP synthesis, urea (used as a basification agent) was added (ca.100-fold urea excess) to a solution of HAuCl4 (5 × 10−4 M). The support (3–5 g, γ-Al2O3, TiO2, CeO2 and α-Fe2O3) was added and the suspension was stirred and heated to 353 K for 16 h. The pH of the suspension progressively increased to reach ca. 7 after 16 h as a result of thermally-induced urea decomposition| |  | (6) |
The solids obtained were separated by centrifugation, washed three times with deionized water (with centrifugation between each washing) and dried under vacuum at 298 K for 12 h. In the case of the IMP synthesis, the support was dispersed in appropriate volumes of HAuCl4 solution (Aldrich, 25 × 10−3 g cm−3, pH = 2) and the resulting slurry was vigorously stirred (600 rpm) and slowly heated (2 K min−1) to 353 K in a He flow. The solid residue was dried in He at 383 K for 3 h. For comparison purposes, a 10 mol% Au on Fe2O3 was synthesized by IMP. The samples prepared by impregnation or deposition-precipitation are denoted in this paper by the suffix “-IMP” and “-DP”, respectively, with the associated Au content (1 or 10 mol%) specified in parentheses. After preparation, the samples were sieved to 75 μm average particle diameter (ATM fine test sieves) and stored at 278 K (in the dark). The Au loading was determined by inductively coupled plasma-optical emission spectrometry (ICP-OES, Vista-PRO, Varian Inc.) from the diluted extract of aqua regia. The catalytic behaviour of a 1 mol% Au/TiO2 reference catalyst supplied by the World Gold Council (type A, lot number Au-TiO2 #02-7, sample number 110) was also considered. The preparation and characterization details of this reference sample have been fully described elsewhere.103,104 Prior to use in catalysis, the samples were activated in 60 cm3 min−1 H2 at 2 K min−1 to 423–603 ± 1 K. Samples for off-line analysis were passivated in 1% v/v O2/He at room temperature.
3.2
Catalyst characterization
Temperature programmed reduction (TPR), H2 chemisorption, temperature programmed desorption (TPD) and BET surface area were determined using the commercial CHEM-BET 3000 (Quantachrome) unit. The samples were loaded into a U-shaped Quartz cell (10 cm × 3.76 mm i.d.) and heated in 17 cm3 min−1 (Brooks mass flow controlled) 5% v/v H2/N2 at 2 K min−1 to 423–603 ± 1 K. The effluent gas passed through a liquid N2 trap and changes in H2 consumption were monitored by a thermal conductivity detector (TCD) with data acquisition/manipulation using the TPR WinTM software. The reduced samples were maintained at the final temperature in a flow of H2 until the signal returned to baseline, swept with a 65 cm3 min−1 flow of N2 for 1.5 h, cooled to room temperature and subjected to H2 chemisorption (at 298 K) using a pulse (10 μl) titration procedure. In a series of blank tests, chemisorption measurements on each support (Al2O3, TiO2, CeO2 or Fe2O3) did not result in any detectable hydrogen uptake. Hydrogen TPD was conducted in a N2 flow (65 cm3 min−1) at 50 K min−1 to 873 K with an isothermal hold until the signal returned to the baseline. BET areas were recorded with a 30% v/v N2/He flow using pure N2 (99.9%) as internal standard. At least 2 cycles of N2 adsorption–desorption in the flow mode were employed to determine total surface area using the standard single point method. BET surface area and H2 uptake values were reproducible to within ±5%; the values quoted represent the mean.
Powder X-ray diffractograms were recorded on a Bruker/Siemens D500 incident X-ray diffractometer using Cu Kα radiation. The samples were scanned at a rate of 0.02° step−1 over the range 20° ≤ 2θ ≤ 90° (scan time = 5 s step−1). Diffractograms were identified using the JCPDS-ICDD reference standards, i.e. α-Fe2O3 (33-0664), Fe3O4 (19-0629) and Au (04-0784). Au particle morphology and size were determined by transmission electron microscopy analysis; JEOL JEM 2011 HRTEM unit with a UTW energy dispersive X-ray detector (Oxford Instruments) operated at an accelerating voltage of 200 kV using Gatan DigitalMicrograph 3.4 for data acquisition/manipulation. The specimens were prepared by dispersion in acetone and deposited on a holey carbon/Cu grid (300 Mesh). Up to 1000 individual Au particles were counted for each catalyst and the surface area-weighted metal diameter (dTEM) was calculated from
| |  | (7) |
where
ni is the number of particles of diameter
di. The size limit for the detection of Au particles is
ca. 1 nm.
