Covalent versus non-covalent (biocatalytic) approaches for enantioselective sulfoxidation catalyzed by corrole metal complexes†
Izana
Nigel-Etinger
,
Atif
Mahammed
and
Zeev
Gross
*
Schulich Faculty of Chemistry, Technion, Israel Institute of Technology, Haifa 32000, Israel. E-mail: chr10zg@techunix.technion.ac.il; Fax: +972-4-8295703; Tel: +972-4-8293954
First published on 13th May 2011
Abstract
Oxidation of thioanisoles, catalyzed by chiral manganese(III) and iron(III) corroles, provides the corresponding sulfoxides in moderate chemical yields and low enantioselectivities. Biocatalysis by non-chiral albumin-associated manganese(III) corroles proceeds much better and allows for the enantioselective synthesis of the pharmacologically important R-modafinil, in 88% yield and 73% ee.
Metal-catalyzed asymmetric synthesis remains at the forefront of modern research because of its far reaching importance and the scientific challenges involved in the design of efficient systems.1 A common scenario in the field is to optimize the catalyst's structure by “covalent chemistry”, i.e., synthesis of a large series of chiral ligands that differ slightly in their chemical structure and three-dimensional morphology.2 A less frequently adopted methodology is to rely on easily accessible (and often more potent) non-chiral metal complexes for the catalysis and on biomolecules for inducing chirality.3 This “biocatalysis” resembles Nature's style, where the non-covalently bound heme b (in cytochrome P450 and chloroperoxidases, for example) is used for catalyzing a large variety of asymmetric transformations.4 Relevant examples for utilization of the principles learned from nature include modification of the natural protein,5 replacement of natural prosthetic groups by synthetic ones,6 and the combination of non-natural metal-based catalysts with proteins whose normal function does not involve any catalytic process.7
The last decade has evidenced a new era in the chemistry of corroles, which started with the disclosure of the first practical synthesis of these long known but hardly accessible macrocycles.8 These advances were immediately used for the utilization of the corresponding metal complexes in various applications, of which catalysis was the first one.9 Metallocorroles were introduced as catalysts for hydroxylation,9aepoxidation,9a,10 cyclopropanation,11 and aziridination of hydrocarbons,12 as well as for safe decomposition of reactive oxygen and nitrogen species,13 an aspect of tremendous medicinal importance.14 Several chiral corroles have been reported,15 but the singular success regarding asymmetric catalysis by metallocorroles relied on biocatalysis.16
The choice of chiral corroles for the present study was guided by the literature about porphyrins with moieties from the “chiral pool” attached to their meso-aryl groups.17 The adopted option relies on the palladium-catalyzed amidation of C–Br bonds in corroles 1–3 (Scheme 1), because Zhang et al. have reported analogous reactions on porphyrins and also demonstrated that the corresponding metal complexes induce chirality in a very efficient manner.18 The precursor corroles were obtained by the synthetic approaches that were introduced in recent years:19 cyclocondensation of 2,6-dibromobenzaldehyde with pyrrole for 3 (3% yield), reaction of 2,6-dibromophenyl-substituted-dipyrromethane with penta-fluorobenzaldehyde for 2 (16% yield), and reaction of pentafluorophenyl-substituted-dipyrromethane with 2,6-dibromobenzaldehyde for 1 (6% yield). The C–Br activation step on the two latter corroles was also carried out successfully, although amidation of the corroles required 100 and 160 h for 1 and 2, respectively, rather than 60 h for 5,10,15,20-tetra(2,6-dibromophenyl)porphyrin.18 Chromatographic workup resulted in 20% and 28% isolated yields of 1C and 2C, respectively.20Metallation was performed only on 2C (to its manganese(III) and iron(III) complexes 2C–Mn and 2C–Fe in 91% and 70% yields, respectively), because of its better synthetic accessibility and since it contains a larger number of chiral moieties.
