Effects of tungsten oxide on soot oxidation activity and sulfur poisoning resistance of Pt/Al2O3 catalyst
Xiaodong
Wu
*,
Shuang
Liu
and
Duan
Weng
Laboratory of Advanced Materials, Department of Materials Science and Engineering, Tsinghua University, Beijing 100084, China. E-mail: wuxiaodong@tsinghua.edu.cn; Fax: +86-10-62792375; Tel: +86-10-62792375
First published on 28th April 2011
Abstract
Tungsten oxide was impregnated into the alumina-supported platinum catalyst in order to improve the soot oxidation activity and the resistance to sulfur dioxide. The catalysts were characterized by X-ray diffraction (XRD), Raman, UV-vis spectroscopy, Brunauer–Emmett–Teller (BET), Fourier transform infrared (FT-IR) spectroscopy, temperature-programmed desulfation, NOx temperature-programmed desorption (NOx-TPD), NO temperature-programmed oxidation (NO-TPO) and soot temperature-programmed oxidation (soot-TPO). The deposition of WOx was found to reduce the availability of Pt active sites on the fresh catalyst. This, as well as the acidic property of tungsten oxide, decreases the NO oxidation and the NOx adsorption abilities. However, a higher soot oxidation activity was achieved on the WOx-modified catalyst in the presence of NO and O2, which is associated with the presence of more platinum in the metallic state by interacting with the electronegative tungsten oxide. The acidity of tungsten oxide and the improved oxidation-resistance of platinum may be critical to the NO ↔ NO2 recycling efficiency and decomposition of surface oxygen complexes. After the sulfur poisoning treatment, fewer sulfates are formed on the WOx-modified catalyst and decompose at lower temperatures. The IR spectra of CO adsorption indicate that less platinum active sites on this catalyst are affected by sulfates and a higher level of Pt dispersion is obtained, which is responsible for the high NO oxidation and soot oxidation activities.
1. Introduction
The airborne particulate emissions (soot) generated from incomplete combustion of biomass and fossil fuels can adsorb carcinogenic and toxic substances such as polycyclic aromatic hydrocarbons (PAHs) and metals.1–4 The removal of soot is a topic of ongoing research due to the environmental and health impacts of these carbon nanoparticles. Among various technical approaches, the combustion of soot using oxidation catalysts is one of the most promising methods. Among the catalysts investigated, Pt exhibits a high level of catalytic activity by oxidizing NO in the exhaust gas to NO2, which has a remarkable effect on the oxidation of soot to CO2.5–8However, Pt based catalysts will be deactivated a lot during exposure in O2 and NO + O2. This deactivation in catalytic combustion is attributed to the oxidation of Pt to PtO or PtO2, which are less active than metallic platinum. Such a phenomenon has been reported on platinum catalysts for propane combustion9 and NO2 ↔ NO + 1/2O2 reactions.10,11 A realistic theory developed by Yoshida's group states that electrons are transferred from Pt to oxygen when forming platinum oxide and in the case of an acid support there is a higher electron density in Pt because of the electrophilic nature of the acidic support, which results in a larger number of metallic platinum.12,13
Sulfur poisoning is a critical problem for the application of soot oxidation catalysts. It has been reported that Pt catalysts supported on non-basic metal oxides (Ta2O5, Nb2O5, WO3, SnO2 and SiO2) show high activities towards the oxidation of soot in the presence of SO2, which is attributed to their non-basicity and negligible affinity towards SO3 (or H2SO4), resulting in less poisoning of the supported Pt and also in an oxidation of soot by NO2 with a catalysis of SO3.14,15NO2 is more powerful oxidant than O2. The oxidation of NO to NO2 is an important step in the mechanism of soot catalytic oxidation in the presence of NO. Dawody et al.16 studied the effect of metal oxide additives (WO3, MoO3, V2O5 and Ga2O3) on enhancing NO oxidation and/or suppressing SO2 oxidation in alumina supported Pt catalysts. They found that WO3 has the highest promoting effect on the NO oxidation activity in a sulfur free atmosphere, while MoO3 seems to be the most promising SO2 oxidation inhibitor of the tested metal oxides. The inhibition mechanism to sulfur poisoning is presented for Pt–WO3/TiO2 catalyst that WO3 competes with SO3 and displaces SO3 on the basic sites of TiO2 surface and tends to cover it, thus limiting its sulfation.17
To the best of our knowledge, few researches have involved the effect of acidic supports or additives on the soot oxidation activity of platinum catalysts in the presence of NO and O2. In the present study, tungsten oxide was introduced to Pt/Al2O3 catalyst to investigate the promotion effects on the soot oxidation activity and the resistance to sulfur poisoning. In order to explore possible modification mechanisms, the catalysts were characterized by a series of structural and surface property measurements.