Reactions were carried out under atmospheric pressure at T = 423 K, in situ immediately after activation, in a fixed bed vertical continuous flow glass reactor (l = 600 mm; i.d. = 15 mm). The catalytic reactor and operating conditions to ensure negligible heat/mass transport limitations have been fully described elsewhere105 but some features, pertinent to this study, are given below. A layer of borosilicate glass beads served as a preheating zone, ensuring that the m-DNB was vaporized and reached reaction temperature before contacting the catalyst. Isothermal conditions (±1 K) were ensured by diluting the catalyst bed with ground glass (75 μm); the ground glass was mixed thoroughly with the catalyst before insertion into the reactor. Temperature was continuously monitored by a thermocouple inserted in a thermowell within the catalyst bed. m-DNB was delivered at a fixed calibrated flow rate to the reactor via a glass/teflon air-tight syringe and teflon line using a microprocessor controlled infusion pump (Model 100 kd Scientific). A co-current flow of m-DNB and ultra pure H2 (<1% v/v –NO2/H2) was maintained at a GHSV = 2 × 104 h−1 with an inlet –NO2 molar flow (F–NO2) over the range 6 × 10−2–20 × 10−2 mmol–NO2 h−1. The H2 inlet was well in excess of the stoichiometric requirement (PH2 = 0.92 atm) and the flow rate was monitored using a Humonics (Model 520) digital flowmeter. The molar metal (nAu) to F–NO2 ratio spanned the range 29 × 10−4–111 × 10−4 h. In a series of blank tests, passage of m-DNB in a stream of H2 through the empty reactor or over the support alone, i.e. in the absence Au, did not result in any detectable conversion. The reactor effluent was frozen in a liquid nitrogen trap for subsequent analysis, which was made using a Perkin-Elmer Auto System XL gas chromatograph equipped with a programmed split/splitless injector and a flame ionization detector, employing a DB-1 50 m × 0.20 mm i.d., 0.33 μm film thickness capillary column (J&W Scientific), as described elsewhere.106 Data acquisition and manipulation were performed using the TotalChrom Workstation Version 6.1.2 (for Windows) chromatography data system and the overall reactant/product molar fractions were obtained using detailed calibration plots (not shown). m-DNB (Aldrich, ≥98% w/w purity) and the 1-butanol solvent (Riedel-de Häen, ≥99.5%) were used without further purification. The extent of nitro-group reduction (x–NO2) is given by| |  | (8) |
where [m-DNB], [m-NAN] and [m-PDM] are, respectively, the concentrations of m-DNB, m-NAN and m-PDM; the subscripts in and out refer to the inlet and outlet streams. Selectivity in terms of (for example) m-NAN (Sm-NAN) is given by | |  | (9) |
Repeated reactions with the same batch of catalysts delivered conversion/selectivity values that were reproducible to within ±6%.
4. Conclusions
We have demonstrated that 1 mol% Au on TiO2, Fe2O3, CeO2 and Al2O3 prepared by DP generated smaller (surface area weighted) mean Au particle sizes (≤2.8 nm) when compared with synthesis by IMP (≥3.5 nm). Moreover, regardless of the preparation method, larger Au particles were formed on the non-reducible (Al2O3) oxide. TPR of Au/TiO2, Au/CeO2 and Au/Al2O3 generated H2 consumption peaks with Tmax in the range of 371–457 K that we attribute to the reduction of Au3+ to Au0. XRD analysis has established the presence of metallic gold in Au/Fe2O3pre-TPR and the activation of Au/Fe2O3 to 423/673 K resulted in a partial/full reduction of the support from hematite to magnetite. This effect was more pronounced for Au particle size <5 nm and H2TPD analysis suggests the involvement of spillover hydrogen in the reduction step. A decrease in Au size (from 9 to 3 nm) was accompanied by an increase in specific hydrogenation rate. A lower specific rate recorded for Au particles <2 nm can be attributed to a quantum size effect. Hydrogenation activity was invariant with time on-stream for all the supported Au systems. Au/TiO2 and Au/Fe2O3 promoted the exclusive hydrogenation of m-DNB to m-NAN (partial –NO2group reduction), Au/CeO2 delivered m-PDM (full –NO2group reduction) as the sole product and Au/Al2O3 generated a mixture of m-NAN and m-PDM.