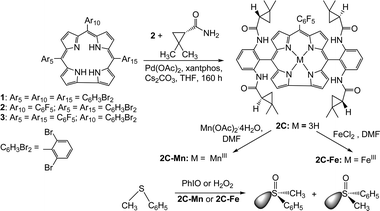 | ||
| Scheme 1 Synthesis of chiral corrole 2 and the corresponding metal complexes and catalysis thereby. | ||
The investigated reaction was asymmetric oxidation of sulfides to the corresponding sulfoxides (Scheme 1, bottom), since metallocorroles are much better suited for catalyzing sulfoxidation than any other oxidation reaction,9–12,21 and also because there are quite a few approved drugs that carry chiral sulfoxide moieties.22 Results with 0.5 mol% catalyst (2C–Mn or 2C–Fe) and equimolar amounts of PhIO and thioanisole led to reasonable chemical yields of sulfoxide, but <5% enantioselectivity and complete bleaching of the catalyst (revealed from UV-vis examination at the end of catalysis). Addition of a variety of amines did not solve these problems, but a tenfold raise in the amount of the substrate and twofold reduction of the oxidant contributed to the survival of the chiral complexes (Table S1, ESI†). The largest ee (15%) was obtained in CH2Cl2 with 2C–Mn as a catalyst, while 2C–Fe was very unstable towards PhIO and hence provided even lower ee and chemical yields under identical reaction conditions.
Very different results were obtained with hydrogen peroxide rather than PhIO as an oxidant. There was almost no reaction with 2C–Mn in any of the six examined solvents, but 2C–Fe provided a 70% chemical yield (40% at 5 °C) and 24% ee (27% at 5 °C) with THF as a solvent (Table S2, ESI†). The differences in performance of 2C–Fe and 2C–Mn with H2O2 and PhIO as oxidants are consistent with recent findings: iron(III) corroles react order of magnitudes faster with H2O2 than manganese(III) corroles.13a In addition, the catalase-like disproportionation of H2O2 by the iron(III) corroles,13a which does not exist with PhIO as an oxidant, apparently prevents bleaching of the catalyst as in the case of heme-based enzymes.4 All this acquired information may be used for the design of more selective and more robust catalysts, but this requires de novo (multistep) synthesis. Instead of doing so, the attention was driven toward the earlier mentioned non-covalent and biocatalytic approach.
A series of corroles with sulfonic acid head groups was prepared (Scheme 2), since these amphipolar derivatives interact strongly with serum albumins via a combination of hydrophobic and hydrophilic interactions and coordination of the chelated metal to basic amino acid residues.14e,16b Condensation of pentafluorophenyl-bridged dipyrromethane with aromatic aldehydes was used for the synthesis of eight free-base corroles that differ only in their C10–Ar substituent, as to allow the determination of possible steric and electronic effects on catalysis efficiency and selectivity. Corroles 1 (and 2 from Scheme 1) contain the large ortho-bromophenyl groups, while the electron-withdrawing effects of the aryl groups differ the most between corroles 1 and 7. The main motivation for the preparation of corroles 5–7 was to test if the presence of nitrogen or oxygen atoms on the aryl groups influences selectivity due to possible interactions with amino acids of the conjugated albumins.