2. Experimental
2.1 Catalyst preparation
The Pt/Al2O3 (PtAl) catalyst was prepared by impregnating Pt(NO3)2 (27.82 wt%, Heraeus) on γ-Al2O3 powders (BASF, 150 m2 g−1). The nominal loading amount of Pt was 1 wt%. The impregnated powders were submitted to drying at 110 °C overnight and calcination at 500 °C for 2 h. The WOx-modified catalyst (WPtAl) was prepared by impregnating 5(NH4)2O·12WO3·5H2O (99%, Juneng) on the Pt supported alumina catalyst with a weight ratio of WO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al2O3 = 1:9. Both catalysts were calcined at 500 °C for 2 h. The catalysts were exposed to 100 ppm SO2/air at 350 °C for 50 h to obtain the so-called sulfated catalysts with a definition of “–SA”. For comparison, the catalysts were also treated in a dry air flow to obtain the aged catalysts with a suffix of “–A”.
:Al2O3 = 1:9. Both catalysts were calcined at 500 °C for 2 h. The catalysts were exposed to 100 ppm SO2/air at 350 °C for 50 h to obtain the so-called sulfated catalysts with a definition of “–SA”. For comparison, the catalysts were also treated in a dry air flow to obtain the aged catalysts with a suffix of “–A”.
2.2 Catalyst characterization
The powder X-ray diffraction (XRD) patterns were determined by a Japan Science D/max-RB diffractometer employing Cu-Kα radiation (λ = 0.15418 nm). The X-ray tube was operated at 40 kV and 30 mA. The X-ray diffractograms were recorded at 0.02° intervals in the range of 20° ≤ 2θ ≤ 80° with a scanning velocity of 4° min−1.The Raman spectra were obtained with a LabRAM HR 800 (HORIBA Jobin Yvon, France) spectrometer at room temperature (RT) and atmospheric pressure. An argon ion laser beam with the wave length of 488 nm was focused on a spot of 1 μm in diameter.
UV-vis spectra were measured in diffuse reflectance mode using a Hitachi U-3010 spectrometer equipped with an integrating sphere. A BaSO4 pellet was used as a reference. The spectra were measured in the region of 200–800 nm at room temperature.
The specific surface areas of the samples were measured using the N2 adsorption isotherm at −196 °C by the four-point Brunauer–Emmett–Teller (BET) method using an automatic surface analyzer (F-Sorb 3400, Gold APP Instrument). The samples were degassed at 200 °C for 2 h prior to the measurements.
Infrared (IR) spectra of ammonia adsorbed on the catalysts were recorded on a Nicolet 6700 FTIR spectrometer equipped with a MCT detector. The catalyst was purged by nitrogen at 500 °C for 0.5 h. A gas mixture of 1000 ppm NH3/N2 was dosed in a diffuse reflectance IR cell at RT for 30 min, and then the catalyst was flushed with N2. The spectra were collected by accumulating 32 scans at a resolution of 4 cm−1.
IR spectra of CO adsorption were recorded on the same apparatus. After pretreatment in nitrogen at 500 °C for 0.5 h, the catalyst was cooled down to RT. The spectra were taken after exposing the catalyst to 1% CO/N2 for 30 min and then purging with nitrogen.
The temperature-programmed desulfation tests of the sulfated catalysts were performed in fixed-bed reactor with the effluent gases monitored by a mass spectrometer (OmniStar TM). Fifty milligrams of catalyst powders were sandwiched by quartz wool and placed in a tubular quartz reactor (i.d. = 10 mm). The reactor temperature was raised up to 1000 °C at a heating rate of 10 °C min−1 in He (50 mL min−1).
The NOx temperature-programmed desorption (TPD) tests were performed a fixed-bed reactor with the effluent gases monitored by an infrared spectrometer (Thermo Nicolet 380). One hundred milligrams of catalyst powders were diluted with 300 mg of silica pellets, and then were sandwiched by quartz wool in a tubular quartz reactor. Prior to the test, the sample was exposed in 1000 ppm NO/10% O2/N2 (500 mL min−1) from room temperature (RT) to 300 °C at a heating rate of 10 °C min−1, cooled down to RT in the same atmosphere and flushed by N2. Afterwards, the NO and NO2 desorption profiles were obtained by ramping the reactor from RT to 600 °C at a heating rate of 10 °C min−1 in a 10% O2/N2 stream.
The NO temperature-programmed oxidation (TPO) tests were carried out in the same apparatus to that used in NOx-TPD tests. A gas mixture of 1000 ppm NO/10% O2/N2 was fed at a flow rate of 500 mL min−1. The reactor temperature was ramped to 650 °C at a heating rate of 10 °C min−1.