Acknowledgements
We acknowledge the World Gold Council for providing the Au/TiO2-Ref catalyst. This work was financially supported by EPSRC through Grant 0231 110525. EPSRC support for free access to the TEM/SEM facility at the University of St Andrews is also acknowledged.
References
-
P. F. Vogt and J. J. Gerulis, Ullmann's Encyclopedia of Industrial Chemistry. “Aromatic Amines”, Wiley-VCH, Verlag GmbH & Co. KGaA, Weinheim, 2005, pp. 2–21 Search PubMed.
-
B. Kammermeier, Ullmann's Encyclopedia of Industrial Chemistry. “Reduction”, Wiley-VCH, Verlag GmbH & Co. KGaA, Weinheim, 2005, pp. 17–21 Search PubMed.
- N. Yao, J. Chen, J. Zhang and J. Zhang, Catal. Commun., 2008, 9, 1510–1516 CrossRef CAS.
- X. D. Wang, M. H. Liang, J. L. Zhang and Y. Wang, Curr. Org. Chem., 2007, 11, 299–314 CrossRef CAS.
- M. Takasaki, Y. Motoyama, K. Higashi, S.-H. Yoon, I. Mochida and H. Nagashima, Org. Lett., 2008, 10, 1601–1604 CrossRef CAS.
-
R. A. Sheldon and H. van Bekkum, Fine Chemicals through Heterogeneous Catalysis. “Aromatic Nitro Compounds”, Wiley-VCH, Weinheim (Germany), 2001, pp. 389–407 Search PubMed.
- S. Zhao, H. Liang and Y. Zhou, Catal. Commun., 2007, 8, 1305–1309 CrossRef CAS.
- M. M. Telkar, J. M. Nadgeri, C. V. Rode and R. V. Chaudhari, Appl. Catal., A, 2005, 295, 23–30 CrossRef CAS.
- E. P. Maris, W. C. Ketchie, M. Murayama and R. J. Davis, J. Catal., 2007, 251, 281–294 CrossRef CAS.
- A. Corma, P. Serna and H. García, J. Am. Chem. Soc., 2007, 129, 6358–6359 CrossRef CAS.
- R. Zanella, C. Louis, S. Giorgio and R. Touroude, J. Catal., 2004, 223, 328–339 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Nature, 1995, 376, 238–240 CrossRef.
- P. Claus, Appl. Catal., A, 2005, 291, 222–229 CrossRef CAS.
- A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef.
- D. He, H. Shi, Y. Wu and B.-Q. Xu, Green Chem., 2007, 9, 849–851 RSC.
- A. Corma, P. Concepción and P. Serna, Angew. Chem., Int. Ed., 2007, 46, 7266–7269 CrossRef CAS.
- M. Boronat, P. Concepción, A. Corma, S. González, F. Illas and P. Serna, J. Am. Chem. Soc., 2007, 129, 16230–16237 CrossRef CAS.
- A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS.
- L. Liu, B. Qiao, Y. Ma, J. Zhang and Y. Deng, Dalton Trans., 2008, 2542–2548 RSC.
- Y. Chen, J. Qiu, X. Wang and J. Xiu, J. Catal., 2006, 242, 227–230 CrossRef CAS.
- H. Sakurai and M. Haruta, Appl. Catal., A, 1995, 127, 93–105 CrossRef CAS.
- J. A. Lopez-Sanchez and D. Lennon, Appl. Catal., A, 2005, 291, 230–237 CrossRef CAS.
-
G. C. Bond, C. Louis and D. T. Thompson, Catalysis by Gold, Imperial College Press, London, 2006, pp. 65–59 Search PubMed.
- J. Radnik, C. Mohr and P. Claus, Phys. Chem. Chem. Phys., 2003, 5, 172–177 RSC.
- B. K. Min, W. T. Wallace and D. W. Goodman, Surf. Sci., 2006, 600, L7–L11 CrossRef CAS.
- A. Y. Stakheev and L. M. Kustov, Appl. Catal., A, 1999, 188, 3–35 CrossRef CAS.
- M. A. Centeno, I. Carrizosa and J. A. Odriozola, Appl. Catal., A, 2003, 246, 365–372 CrossRef CAS.