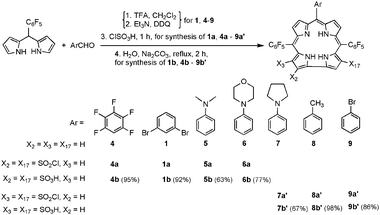 | ||
| Scheme 2 Syntheses of free base corroles and the functionalization of their β-pyrrole positions, with values in parentheses denoting the NMR/HPLC-determined selectivity for the indicated isomer relative to the other bis-substituted corrole. | ||
The triarylcorroles shown in Schemes 1 and 2 were transformed to amphipolar derivatives by the chlorosulfonation/hydrolysis route originally developed for 5,10,15-trispentafluorophenylcorrole 4.15 The selectivity for bis-substituted products was very high in all cases, but the substitution pattern of the main product varied within the series. This conclusion was deduced from the 19F-NMR spectra of chlorosulfonated corroles. Substitution on C3 and C17 (such as in 9a′) leads to derivatives with two magnetically equivalent C6F5 groups and only one set of ortho-, meta- and para-F resonances, while two such sets are obtained for the lower symmetry C2,C17-bis-substituted corrole such as 1a (Fig. S5, ESI†). All corroles except of 2 displayed quite a pronounced selectivity for only one of the two possible isomers (Scheme 2). Hydrolysis of the chlorosulfonated corroles (e.g., the transformation of 1a into 1b) was followed by metallation as to provide the corresponding manganese(III) complexes (e.g.1b–Mn) with two sulfonic acid head groups on their respective corroles. HPLC analysis revealed that the isomer ratios in the metal complexes are similar to those determined by NMR for the precursors.
For a fast evaluation of catalytic efficiency, the amphipolar manganese(III) corroles (but not the iron(III) derivatives because of their pronounced catalase-like activity)13a,23 were first used as isomeric mixtures at a 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶50∶1.5∶1 H2O2/thioanisole/albumin/catalyst molar ratio in a pH 7.0 buffer solution (Table S3, ESI†). The more interesting results (Table 1) revealed yields that varied between 20% and 71%, that the highest %ee were obtained with the complexes derived from corroles 4 and 1, and a R rather than S selectivity in the reaction catalyzed by the complex derived from 2. Accordingly, these three derivatives were further purified by HPLC as to provide 4b–Mn, 1b–Mn, and 2b′–Mn, respectively, free of their other isomer.
∶50∶1.5∶1 H2O2/thioanisole/albumin/catalyst molar ratio in a pH 7.0 buffer solution (Table S3, ESI†). The more interesting results (Table 1) revealed yields that varied between 20% and 71%, that the highest %ee were obtained with the complexes derived from corroles 4 and 1, and a R rather than S selectivity in the reaction catalyzed by the complex derived from 2. Accordingly, these three derivatives were further purified by HPLC as to provide 4b–Mn, 1b–Mn, and 2b′–Mn, respectively, free of their other isomer.
| Catalyst | HSA | BSA | PSA | RSA | SSA |
|---|---|---|---|---|---|
|
a Reaction conditions: H2O2∶thioanisole∶albumin∶catalyst (0.2 mM) = 5∶50∶ 1.5∶ 1; pH 7.0; T = 24 °C; reaction time = 1.5 h.
b Together with other isomer.
c Free of other isomer.
|
|||||
| 4b– Mn b | 68, 0, 0 | 52, S, 45 | 71, S, 37 | 71, S, 40 | 58, S, 28 |
| 1b– Mn b | 30, S, 15 | 55, S, 31 | 70, S, 28 | 70, S, 58 | 56, S, 25 |
| 2b′– Mn b | 40, R, 12 | 23, R, 5 | 30, R, 7 | 30, 0, 0 | 20, R, 2 |
| 4b– Mn c | 49, 0, 0 | 46, S, 55 | 60, S, 47 | 57, S, 52 | 50, S, 36 |
| 1b– Mn c | 26, S, 30 | 50, S, 40 | 63, S, 26 | 56, S, 65 | 52, S, 20 |
| 2b′– Mn c | 16, S, 5 | 20, S, 14 | 22, S, 10 | 14, S, 12 | 0, S, 9 |
Despite the quite minute structural differences between the isomers, the results obtained with the purified and non-purified complexes differ quite significantly (Table 1). The R selectivity obtained with the 1∶2 2b–Mn∶2b′–Mn mixture changed to S selectivity when pure 2b′–Mn was applied. For the purified 4b–Mn corrole, the ee increased from 37% to 47% and from 40% to 52% ee, with PSA and RSA, respectively. Using RSA with purified 1b–Mn provided the highest ee, 65%. Encouraged by these results, the decision was made to focus on a sulfoxide with pharmacological activity rather than on further optimization of the system or extension to substituted thioanisoles.