2.3 Activity measurement
Printex-U (Degussa) was used as a model soot. Its particle size was 25 nm and the specific surface area was 100 m2 g−1. Ten milligrams of soot and one hundred milligrams of catalyst powders were mixed by a spatula for 2 min for “loose contact” conditions. In order to prevent reaction runaway, 110 mg of the soot-catalyst mixture was diluted with 300 mg of silica pellets. The inlet gas mixture was 1000 ppm NO/10% O2/N2 with a total flow rate of 500 mL min−1. The activities of the catalysts for soot oxidation were evaluated in a temperature-programmed oxidation (TPO) reaction apparatus. The activities of the catalysts for soot oxidation were evaluated in the same apparatus to that used in NO-TPO tests. T50 represented the temperature at which 50% of soot is oxidized. The downstream CO2/(CO2 + CO) ratio during soot oxidation was defined as the selectivity to CO2. The soot oxidation activities of the catalysts in a NOx-free atmosphere were also measured in 10% O2/N2 with the catalyst-soot mixture diluted by 600 mg of silica pellets to prevent reaction runaway.3. Results
3.1 Solid properties
The structural features of the catalysts based on XRD and Raman measurements are listed in Table 1. Only typical peaks of γ-Al2O3 (2θ = 33.0°, 37.3°, 39.6°, 45.7°, 60.8° and 67.0°) are observed in the X-ray diffraction patterns of the fresh, aged and sulfated catalysts (not shown), indicating that tungsten oxide, platinum species and possible sulfates are either in high dispersion or at low degree of crystallinity. This observation agrees with the Raman results (not shown) that no obvious bands at ca. 804, 714, 327 and 267 cm−1 assigned to the octahedral crystalline WO3 are observed, illustrating that WOx species are well dispersed on the high-surface-area alumina support in despite of the impregnation method adapted.18 Neither the thermal ageing nor the sulfation ageing at a temperature as low as 350 °C seems to result in any obvious changes in the structural properties of the catalysts.Further investigation has been done by applying UV-vis diffuse reflectance spectroscopy and Kubelka–Munk function multiplied by the photon energy as described in ref. 19. This method is well known to be sensitive to identify the dispersion of metal oxides and has been used to characterize the average particle size of nanocrystalline semiconductors. As listed in Table 1, the WOx absorption edge derived from the UV-vis spectra shifts to lower energies after the ageing treatments, indicating the growth of WOx domains at higher WOx surface densities. However, these edge energy values are all larger than 3.5 eV, indicating the presence of isolated WOx species in distorted octahedral symmetry and without bridging W–O–W bonds.19,20 The BET surface area of the catalysts follows the order of the fresh sample > the aged sample > the sulfated sample. That is, the impregnation of tungsten oxide as well as the formation of surface sulfates leads to a slight decrease in the surface area of the catalysts due to the blocking effect on the support pores and the additional calcination. WOx species remain reasonably well dispersed on the high-surface-area support after the treatments at 350 °C.
Ex situ infrared spectroscopy is an effective technique to detect the formation of sulfates. Fig. 1 shows the IR spectra of the fresh and sulfated catalysts. The band at 1040 cm−1 is assigned to the OH-bending vibration of hydrated alumina surface,21 and that at 1635 cm−1 is caused by adsorbed water. After sulfation, three intense adsorption bands at 1300, 1180 and 1070 cm−1 are observed, associated with a tri-coordinated sulfate and surface sulfate species on the precious metal and alumina support.22 The last band may also be partly attributed to sulfite species.23 As shown in Fig. 2, SO2 is released in a broad temperature range between 200 and 1000 °C for PtAl–SA in the absence of any reductants. SO2 release also starts at 200 °C for WPtAl–SA, but it is almost completed before 800 °C. The desulfation process is strongly limited by the surface sulfur species mobility, which is related to sulfur transfer to Pt sites,23 different types of adsorbed sulfur species and depend on temperature.24 Thus, in combination with Fig. 1, it is possible to involve a role of less oxidized form of SOx such as sulfites on the WOx-modified catalyst and that of less coverage of Pt sites. The estimated total sulfur amount released from WPtAl–SA is significantly less than that from PtAl–SA. These suggest an enhanced resistance to sulfur dioxide for the WOx-modified catalyst. The formation of bulk aluminum sulfate and the coverage of platinum species by sulfates are limited on WOx/Pt/Al2O3, and thus the loss of oxidation activity may not involve these species to any significant extent.