- B. Campo, C. Petit and M. A. Volpe, J. Catal., 2008, 254, 71–78 CrossRef CAS.
- C. Milone, R. Ingoglia, L. Schipilliti, C. Crisafulli, G. Neri and S. Galvagno, J. Catal., 2005, 236, 80–90 CrossRef CAS.
- C. Milone, C. Crisafulli, R. Ingoglia, L. Schipilliti and S. Galvagno, Catal. Today, 2007, 122, 341–351 CrossRef CAS.
- B. C. Campo, S. Ivanova, C. Gigola, C. Petit and M. A. Volpe, Catal. Today, 2008, 133–135, 661–666 CrossRef CAS.
- J. E. Bailie and G. J. Hutchings, Chem. Commun., 1999, 2151–2152 RSC.
- F. Cárdenas-Lizana, S. Gómez-Quero and M. A. Keane, Catal. Commun., 2008, 9, 475–481 CrossRef CAS.
- F. Cárdenas-Lizana, S. Gómez-Quero and M. A. Keane, ChemSusChem, 2008, 1, 215–221 CrossRef CAS.
- F. Cárdenas-Lizana, S. Gómez-Quero, N. Perret and M. A. Keane, Gold Bull., 2009, 42, 124–132 CrossRef CAS.
- F. Cárdenas-Lizana, S. Gómez-Quero, H. Idriss and M. A. Keane, J. Catal., 2009, 268, 223–234 CrossRef CAS.
- F. Cárdenas-Lizana, Z. Martínez De Pedro, S. Gómez-Quero and M. A. Keane, J. Mol. Catal., 2010, 326, 48–54 Search PubMed.
- M. Trueba and S. P. Trasatti, Eur. J. Inorg. Chem., 2005, 3393–3403 CrossRef CAS.
- L. Ilieva, J. W. Sobczak, M. Manzoli, B. L. Su and D. Andreeva, Appl. Catal., A, 2005, 291, 85–92 CrossRef CAS.
- L. Ilieva, G. Pantaleo, I. Ivanov, A. M. Venezia and D. Andreeva, Appl. Catal., A, 2006, 65, 101–109 CAS.
- M. M. Mohamed, T. M. Salama, R. Ohnishi and M. Ichikawa, Langmuir, 2001, 17, 5678–5684 CrossRef CAS.
- E. Bus, R. Prins and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2007, 9, 3312–3320 RSC.
- S. Y. Liu and S. M. Yang, Appl. Catal., A, 2008, 334, 92–99 CrossRef CAS.
- L. Delannoy, N. Weiher, N. Tsapatsaris, A. M. Beesley, L. Nchari, S. L. M. Schroeder and C. Louis, Top. Catal., 2007, 44, 263–273 CrossRef CAS.
- C. Baatz and U. Prüße, J. Catal., 2007, 249, 34–40 CrossRef CAS.
- A. C. Gluhoi, X. Tang, P. Marginean and B. E. Nieuwenhuys, Top. Catal., 2006, 39, 101–110 CrossRef CAS.
- S. D. Yim and I.-S. Nam, J. Catal., 2004, 221, 601–611 CrossRef CAS.
- V. Idakiev, T. Tabakova, Z.-Y. Yuan and B.-L. Su, Appl. Catal., A, 2004, 270, 135–141 CrossRef CAS.
- A. Pineau, N. Kanari and I. Gaballah, Thermochim. Acta, 2006, 447, 89–100 CrossRef CAS.
- M. Khoudiakov, M. C. Gupta and S. Deevi, Appl. Catal., A, 2005, 291, 151–161 CrossRef CAS.
- B. A. A. Silberova, G. Mul, M. Makkee and J. A. Moulijn, J. Catal., 2006, 243, 171–182 CrossRef.
- J. Li, Y. Zhan, X. Lin and Q. Zheng, Acta Phys. Chim. Sin., 2008, 24, 932–938 Search PubMed.
- G. Y. Wang, H. L. Lian, W. X. Zhang, D. Z. Jiang and T. H. Wu, Kinet. Catal., 2002, 43, 433–442 CrossRef CAS.
- G. C. Bond and D. T. Thompson, Catal. Rev. Sci. Eng., 1999, 41, 319–388 CrossRef CAS.