Due to a growing interest in the FDA-approved modafinil and its R enantiomer armodafinil, we checked the oxidation of its non-chiral precursor by hydrogen peroxide in our biocatalytic system (Scheme 3). The results obtained with four purified amphipolar metal complexes conjugated to five different albumins (Table S4, ESI†) revealed that: (a) all catalysts display selectivity for the desired R enantiomer; (b) the best catalyst for thioanisole sulfoxidation (1b–Mn) provides only modest results; (c) the highest chemical yield (24%) and enantioselectivity (66%) are obtained by the RSA/4b–Mn conjugate; (d) chemical yields are surprisingly low (3–24%).
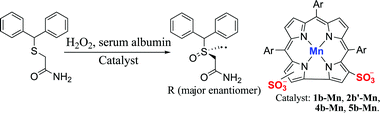 | ||
| Scheme 3 Asymmetric synthesis of R-modafinil. | ||
An important clue regarding the factor responsible for the low yields was observation of gas bubbles, signaling disproportionation of H2O2 to O2 and H2O. Realizing that the sulfide and H2O2 may compete for the same high-valent reaction intermediate, this shortcoming was resolved by gradual addition of H2O2 (8 portions, 1/h) to a 200∶50∶1.5∶1 ratio of oxidant∶substrate∶RSA∶4b–Mn (0.2 mM). HPLC examinations revealed a 90% yield and 70% ee; and recrystallization from EtOAc/hexane provided product free of unreacted sulfide in 75% ee. Up to 15% acetonitrile as an organic co-solvent had no negative effect on the outcome, which allowed for performing a somewhat larger scale reaction. Starting with 25 mg sulfide and the same ratio of reagents in a 15% CH3CN/buffer solution provided HPLC-based 88% yield and 73% ee. Recrystallization led to pure armodafinil in an isolated yield of 58% (16 mg) with 75% ee.
The covalent approach requires the preparation of chiral corroles, which was achieved by palladium-catalyzed amidation of ortho-bromo-phenyl-substituted derivatives. The iron(III) and manganese(III) complexes of the most accessible corrole were tested as catalysts for the asymmetric oxidation of thioanisole by PhI and H2O2. Chemical stability of the catalysts was one major problem; and the results were limited to 24–27% ee under low conversion conditions. On the other hand, the non-covalent approach was found to be much more fruitful: 8 amphipolar corroles were easily obtained, 5 cheap and accessible proteins were used for supplying a chiral environment, economical and environmentally benign hydrogen peroxide was used as an oxidant, and water was the solvent. This extremely simple biocatalytic system allows for a very fast screening, resulting in the identification of conditions where asymmetric oxidation of thioanisole may be achieved with 65% ee and high conversion. The most novel result is that the pharmacologically important armodafinil is produced by the same approach in 75% ee with 2 mol% of the catalyst. The flexibility of the system allows for many other easily performed optimizations (pH, additives, temperatures, etc.), which suggests that it might be of practical utility as well.
This research was supported by Technion VPR funds.
Notes and references
- (a) Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley–VCH, New York, 2010, 3rd edn, pp. 1–998 Search PubMed; (b) K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2024 CrossRef CAS; (c) R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008 CrossRef CAS.
- (a) A. Pfaltz and W. J. Drury, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5723 CrossRef CAS; (b) H. Tanaka, H. Nishikawa, T. Uchida and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 12034–12041 CrossRef CAS; (c) V. Mojr, V. Herzig, M. Budesinsky, R. Cibulka and T. Kraus, Chem. Commun., 2010, 46, 7599–7601 RSC.
- (a) Y. Lu, N. Yeung, N. Sieracki and N. M. Marshall, Nature, 2009, 460, 855 CrossRef CAS; (b) F. Pezzotti and M. Therisod, Tetrahedron: Asymmetry, 2007, 18, 701–704 CrossRef CAS.