 | ||
| Fig. 1 IR spectra of the fresh and sulfated catalysts. | ||
 | ||
| Fig. 2 SO2 profiles during temperature-programmed desulfation of the sulfated catalysts in inert He. | ||
3.2 NH3/CO adsorption and NOx desorption
The impregnation of tungsten oxide is expect to enhance the acidity of the catalyst significantly. Fig. 3 shows the IR spectra obtained after NH3 adsorption on the catalysts at room temperature. Typical bands at 3400–3348, 3276 and 3185 cm−1 are assigned to υas(NH), υs(NH) and δas(NH) of Lewis-bound ammonia, respectively. Bands at 3030 and 2810 cm−1 are due to Brønsted-bound ammonia.24 These bands appear on the WOx-modified catalysts and the sulfated Pt/Al2O3. That is, the addition of WOx, as well as the sulfation treatment, induces both Lewis and Brønsted acidity. The sharp band at 1452 cm−1, together with a shoulder at 1681 cm−1, is characteristic of Brønsted-bound ammonia.25 Bands at 1250 and 1178 cm−1 are corresponding to the symmetric deformation mode of ammonia coordinated to Lewis acid sites. These bands are also observed in weak intensity on the fresh and aged Pt/Al2O3 catalysts due to the electroneutral nature of alumina. The band at 1020 cm−1 is related to the OH-bending vibration of alumina.21 It is more intense for the WOx-modified catalyst due to the fundamental overtone of the W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode of surface wolframyl species.25,26 These suggest that the modification of tungsten oxide results in abundant Lewis and Brønsted sites on Pt/Al2O3 catalyst, and Brønsted-bound ammonia is more labile. The thermal ageing does not affect obviously the acidity of the WOx-modified catalyst. After the sulfation, both Pt/Al2O3 and the WOx-modified catalyst exhibit sharper bands assigned to Lewis- and Brønsted-bound ammonia coordinated to surface sulfates.
O stretching mode of surface wolframyl species.25,26 These suggest that the modification of tungsten oxide results in abundant Lewis and Brønsted sites on Pt/Al2O3 catalyst, and Brønsted-bound ammonia is more labile. The thermal ageing does not affect obviously the acidity of the WOx-modified catalyst. After the sulfation, both Pt/Al2O3 and the WOx-modified catalyst exhibit sharper bands assigned to Lewis- and Brønsted-bound ammonia coordinated to surface sulfates.
 | ||
| Fig. 3 IR spectra of the adsorbed species from contact with ammonia on catalysts at RT. | ||
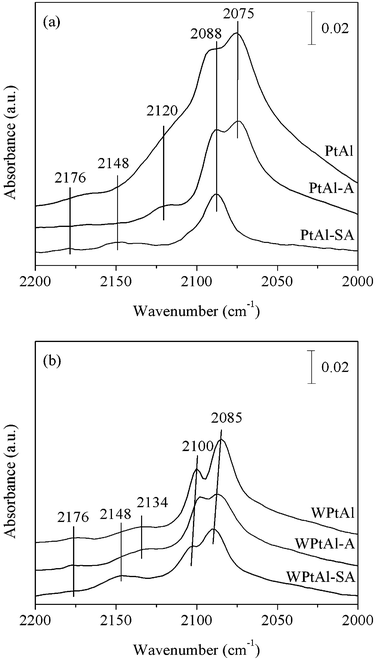
The oxidation state of platinum and the reactivity of active sites can be affected by the metal oxide additive. Fig. 4 presents the IR spectra of CO linearly adsorbed on the catalysts at room temperature under a N2 stream after CO adsorption. Band positions for adsorbed CO are sensitive to platinum dispersion, the oxidation state of platinum and charge transfer in the vicinity of Pt adsorption sites.27 Three overlapped bands are observed at 2120, 2088 and 2075 cm−1 on PtAl, which are assigned to CO linearly adsorbed on Pt electron-deficient clusters (Ptδ+), poorly dispersed Pt0 crystallites and highly dispersed Pt0 clusters, respectively.28 These bands shift to higher wavenumbers (2134, 2100 and 2085 cm−1) on WPtAl due to more electron-deficient state of platinum on the acidic support.12 The intensities of CO adsorption bands on WPtAl are lower than those obtained on PtAl perhaps due to the lowered reducibility of dispersed platinum particles on the WOx/Al2O3 support produced by a strong interaction between Pt and surface WOx species29 and blocking of active sites by WOx deposition. The weak band at 2176 cm−1 is associated with CO linearly adsorbed on Pt3+.
 | ||
| Fig. 4 IR spectra of the (a) PtAl and (b) WPtAl series catalysts after exposure to CO. | ||
Both the catalysts maintain relatively sharp bands after the long-term thermal ageing treatment. It is noted that the band assigned to CO linearly adsorbed on highly dispersed Pt0 decreases more rapidly in intensity for WPtAl–A. It implies a more significant effect of the thermal ageing on the sintering of platinum on this catalyst, which may be associated with more platinum species in the metallic state with the interaction between Pt and WOx. The sulfation inhibits obviously the CO adsorption on the Pt/Al2O3 catalyst after exposure to sulfur dioxide. The band assigned to highly dispersed Pt0 disappears, which is related to the promoted sintering of Pt and the coverage of Pt particles by sulfates. Comparatively, the sintering of Pt is much less significant on WPtAl–SA. Slight changes are observed in the spectrum of WPtAl–SA. The band due to highly dispersed Pt–CO shifts to 2090 cm−1. Additionally, the band at 2134 cm−1 assigned to Pt2+–CO decreases in intensity, while that at 2148 cm−1 assigned to Pt2+–(CO)2 increases.30 These imply a less electron-deficient state of platinum due to a weakened synergistic effect between platinum and tungsten oxide after exposure to sulfur dioxide.