- S. H. Overbury, V. Schwartz, D. R. Mullins, W. Yan and S. Dai, J. Catal., 2006, 241, 56–65 CrossRef CAS.
- J. Huang, W.-L. Dai, H. Li and K. Fan, J. Catal., 2007, 252, 69–76 CrossRef CAS.
- G. Yuan, C. Louis, L. Delannoy and M. A. Keane, J. Catal., 2007, 247, 256–268 CrossRef CAS.
- N. A. Hodge, C. J. Kiely, R. Whyman, M. R. H. Siddiqui, G. J. Hutchings, Q. A. Pankhurst, F. E. Wagner, R. R. Rajaram and S. E. Golunski, Catal. Today, 2002, 72, 133–144 CrossRef CAS.
- A. Venugopal and M. S. Scurrell, Appl. Catal., A, 2004, 258, 241–249 CrossRef CAS.
- Z. Hao, L. An, H. Wang and T. Hu, React. Kinet. Catal. Lett., 2000, 70, 153–160 CrossRef CAS.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- M. Okumura, T. Akita and M. Haruta, Catal. Today, 2002, 74, 265–269 CrossRef CAS.
- S. Gómez-Quero, F. Cárdenas-Lizana and M. A. Keane, Ind. Eng. Chem. Res., 2008, 47, 6841–6853 CrossRef CAS.
- E. Bus, J. T. Miller and J. A. van Bokhoven, J. Phys. Chem. B, 2005, 109, 14581–14587 CrossRef CAS.
- H. Berndt, I. Pitsch, S. Evert, K. Struve, M.-M. Pohl, J. Radnik and A. Martin, Appl. Catal., A, 2003, 244, 169–179 CrossRef CAS.
- A. G. Sault, R. J. Madix and C. T. Campbell, Surf. Sci., 1986, 169, 347–356 CrossRef CAS.
- C. Amorim and M. A. Keane, J. Colloid Interface Sci., 2008, 322, 196–208 CrossRef CAS.
- W. C. Conner and J. L. Falconer, Chem. Rev., 1995, 95, 759–788 CrossRef CAS.
- D. A. Panayotov and J. T. Yates, J. Phys. Chem. C, 2007, 111, 2959–2964 CrossRef CAS.
- S. E. Collins, J. M. Cíes, E. del Río, M. López-Haro, S. Trasobares, J. J. Calvino, J. M. Pintado and S. Bernal, J. Phys. Chem. C, 2007, 111, 14371–14379 CrossRef CAS.
- M. Snåre, I. Kubičková, P. Mäki-Arvela, K. Eränen and D. Y. Murzin, Ind. Eng. Chem. Res., 2006, 45, 5708–5715 CrossRef.
- F. Benseradj, F. Sadi and M. Chater, Appl. Catal., A, 2002, 228, 135–144 CrossRef CAS.
- S. Velu and S. K. Gangwal, Solid State Ionics, 2006, 177, 803–811 CrossRef CAS.
- F. Boccuzzi, A. Chiorino, M. Manzoli, D. Andreeva and T. Tabakova, J. Catal., 1999, 188, 176–185 CrossRef.
- B. Pawelec, A. M. Venezia, V. La Parola, S. Thomas and J. L. G. Fierro, Appl. Catal., A, 2005, 283, 165–175 CrossRef CAS.
- X. Zhang, H. Shi and B.-Q. Xu, Catal. Today, 2007, 122, 330–337 CrossRef CAS.
- C. Milone, M. L. Tropeano, G. Gulino, G. Neri, R. Ingoglia and S. Galvagno, Chem. Commun., 2002, 868–869 RSC.
- T. V. Choudhary, C. Sivadinarayana, A. K. Datye, D. Kumar and D. W. Goodman, Catal. Lett., 2003, 86, 1–8 CrossRef CAS.
- Y. Azizi, C. Petit and V. Pitchon, J. Catal., 2008, 256, 338–344 CrossRef CAS.
- E. Klemm, B. Amon, H. Redlingshöfer, E. Dieterich and G. Emig, Chem. Eng. Sci., 2001, 56, 1347–1353 CrossRef CAS.
- V. Vishwanathan, V. Jayasri, P. M. Basha, N. Mahata, L. M. Sikhwivhilu and N. J. Coville, Catal. Commun., 2008, 9, 453–458 CrossRef CAS.