- (a) J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3569 CrossRef CAS; (b) L. P. Hager, J. Biol. Chem., 2010, 285, 14852 CrossRef CAS; (c) F. van de Velde, I. W. C. E. Arends and R. A. Sheldon, Top. Catal., 2000, 13, 259–265 CrossRef CAS.
- (a) Y. Watanabe and T. Hayashi, Prog. Inorg. Chem., 2005, 54, 449 CrossRef CAS; (b) R. Feingersch, J. Shainsky, T. K. Wood and A. Fishman, Appl. Environ. Microbiol., 2008, 74, 1555 CrossRef CAS.
- T. Matsuo, A. Hayashi, M. Abe, T. Matsuda, Y. Hisaeda and T. Hayashi, J. Am. Chem. Soc., 2009, 131, 15124 CrossRef CAS.
- (a) C. M. Thomas and T. R. Ward, Chem. Soc. Rev., 2005, 34, 337 RSC; (b) C. Letondor, N. Humbert and T. R. Ward, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4683 CrossRef CAS; (c) E. Sansiaume, R. Ricoux, D. Gori and J. P. Mahy, Tetrahedron: Asymmetry, 2010, 21, 1593–1600 CrossRef CAS; (d) M. T. Reetz and N. Jiao, Angew. Chem., Int. Ed., 2006, 45, 2416–2419 CrossRef CAS; (e) A. Pordea, M. Creus, J. Panek, C. Duboc, D. Matis, M. Novic and T. R. Ward, J. Am. Chem. Soc., 2008, 130, 8085–8088 CrossRef CAS.
- (a) Z. Gross, N. Galili and I. Saltsman, Angew. Chem., Int. Ed., 1999, 38, 1427 CrossRef CAS; (b) I. Aviv-Harel and Z. Gross, Chem.–Eur. J., 2009, 15, 8382 CrossRef CAS.
- (a) Z. Gross, L. Simkhovich and N. Galili, Chem. Commun., 1999, 599 RSC; (b) I. Aviv and Z. Gross, Chem. Commun., 2007, 1987 RSC.
- (a) Z. Gross, G. Golubkov and L. Simkhovich, Angew. Chem., Int. Ed., 2000, 39, 4045 CrossRef CAS; (b) J. P. Collman, L. Zeng and R. A. Decreau, Chem. Commun., 2003, 2974 RSC; (c) S. H. Wang, B. S. Mandimutsira, R. Todd, B. Ramdhanie, J. P. Fox and D. P. Goldberg, J. Am. Chem. Soc., 2004, 126, 18 CrossRef CAS; (d) R. Zhang, D. N. Harischandra and M. Newcomb, Chem.–Eur. J., 2005, 11, 5713 CrossRef CAS.
- (a) I. Luobeznova, M. Raizman, I. Goldberg and Z. Gross, Inorg. Chem., 2006, 45, 386 CrossRef CAS; (b) L. Simkhovich, A. Mahammed, I. Goldberg and Z. Gross, Chem.–Eur. J., 2001, 7, 1041 CrossRef CAS; (c) L. Simkhovich and Z. Gross, Inorg. Chem., 2004, 43, 6131; (d) L. Simkhovich, P. Lyer, I. Goldberg and Z. Gross, Chem.–Eur. J., 2002, 8, 2595 CrossRef CAS.
- L. Simkhovich and Z. Gross, Tetrahedron Lett., 2001, 42, 8089 CrossRef CAS.
- (a) A. Mahammed and Z. Gross, Chem. Commun., 2010, 46, 7040 RSC; (b) M. Eckshtain, I. Zilbermann, A. Mahammed, I. Saltsman, Z. Okun, E. Maimon, H. Cohen, D. Meyerstein and Z. Gross, Dalton Trans., 2009, 7879 RSC; (c) A. Mahammed and Z. Gross, Angew. Chem., Int. Ed., 2006, 45, 6544 CrossRef CAS; (d) Z. Gershman, I. Goldberg and Z. Gross, Angew. Chem., Int. Ed., 2007, 46, 4320 CrossRef.