Fig. 5 shows the NOx–TPD profiles of PtAl and WPtAl. The catalysts were exposed to a flow of 1000 ppm NO/10% O2/N2 from RT to 350 °C and were then cooled down to RT prior to the TPD tests. Two NO2 desorption peaks are observed at 120 and 315 °C on PtAl, which are associated with the desorption of weakly adsorbed NO2 and the decomposition of nitrates, respectively. A low-temperature NO desorption peak appears at 120 °C ascribed to the desorption of weakly adsorbed NO and/or decomposition of surface nitrites. It is noted that the high-temperature NO desorption peak lags just a few degrees (45 °C) above the high-temperature NO2 peak. Thus, it is reasonable to suggest that the desorption of NO at high temperatures arises from the thermodynamic-driven decomposition of NO2 and/or NO2 dissociation on reducible metal sites during the TPD tests under N2.31 The NO2 desorption peaks shift towards lower temperatures at 85 and 270 °C for WPtAl, indicating a reduced stability of nitrates due to the increase of acidity of the catalysts by the addition of acidic WOx. Meanwhile, little NO is released from this catalyst, suggesting that NOx stores mainly in the form of nitrates on the WOx-modified catalyst.32 It may be attributed to more appearance of metallic platinum state with the introduction of tungsten oxide. The amounts of NO and NO2 desorbed from PtAl are estimated to be 0.16 and 0.15 mmol g−1catalyst, which decreases to 0.03 and 0.12 mmol g−1catalyst for WPtAl, respectively.
 | ||
| Fig. 5 NOx–TPD profiles of PtAl and WPtAl. | ||
3.3 Oxidation of soot and NO
Fig. 6 shows the soot conversions with the catalysts in the soot-TPO measurements in 1000 ppm NO/10% O2/N2 and 10% O2/N2. The T50 of the catalysts follows the order of WPtAl (461 °C) < WPtAl–A (469 °C) < WPtAl–SA (473 °C) = PtAl (473 °C) ≈ PtAl–A (474 °C) < PtAl–SA (489 °C) in the presence of NO as shown in Fig. 6a. The deactivation of Pt/Al2O3 is mainly caused by exposure to SO2, while the loss in soot oxidation activity of WOx/Pt/Al2O3 seems to be more significantly affected by the thermal treatment. These catalysts always yield a high CO2 selectivity (>98%). It is noted that not only the sulfur resistance of Pt/Al2O3 catalyst is improved by the introduction of tungsten oxide, but also the soot oxidation activity of the fresh catalyst in the presence of NO is obviously promoted. Contrarily, the soot oxidation activity of the fresh catalyst in the absence of NO is slightly weakened by the addition of tungsten oxide with a T50 shift from 150 to 153 °C as shown in Fig. 6b. The sequences of soot oxidation activity of different catalysts in NO + O2 and O2 were confirmed reliable by repeating soot-TPO tests. That is, the presence of NO influences the soot catalytic oxidation in a different way for the catalysts with and without tungsten oxide. | ||
| Fig. 6 Soot conversions during temperature-programmed oxidation of soot with the catalysts in (a) 1000 ppm NO/10% O2/N2 and (b) 10% O2/N2. | ||
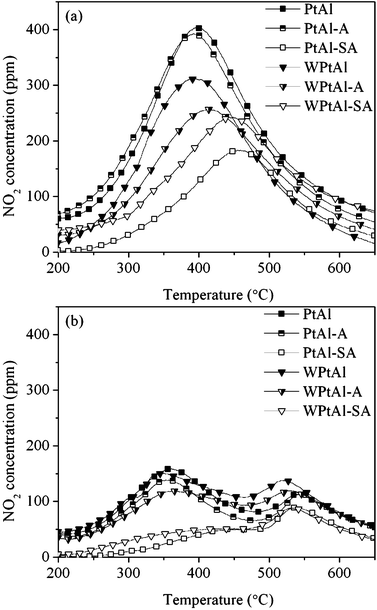
The availability of NO2 as the strong oxidant within the reaction temperature interval has been considered as a critical factor to determine the catalytic activity for soot oxidation in the presence of NO.33,34 It is generally reported that the more NO2 is produced, the higher the soot oxidation activity is attained. Fig. 7a shows the evolution of the outlet NO2 concentration during the NO–TPO measurements. The NO2 production ability of the catalysts follows the sequence of PtAl (2.11 mmol g−1catalyst) > PtAl–A (2.09 mmol g−1catalyst) > WPtAl (1.47 mmol g−1catalyst) > WPtAl–A (1.40 mmol g−1catalyst) > WPtAl–SA (1.38 mmol g−1catalyst) > PtAl–SA (0.83 mmol g−1catalyst). It is seen that Pt/Al2O3 experiences a severe loss in NO oxidation activity after the sulfation treatment, while the sulfur poisoning does not appear to be serious for the WOx-modified catalyst, which is consistent with the soot-TPO results. It is also noted that the fresh WOx/Pt/Al2O3 exhibits a higher soot oxidation activity despite its lower NO oxidation activity with respect to Pt/Al2O3. This paradoxical phenomenon implies that the soot catalytic oxidation activity of the catalysts does not depend exclusively on the NO2 production ability.