- K. K. Yeong, A. Gavriilidis, R. Zapf and V. Hessel, Catal. Today, 2003, 81, 641–651 CrossRef CAS.
- P. Sangeetha, P. Seetharamulu, K. Shanthi, S. Narayanan and K. S. Rama Raob, J. Mol. Catal. A: Chem., 2007, 273, 244–249 CrossRef CAS.
- S. Diao, W. Qian, G. Luo, F. Wei and Y. Wang, Appl. Catal., A, 2005, 286, 30–35 CrossRef CAS.
- L. Petrov, K. Kumbilieva and N. Kirkov, Appl. Catal., 1990, 59, 31–43 Search PubMed.
- F. Cárdenas-Lizana, S. Gómez-Quero and M. A. Keane, Catal. Lett., 2009, 127, 25–32 CrossRef CAS.
- F. Cárdenas-Lizana, S. Gómez-Quero, C. J. Baddeley and M. A. Keane, Appl. Catal., A, 2010, 387, 155–165 CrossRef CAS.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef.
- C. Mohr, H. Hofmeister and P. Claus, J. Catal., 2003, 213, 86–94 CrossRef.
- L. Stievano, S. Santucci, L. Lozzi, S. Calogero and F. E. Wagner, J. Non-Cryst. Solids, 1998, 232–234, 644–649 CrossRef CAS.
- Q. Guo, K. Luo, K. A. Davis and D. W. Goodman, Surf. Interface Anal., 2001, 32, 161–165 CrossRef CAS.
- H.-G. Boyen, Th. Herzog, G. Kästle, F. Weigl, P. Ziemann, J. P. Spatz, M. Möller, R. Wahrenberg, M. G. Garnier and P. Oelhafen, Phys. Rev. B: Condens. Matter, 2002, 65, 075412 CrossRef.
- P. Claus, A. Brückner, C. Mohr and H. Hofmeister, J. Am. Chem. Soc., 2000, 122, 11430–11439 CrossRef CAS.
- F. Zhao, S.-I. Fujita, J. Suna, Y. Ikushima and M. Arai, Catal. Today, 2004, 98, 523–528 CrossRef CAS.
- V. L. Khilnani and S. B. Chandalia, Org. Process Res. Dev., 2001, 5, 263–266 Search PubMed.
- Y. X. Liu, J. X. Chen and J. Y. Zhang, Chin. J. Chem. Eng., 2007, 15, 63–67 Search PubMed.
- O. Exner and T. M. Krygowski, Chem. Soc. Rev., 1996, 25, 71–75 RSC.
- G. Busca, Phys. Chem. Chem. Phys., 1999, 1, 723–736 RSC.
- T. Okazawa, M. Fujiwara, T. Nishimura, T. Akita, M. Kohyama and Y. Kido, Surf. Sci., 2006, 600, 1331–1338 CrossRef CAS.
- G. Neri, A. M. Visco, S. Galvagno, A. Donato and M. Panzalorto, Thermochim. Acta, 1999, 329, 39–46 CrossRef CAS.
- F. E. Wagner, S. Galvagno, C. Milone, A. M. Visco, L. Stievano and S. Calogero, J. Chem. Soc., Faraday Trans., 1997, 93, 3403–3409 RSC.
- D. Andreeva, V. Idakiev, T. Tabakova, A. Andreev and R. Giovanoli, Appl. Catal., A, 1996, 134, 275–283 CrossRef CAS.
- M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, J. Catal., 1993, 144, 175–192 CrossRef CAS.
-
S. Tsubota, A. Yamaguchi, M. Daté, M. Haruta, Characterization of Reference Gold Catalysts in GOLD2003, 3rd International Conference on Gold Science, Technology and its Applications, Vancouver, Canada, September–October, 2003.
- G. Tavoularis and M. A. Keane, J. Chem. Technol. Biotechnol., 1999, 74, 60–70 CrossRef CAS.
- G. Yuan and M. A. Keane, Chem. Eng. Sci., 2003, 58, 257–267 CrossRef CAS.
Footnotes |
| † Present address: Group of Catalytic Reaction Engineering, Ecole Polytechnique Fédérale de Lausanne (GGRC-ISIC-EPFL), Lausanne CH-1015, Switzerland. |
| ‡ Present address: Van't Hoff Institute for Molecular Sciences, University of Amsterdam, Amsterdam 1090 GS, The Netherlands. |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.