- (a) Z. Okun, L. Kupershmidt, T. Amit, S. Mandel, O. Bar-Am, M. B. H. Youdim and Z. Gross, ACS Chem. Biol., 2009, 4, 910 CrossRef CAS; (b) L. Kupershmidt, Z. Okun, T. Amit, S. Mandel, I. Saltsman, A. Mahammed, O. Bar-Am, Z. Gross and M. B. H. Youdim, J. Neurochem., 2010, 113, 363–373 CrossRef CAS; (c) A. Kanamori, M.-M. Catrinescu, A. Mahammed, Z. Gross and L. A. Levin, J. Neurochem., 2010, 114, 488–498 CAS; (d) A. Haber, A. Mahammed, B. Fuhrman, N. Volkova, R. Coleman, T. Hayek, M. Aviram and Z. Gross, Angew. Chem., Int. Ed., 2008, 47, 7896–7900 CrossRef CAS; (e) A. Haber, M. Aviram and Z. Gross, Chem. Sci., 2011, 2, 295–302 RSC.
- (a) A. Mahammed, I. Goldberg and Z. Gross, Org. Lett., 2001, 3, 3443 CrossRef CAS; (b) L. Simkhovich, P. Iyer, I. Goldberg and Z. Gross, Chem.–Eur. J., 2002, 8, 2595 CrossRef CAS; (c) B. Andrioletti and E. Rose, J. Chem. Soc., Perkin Trans. 1, 2002, 715 RSC; (d) I. Saltsman, I. Goldberg and Z. Gross, Tetrahedron Lett., 2003, 44, 5669 CrossRef CAS; (e) I. Saltsman, L. Simkhovich, Y. S. Balazs, I. Goldberg and Z. Gross, Inorg. Chim. Acta, 2004, 357, 3038 CrossRef CAS.
- (a) A. Mahammed and Z. Gross, J. Am. Chem. Soc., 2005, 127, 2883 CrossRef CAS; (b) A. Mahammed, H. B. Gray, J. J. Weaver, K. Sorasaenee and Z. Gross, Bioconjugate Chem., 2004, 15, 738–746 CrossRef CAS.
- Q. H. Xia, H. Q. Ge, C. P. Ye, Z. M. Liu and K. X. Su, Chem. Rev., 2005, 105, 1603 CrossRef CAS; E. Rose, B. Andrioletti, S. Zrig and M. Quelquejeu-Ehteve, Chem. Soc. Rev., 2005, 34, 573 RSC.
- Y. Chen, K. B. Fields and X. P. Zhang, J. Am. Chem. Soc., 2004, 126, 14718 CrossRef CAS.
- D. T. Gryko, Eur. J. Org. Chem., 2002, 1735 CrossRef CAS.
- For a similar approach reported while this work was in progress, see: M. Broring, C. Milsmann, S. Ruck and S. Kohler, J. Organomet. Chem., 2009, 694, 1011 Search PubMed.
- K. A. Prokop, S. P. de Visser and D. P. Goldberg, Angew. Chem., Int. Ed., 2010, 49, 5091 CAS.
- (a) V. Sverker, L. Vratislav and S. Lennart, Tetrahedron: Asymmetry, 1997, 8, 1967–1970 CrossRef; (b) T. Prisinzano, J. Podobinski, K. Tidgewell, M. Luoa and D. Swenson, Tetrahedron: Asymmetry, 2004, 15, 1053 CrossRef CAS.
- (a) M. Schwalbe, D. K. Dogutan, S. A. Stoian, T. S. Teets and D. G. Nocera, Inorg. Chem., 2011, 50, 1368–1377 CrossRef CAS; (b) A. Mahammed and Z. Gross, Catal. Sci. Technol., 2011, 2 Search PubMed , in press.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cy00046b |
| This journal is © The Royal Society of Chemistry 2011 |