 | ||
| Fig. 7 NO2 profiles during temperature-programmed oxidation of (a) NO and (b) soot. | ||
Fig. 7b shows the corresponding evolution of NO2 during the soot-TPO tests with different catalysts. It is seen by comparison with the NO–TPO profiles (Fig. 7a) that the NO2 gap between these two tests over PtAl is larger than that over WPtAl. The importance of high efficiency of NO ↔ NO2 cycle has been demonstrated by some researches,35–37 which may also account for the high soot oxidation activity of the WOx-modified catalyst with less NO2 produced. The efficiency of NO2 can be defined by eqn (1):
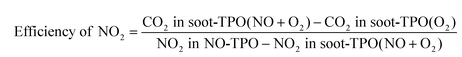 | (1) |
4. Discussion
4.1 Deactivation modes
It has been known that two deactivation modes affect platinum oxidation catalysts, namely thermal ageing and chemical poisoning, which are common to combustion catalysis. In the present study, the thermal ageing has little influence on the soot oxidation activity of Pt/Al2O3, while it results in a more significant loss in the activity of the WOx-modified catalyst. On the other hand, the sulfation treatment deactivates the unmodified catalyst much more severely. That is, the introduction of tungsten oxide greatly changes the deactivation mode of Pt/Al2O3 catalyst.The availability of NO2 as reactant is important for soot catalytic oxidation activity in the presence of NO, which is mainly determined by the number and nature of surface platinum active sites. The deposition of tungsten oxide is supposed to reduce the number of exposed active sites, which can account for the decrease in NO oxidation activity of this catalyst. Although describing the specific sintering and migration of active sites is difficult, it can be implied that the sintering of Pt seems to be more significant on WPtAl–A with a relatively rapid decrease in intensity of the CO adsorption band assigned to highly dispersed Pt0 (Fig. 4b). It may be due to the trend of platinum species existing in metallic state with the interaction between platinum and the acidic oxide additive in a oxidizing atmosphere. This further reduces the number of available Pt sites on WPtAl–A and hereby decreases the NO oxidation activity as shown in Fig. 7a. On the other hand, some aggregation and migration of finely dispersed WOx clusters may occur as indicated by the UV-vis results, indicating the evolvement of the stoichiometry and the decrease of the number of oxygen vacancies.38 This transformation of WOx to WO3 reduces the electron withdrawing ability of WOx and hereby weakens the interaction between platinum and tungsten oxide. As will be discussed in next section, such a weakening in Pt–WOx interaction is unfavorable to soot catalytic oxidation by NOx.
γ-Al2O3 is known to react with acidic molecules such as SO2 because of its amphoteric nature. The SO2 exposure at this low temperature would cause a larger Pt migration and sintering.39 Thus, the poisoning of CO adsorption at Pt sites is at a high level on Pt/Al2O3 induced by sulfation, which is consistent with the deactivation of this catalyst for NO oxidation and soot oxidation. It has been reported by some groups7,8,40 that the sulfation pretreatment or adding SO2 in the reaction atmosphere can promote the soot oxidation activity due to the facilitation of NO oxidation by the Pt–sulfate interfacial active sites with enhanced acidity. However, significant inhibition effects by sulfation on the oxidation activity of Pt/Al2O3 are observed in this case. The sulfates are supposed to form mainly on the alumina support and then to migrate to cover the adjacent Pt species. The covering effect of Pt active sites is more significant than the aggregation of Pt and the beneficial interaction between Pt and acidic sulfates, which is validated by the IR spectra of CO adsorption (Fig. 4a) and explains the severe deactivation of the sulfated Pt/Al2O3 catalyst. On the other hand, it is expected that the sulfation of the support material can be effectively inhibited by the acidic additive WOx. Although it is possible for WOx additive to promote the oxidation of SO2 on Pt/Al2O3,16 both the amount of sulfates formed on WOx/Pt/Al2O3 and the desulfation temperature decrease due to the acidic characteristic of tungsten oxide, which are evidenced by the results of ex situIR spectra and temperature-programmed desulfation. Therefore, the detrimental effects of sulfation on the NO oxidation and soot oxidation activities of the WOx-modified catalyst are much less pronounced in respect to Pt/Al2O3.
4.2 Reaction mechanisms
It has been mentioned that some platinum species on WPtAl are covered by tungsten oxide during catalyst preparation, which is confirmed by CO adsorption and is responsible for the decrease in NO oxidation activity11 and soot oxidation activity in absence of NO. It is unusual to find that the introduction of tungsten oxide obviously enhances the soot oxidation activity of the fresh catalyst in the presence of NO. The lower soot oxidation activity of the fresh Pt/Al2O3 catalyst is also likely due to the formation of platinum oxide, which is less active than metallic platinum, and/or the occupation of active sites by stable nitrates. Contrarily, the high activity of platinum on the acidic support is attributed to the high capacity to maintain the metallic state of platinum and the facility of unstable nitrates on active sites.Another plausible explanation is that the effect of tungsten oxide is attributed to the enhancement of the successive step, i.e. the decomposition of the surface oxygen compounds (SOCs) such as carboxylic anhydrides, lactones, quinine, ceto-enol groups, ethers and phenols. A similar speculated reaction scheme has been proposed to explain the promotion effect of SO3 (or H2SO4) on soot oxidation activity of Pt/SiO2 catalyst.40 These oxygenates are electron-withdrawing groups, which decrease the reactivity of neighbouring carbon for electrophilic reactions such as nitration of aromatics. The decarboxylation of these surface oxygenate groups can be catalyzed by a strong acidic additive like WOx. The specific mechanism should be further explored. For example, the diffusion rate of SOCs from the support to the Pt surface may be critical for the reaction. The presence of a larger number of acidic sites can lead to the formation of a large pool of reactive intermediates with high mobility.41 After these processes of surface oxygenate decomposition, the catalyst surface may restore the reactivity with NO2 and be subjected to a new cycle. Regarding the origin of the higher soot oxidation activity of the WOx-modified catalyst in the presence of NO+O2, the improved efficiency of NO ↔ NO2 cycle acts an important role.
5. Conclusions
Pt/Al2O3 catalyst is modified by impregnation of tungsten oxide to improve the soot oxidation activity and the resistance to sulfur dioxide. A higher soot oxidation activity is obtained over this catalyst in the presence of NO and O2 despite its lower NO oxidation activity. A possible explanation is associated with the improved oxidation-resistance of metallic platinum by interacting with the electronegative tungsten oxide in the oxidizing atmosphere, which is important for achieving high NO ↔ NO2 recycling efficiency. The acidic tungsten oxide may be also favorable for the decomposition of surface oxygen complexes and thus liberate the Pt active sites from oxygen poisoning by NO2. The transformation of WOx to WO3 weakens the interaction between WOx clusters and the adjacent Pt species, which is mainly responsible for the decrease in soot oxidation activity of WOx/Pt/Al2O3 catalyst after the long-time thermal treatment at 350 °C. After exposure to SO2 at the same temperature, the WOx-modified catalyst is much less severely affected by the sulfation with at least some of Pt species kept at a high dispersion level due to the acidic nature of the additive.Acknowledgements
The authors would like to acknowledge Projects 2009AA064801 and 2009AA06Z313 supported by the Ministry of Science and Technology of China.References
- N. Schleicher, S. Norra, F. Chai, Y. Z. Chen, S. L. Wang and D. Stüben, J. Environ. Monit., 2010, 12, 434 RSC.
- M. Kendall, A. Duarte, T. Rocha-Santos, R. Hamiltond and I. Williams, J. Environ. Monit., 2002, 4, 890 RSC.
- J. Lim, C. Lim and L. E. Yu, J. Environ. Monit., 2009, 11, 1614 RSC.
- R. Kamalakkannan, V. Zettel, A. Goubatchev, K. Stead-Dexter and N. I. Ward, J. Environ. Monit., 2004, 6, 175 RSC.
- M. Jeguirim, V. Tschamber and P. Ehrburger, Appl. Catal., B, 2007, 76, 235 CrossRef CAS.
- M. Kalogirou, D. Katsaounis, G. Koltsakis and Z. Samaras, Top. Catal., 2007, 42–43, 247 CrossRef.
- G. Corro, J. L. G. Fierro and F. B. Romero, Catal. Commun., 2006, 7, 867 CrossRef CAS.
- J. Oi-Uchisawa, S. D. Wang, T. Nanba, A. Ohi and A. Obuchi, Appl. Catal., B, 2003, 44, 207 CrossRef CAS.
- Y. Yazawa, N. Kagi, S. Komai, A. Satsuma, Y. Murakami and T. Hattori, Catal. Lett., 2001, 72, 157 CrossRef CAS.
- L. Olsson and E. Fridell, J. Catal., 2002, 210, 340 CrossRef CAS.
- R. Marques, P. Darcy, P. D. Costa, H. Mellottée, J. M. Trichard and G. Djéga-Mariadassou, J. Mol. Catal. A: Chem., 2004, 221, 127 CrossRef CAS.
- Y. Yazawa, N. Takagi, H. Yoshida, S. Komai, A. Satsuma, T. Tanaka, S. Yoshida and T. Hattori, Appl. Catal., A, 2002, 233, 103 CrossRef CAS.
- Y. Yazawa, H. Yoshida and T. Hattori, Appl. Catal., A, 2002, 237, 139 CrossRef CAS.
- J. Oi-Uchisawa, A. Obuchi, R. Enomoto, A. Ogata and S. Kushiyama, Chem. Commun., 1998, 2255 RSC.
- J. Oi-Uchisawa, A. Obuchi, R. Enomoto, S. Liu, T. Nanba and S. Kushiyama, Appl. Catal., B, 2000, 26, 17 CrossRef CAS.
- J. Dawody, M. Skoglundh and E. Fridell, J. Mol. Catal. A: Chem., 2004, 209, 215 CrossRef CAS.
- M. F. Irfan, J. H. Goo, S. D. Kim and S. C. Hong, Chemosphere, 2007, 66, 54 CrossRef CAS.
- X. L. Yang, W. L. Dai, R. H. Gao, H. Chen, H. X. Li, Y. Cao and K. N. Fan, J. Mol. Catal. A: Chem., 2005, 241, 205 CrossRef CAS.
- D. G. Barton, M. Shtein, R. D. Wilson, S. L. Soled and E. Iglesia, J. Phys. Chem. B, 1999, 103, 630 CrossRef CAS.
- J. E. Herrera, J. H. Kwak, J. Z. Hu, Y. Wang, C. H. F. Peden, J. Macht and E. Iglesia, J. Catal., 2006, 239, 200 CrossRef CAS.
- M. D. Nero, C. Galindo, R. Barillon, E. Halter and B. Madé, J. Colloid Interface Sci., 2010, 342, 437 CrossRef.
- M. Skoglundh, A. Ljungqvist, M. Petersson, E. Fridell, N. Cruise, O. Augustsson and E. Jobson, Appl. Catal., B, 2001, 30, 315 CrossRef CAS.
- J. Y. Luo, D. Kisinger, A. Abedi and W. S. Epling, Appl. Catal., A, 2010, 383, 182 CrossRef CAS.
- P. Fabrizioli, T. Bürgi and A. Baiker, J. Catal., 2002, 207, 88 CrossRef CAS.
- M. A. L. Vargas, M. Casanova, A. Trovarelli and G. Busca, Appl. Catal., B, 2007, 75, 303 CrossRef CAS.
- A. S. Khder and A. I. Ahmed, Appl. Catal., A, 2009, 354, 153 CrossRef CAS.
- S. Zafeiratos, G. Papakonstantinou, M. M. Jacksic and S. G. Neophytides, J. Catal., 2005, 232, 127 CrossRef CAS.
- J. A. Anderson, F. K. Chong and C. H. Rochester, J. Mol. Catal. A: Chem., 1999, 140, 65 CrossRef CAS.
- M. G. Falco, J. M. Grau and N. S. Fígoli, Appl. Catal., A, 2004, 264, 183 CrossRef CAS.
- K. Chakarova, M. Mihaylov and K. Hadjiivanov, Microporous Mesoporous Mater., 2005, 81, 305 CrossRef CAS.
- X. D. Wu, F. Lin, H. B. Xu and D. Weng, Appl. Catal., B, 2010, 96, 101 CrossRef CAS.
- L. Chen, J. H. Li, M. F. Ge and R. H. Zhu, Catal. Today, 2010, 153, 77 CrossRef CAS.
- I. Atribak, A. Bueno-López, A. García-García and B. Azambre, Phys. Chem. Chem. Phys., 2010, 12, 13770 RSC.
- X. D. Wu, S. Liu, D. Weng and F. Lin, Catal. Commun., 2011, 12, 345 CrossRef CAS.
- A. Y. Stakheev, A. M. Gololobov, G. N. Baeva, G. O. Bragina and N. S. Telegina, Mendeleev Commun., 2010, 20, 269 Search PubMed.
- A. L. Kustov and M. Makkee, Appl. Catal., B, 2009, 88, 263 CrossRef CAS.
- K. Krishna and M. Makkee, Catal. Today, 2006, 114, 48 CrossRef CAS.
- C. Zhang, A. Boudiba, C. Navio, C. Bittencourt, M. G. Olivier, R. Snyders and M. Debliquy, Int. J. Hydrogen Energy, 2011, 36, 1107 CrossRef CAS.
- L. Olsson and H. Karlsson, Catal. Today, 2009, 147, S290 CrossRef CAS.
- J. Oi-Uchisawa, A. Obuchi, A. Ogata, R. Enomoto and S. Kushiyama, Appl. Catal., B, 1999, 21, 9 CrossRef CAS.
- P. Papaefthimiou, T. Ioannides and X. E. Verykios, Catal. Today, 1999, 54, 81 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |