2-(1-Aryliminopropylidene)quinolylcobalt(II) dichlorides: synthesis, characterization and catalytic behaviour towards ethylene†
Tianpengfei
Xiao
a,
Jingjuan
Lai
ab,
Shu
Zhang
a,
Xiang
Hao
a and
Wen-Hua
Sun
*a
aKey Laboratory of Engineering Plastics and Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: whsun@iccas.ac.cn; Fax: +86 10 62618239; Tel: +86 10 62557955
bCenter of Analysis and Measurement, Shanxi Datong University, Datong 037009, China
First published on 10th March 2011
Abstract
A series of 2-(1-aryliminopropylidene)quinolines and their cobalt(II) chlorides were synthesized and characterized. Activation with methylaluminoxane (MAO) under 10 atm ethylene pressure, all the cobalt procatalysts exhibited good activities for ethylene dimerization at room temperature, however, performed ethylene polymerization at elevated reaction temperatures. Sole ethylene polymerization was achieved at 10 atm ethylene at 90 °C. The polyethylenes obtained indicated that the molecular weight ranged from 92.5 Kg mol−1 to 176.9 Kg mol−1 with narrower molecular weight distributions (2.82–4.17).
1. Introduction
The discovery of bis(imino)pyridylmetal (Fe or Co) chlorides (A, Scheme 1)1 symbolized a milestone of late-transition metal procatalysts in ethylene polymerization and oligomerization.2 Subsequently, bis(imino)pyridylmetal (Fe or Co) procatalysts have been extensively investigated through changing substituents of bis(imino)pyridines,3 for understanding the active species and the mechanisms.4 Meanwhile, tremendous efforts have been spent on designing N-heterocyclic compounds as ligands, and the catalytic behaviour of their metal (Fe or Co) complexes were explored in ethylene reactivity.5 In the past decade, metal (Fe or Co) complexes bearing tridentate ligands have been focused on ethylene oligomerization and polymerization in our group,2g,h and those interesting ligands (Scheme 1) included 2-benzimidazolyl-6-iminopyridines (B),6 2-quinoxalinyl-6-iminopyridines (C),7 2-imino-1,10-phenanthrolines (D),8 2-(benzimidazol-2-yl)-1,10-phenanthrolines (E),9iminoquinolyl derivatives (F),10 2-methyl-2,4-bis-(6-iminopyridin-2-yl)-1H-1,5-benzodiazepines (G),11 2-benzoxazolyl-6-imino-pyridines (H),12 and 2,8-bis(imino)quinolylmetal (Fe or Co) procatalysts (I).13 In general, the late-transition metal procatalysts deactivated and produced more oligomers when the reaction temperature was elevated. However, there were two exceptional cobalt models of H12a and I,13 which favourably performed ethylene polymerization at elevated reaction temperatures. Considering the distorted square-based pyramidal geometry of 2,8-bis[1-(2,6-dimethylphenylimino) ethyl]quinolylcobalt dichloride,13 the bidentate ligands of iminoquinolines are promising. According to previous reports, cobalt complexes bearing bidentate ligands (J, Scheme 1) showed interesting results in ethylene reactivity.14 Therefore, the 2-iminoquinolines are purposely synthesized and used in preparing the title complexes (K, Scheme 1). After activation with methylaluminoxane (MAO), all the cobalt procatalysts exhibited high activity for ethylene dimerization at ambient temperature, but ethylene polymerization was achieved at elevated reaction temperatures. Besides tridentate cobalt procatalysts,12a this is the first example for cobalt complexes with bidentate ligands performing the temperature-switching ethylene reactivity and favouring polymerization at elevated temperatures. Herein, the synthesis and characterization of the title complexes and their catalytic behaviour are reported and discussed. | ||
| Scheme 1 Model procatalysts. | ||
2. Results and discussion
2.1 Synthesis and characterization
The stoichiometric condensation reaction of 2-propionyl quinoline with corresponding anilines with a catalytic amount of p-toluenesulfonic acid in toluene gave 2-(1-arylimino propylidene)quinoline derivatives (L1–L6) in good isolated yields, and further reaction with anhydrous CoCl2 in ethanol formed the cobalt complexes in high yields (Scheme 2). | ||
| Scheme 2 Synthetic procedures. | ||
All organic compounds were routinely characterized by elemental analysis, 1H and 13C NMR and IR spectra, and their cobalt complexes were characterized by elemental analysis, FT-IR and MALDI-TOF mass measurement. According to their IR spectra, the stretching vibrations of C–N in these complexes (1603–1621 cm−1) apparently shift to lower wavenumbers with greatly reduced intensities compared to those of the corresponding ligands (1641–1647 cm−1), indicating the effective coordination interaction between the imino-nitrogen and the cobalt. According to the mass spectra, monomeric cobalt cationic species with losing one chlorine were observed in all cases, indicating the monomeric feature in the solution. The unambiguous molecular structures of C2 and C3 were further confirmed by X-ray diffraction analysis, indicating the monomeric for C3 and the aggregating dimeric for C2 in the solid state. Metal complex halides were sometime present monomeric molecules in solution but dimeric in the solid states through halo-bridges.14a,15
2.2 Crystal structures
Single crystals of complexes C2 suitable for X-ray diffraction analysis were grown by slow layer diffusion of diethyl ether into its DMF solution. However, single crystals of C3 were obtained by diffusing a diethyl ether layer into its solution in methanol. Their structures are depicted in Fig. 1 and 2, respectively, with selected bond lengths and angles. | ||
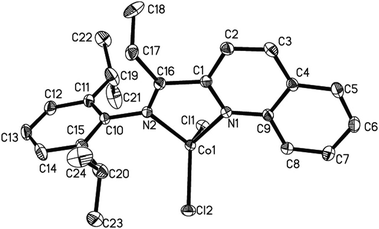
| Fig. 1 Molecular structure of C2. Thermal ellipsoids are shown at the 30% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Co1–N1, 2.154(2); Co1–N2, 2.061(3); Co1–Cl1, 2.2753(10); Co1–Cl2, 2.4376(9); N2–C10, 1.446(4); N2–C16, 1.286(4); N1–C1, 1.330(4); N1–C9, 1.360(4); N2–Co1–N1, 76.95(10); N2–Co1–Cl1, 116.88(8); N2–Co1–Cl2, 97.60(8); N1–Co1–Cl1, 91.88(7); N1–Co1–Cl2, 172.92(7); Cl2–Co1–Cl1, 94.64(4). | ||
 | ||
| Fig. 2 Molecular structure of C3. Thermal ellipsoids are shown at the 30% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Co1–N1, 2.047(2); Co1–N2, 2.035(2); Co1–Cl1, 2.2202(10); Co1–Cl2, 2.2087(9); N2–C10, 1.449(3); N2–C16, 1.288(3); N1–C1, 1.332(3); N1–C9, 1.367(3); N2–Co1–N1, 80.62(9); N2–Co1–Cl1, 113.91(7); N2–Co1–Cl2, 117.03(7); N1–Co1–Cl1, 106.90(7); N1–Co1–Cl2, 117.26(7); Cl2–Co1–Cl1, 115.88(4). | ||
In C2, a centrosymmetric dicobalt center is shown with two chloro-bridges. Each cobalt atom is coordinated by two nitrogen atoms of the ligand, one terminal chlorine atom (Cl1), and two bridged chlorine atoms (Cl2 and Cl2A). The five-coordinate cobalt center forms a distorted trigonal bipyramid coordination. The Co1–N1 bond length is 2.154(2) Å, longer than that of Co1–N2 bond. The distance between the cobalt and the terminal chlorine (Co1–Cl1) is 2.2753(10) Å, whereas, the bond lengths between the cobalt and another two bridging chlorines are different significantly (Co1–Cl2 = 2.4376(9) and Co1–Cl2A = 2.3451(10) Å). The Co⋯Co length is 3.460 Å, indicating no direct metal–metal interaction. The Co1 atom deviates by 0.095 Å from the plane of the Cl2–N1–Cl2 atoms. The dihedral angle between the plane of Cl1–Co1–Cl2 and the plane of N1–Co1–N2 is 62.77°, significantly different from that of complex C3. The dihedral angle between the quinolinyl plane and the plane of the phenyl ring is 85.31°.
The four-coordinate complex C3 (Fig. 2) shows a distorted tetrahedral geometry around the cobalt. Two Co–N bonds have nearly similar bond lengths. The Co1–Cl1 bond length [2.2202(10) Å] are longer than the bond lengths of Co1–Cl2 [2.2087(9) Å], and both of the Co–Cl bond length are shorter than those of complex C2. The dihedral angle formed by Cl1–Co1–Cl2 and N1–Co1–N2 is 86.54°. In the five-membered cobaltacycle N2–C16–C1–N1–Co1, all atoms almost lie in the same plane with the largest deviation of 0.136 Å for N1 and 0.046 Å for Co, while the sum of internal ring angles (539°) is close to the idea value of 540°. The plane formed by the quinolinyl and the plane of phenyl ring on imino group are nearly vertical with the dihedral angle of 83.34°.
2.3 Ethylene dimerization and polymerization
| Entry | Al/Co | T/°C | Butene | Polymer | |||
|---|---|---|---|---|---|---|---|
| Product yield/ga | Activityb | α-C4/∑C4 (%)a | Product yield/mg | Activityc | |||
| Condition: 5 μmol Complex, 30 min, 100 mL toluene.a Determined by GC.b 105 g mol−1 (Co) h−1.c 104 g mol−1 (Co) h−1. | |||||||
| 1 | 500 | 20 | 0.48 | 1.9 | 32.2 | — | — |
| 2 | 750 | 20 | 1.08 | 4.3 | 40.1 | — | — |
| 3 | 1000 | 20 | 4.75 | 19 | 59.8 | — | — |
| 4 | 1250 | 20 | 1.93 | 7.7 | 47.9 | — | — |
| 5 | 1500 | 20 | 1.58 | 6.3 | 46.4 | — | — |
| 6 | 1000 | 40 | 1.45 | 5.8 | 44.3 | — | — |
| 7 | 1000 | 60 | 1.18 | 4.7 | 40.8 | — | — |
| 8 | 1000 | 80 | 1.08 | 4.3 | 41.2 | 15 | 0.63 |
| 9 | 1000 | 90 | — | — | — | 23 | 0.91 |
As shown in Table 1, the optimum condition for ethylene dimerization was using the Al/Co molar ratio of 1000 at room temperature. Changing the Al/Co molar ratio or elevating the reaction temperature caused the catalytic activity to gradually decrease. Such phenomena have been commonly observed in catalytic systems of late-transition metal procatalysts. Interestingly, both butenes and polyethylene were observed with low activity at 80 °C. Surprisingly, further increasing temperature up to 90 °C, polyethylenes were only obtained without noticing any oligomers. Therefore the detail study was carried out in both ethylene dimerization and polymerization. The ethylene dimerization by all procatalysts with the Al/Co molar ratio of 1000 at 20 °C and 10 atm was preceded, and their results were tabulated in Table 2.
According to Table 2 (entries 1–5), similar to cobalt procatalysts in the literature,14b–f current procatalysts showed high activity in ethylene dimerization. The catalytic activities were in the order C3 > C2 > C1 and C5 > C4, and all higher than C6. The more bulky substituents of ligands protected the active sites, therefore active species was maintained.3c The tendency of catalytic activities with substituent influences was not consistent with the calculation simulation for N⁁N⁁N-tridentate late-transition metal procatalysts,14e,16 which indicated the higher net charge of the metal centre performed better catalytic activity. In the current system, the bidentate ligands provided less electronic donations to the metal center. Regarding observations by C2vs.C5 and C1vs.C4, the procatalysts bearing ligands with an additional methyl group showed better activity because of a better solubility in solution, consistent with the literature.1
| Entry | Complex | Al/Co | T/°C | Product yield/mg | Activitya | T m/°Cb | M w c × 10−4 | M w/Mnc |
|---|---|---|---|---|---|---|---|---|
| Condition: 5 μmol Co, 100 mL toluene.a 104 g mol−1 (Co) h−1.b Determined by DSC.c Determined by GPCvs.polystyrene standards. | ||||||||
| 1 | C3 | 1000 | 90 | 23 | 0.91 | 131.6 | — | — |
| 2 | C3 | 2000 | 90 | 35 | 1.4 | 132.7 | 10.27 | 3.40 |
| 3 | C3 | 2500 | 90 | 58 | 2.3 | 133.2 | 11.69 | 3.57 |
| 4 | C3 | 3000 | 90 | 130 | 5.2 | 133.4 | 12.54 | 3.79 |
| 5 | C3 | 3500 | 90 | 108 | 4.3 | 133.7 | 13.51 | 3.99 |
| 6 | C3 | 4000 | 90 | 103 | 4.1 | 133.0 | 11.32 | 4.17 |
| 7 | C3 | 3000 | 100 | 98 | 4.0 | 133.1 | 12.97 | 4.14 |
| 8 | C1 | 3000 | 90 | 105 | 4.2 | 133.0 | 11.16 | 3.87 |
| 9 | C2 | 3000 | 90 | 115 | 4.6 | 132.8 | 9.54 | 3.98 |
| 10 | C4 | 3000 | 90 | 113 | 4.5 | 132.9 | 10.73 | 3.97 |
| 11 | C5 | 3000 | 90 | 123 | 4.9 | 132.4 | 9.25 | 2.82 |
| 12 | C6 | 3000 | 90 | 98 | 4.0 | 132.7 | 10.41 | 3.72 |
According to their GPC data, the polyethylenes obtained had similar molecular weights and narrow molecular weight distributions (2.82–4.17), and the similar active species were assumed for those cobalt procatalysts. The higher temperature (entry 7, Table 3) and more MAO (entries 5 and 6, Table 3) used, resulted in polyethylenes with broader molecular weight distributions because of multi-active species that formed.13,18 Considering molecular weights of polyethylenes formed and the activity data, even imaging an active species producing one polymeric chain, there were less than 10% of cobalt complexes transferred into active species for such polymerization. In the dimerization process at ambient temperature, the activities were obtained one-order higher. Therefore, there are more active species formed at low reaction temperature, indicating that either less active species are formed or the ligands can not sufficiently protect the active species at elevated temperatures.5a Though bidentate cobalt complexes bearing iminopyridine ligands14a and α-diimine ligands14g,h were investigated, the current cobalt procatalysts showed the highest activity in ethylene polymerization, especially at 90 °C.
In general, ethylene oligomerization and polymerization are considered as the competitive reactions of chain propagation and chain transfer. In the current catalytic systems two different active species formed at different reaction temperatures. Though most bidentate cobalt complexes show lower activity in ethylene polymerization,14a,g,h newly active species were potentially formed in the current cobalt system when the catalytic system was heated up to above 80 °C in the presence of MAO. The bidentate cobalt complexes were stable in toluene for three hours at 100 °C without noticing any change; however, the colour of the cobalt solution changed with adding 1000 equiv. of MAO. Considering a higher amount of butenes (in terms of weight) formed at a lower temperature than the amount of polyethylenes at a high temperature, suggests the active species was easily formed at the lower reaction temperature with possibly repeating dimerization. However, one of several cobalt complex molecules would be transferred as an active species acting for ethylene polymerization, which could be one cobalt species (with most cobalt deactivated) or several cobalt complexes aggregating together as an multi-nuclear species. Therefore, two kinds of characteristic active species were assumed at different reaction temperatures.
3. Conclusion
The 2-(1-aryliminopropylidene)quinolylcobalt(II) dichlorides, when activated with MAO, showed high activities in ethylene dimerization at ambient temperature, and considerable activities in ethylene polymerization at 90 °C. Two kinds of active species are potentially formed and performed in ethylene reactivity at different temperatures. It is assumed that only partially active species could be formed at higher temperatures. In the current cobalt procatalysts, the more bulky substituents of the ligands that were used, the better the activities observed. The bidentate cobalt complexes in ethylene reactivity would stimulate a more extensive study.4. Experimental
4.1 General considerations
All manipulations of air- and moisture-sensitive compounds were performed under a nitrogen atmosphere using standard Schlenk techniques. Toluene was refluxed over sodium benzophenone and distilled under argon prior to use. Methylaluminoxane (MAO, a 1.46 M solution in toluene) and modified methylaluminoxane (MMAO, 1.93 M in heptane, 3A) were purchased from Akzo Nobel Corporation. Other reagents were purchased from Aldrich or Acros Chemicals. 1H and 13C NMR spectra were recorded on a Bruker DMX 300 MHz or a Bruker DMX 400 MHz instrument at ambient temperature using TMS as an internal standard. IR spectra were recorded on a Perkin-Elmer System 2000 FT-IR spectrometer. Elemental analysis was carried out using an HPMOD 1106 microanalyzer. MALDI-TOF mass spectra were obtained with a Bruker Autoflex III matrix-assisted laser desorption/ionization time of-flight mass spectrometer, with DHB as the matrix. GC analysis was performed with a VARIAN CP-3800 gas chromatograph equipped with a flame ionization detector and a 30 m (0.2 mm i.d., 0.25 μm film thickness) CP-Sil 5 CB column. The yield of oligomers was calculated by referencing with the mass of the solvent on the basis of the prerequisite that the mass of each fraction was approximately proportional to its integrated areas in the GC trace. Selectivity for the linear α-olefin was defined as (amount of linear α-olefin of all fractions)/(total amount of oligomer products) in percent. The molecular weights and the polydispersities of the polymer samples were determined at 150 °C by a PL-GPC 220 type high-temperature chromatograph equipped with three PLgel 10 mm Mixed-B LS type columns. 1,2,4-Trichlorobenzene (TCB) was employed as the solvent at a flow rate of 1.0 mL min−1. The calibration was made by using a polystyrene standard.4.2 Syntheses
4.3 General procedure for ethylene dimerization and polymerization
Ethylene dimerization or/and polymerization at 10 atm of ethylene pressure was carried out in a stainless steel autoclave (250 mL capacity) equipped with a mechanical stirrer and a temperature controller. The toluene solution of the catalytic precursor and toluene (the total volume was 100 mL) were added into the autoclave under nitrogen atmosphere. The required amount of co-catalyst then was injected into the reactor via a syringe. The ethylene pressure was tuned to 10 atm, and maintained at this level by constant feeding of ethylene at the desired reaction temperature. After the reaction mixture was stirred for the desired period, the mixture was cooled to room temperature by the ice bath, then the pressure was released. A small amount of the reaction solution was collected, which was then analyzed by gas chromatography (GC) for determining the composition and mass distribution of oligomers obtained. While the reaction solution was quenched with 5% hydrochloride acid in ethanol, the precipitated polymer was collected by filtration, and was adequately washed with ethanol and water, and was then dried in a vacuum until constant weight.4.4 Crystal structure determinations
Single-crystal X-ray diffraction study for C2 and C3 was carried out on a Rigaku RAXIS Rapid IP diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). Cell parameters were obtained by global refinement of the positions of all collected reflections. Intensities were corrected for Lorentz and polarization effects and empirical absorption. The structures were solved by direct methods and refined by full-matrix least squares on F2. All non-hydrogen atoms were refined anisotropically. Structure solution and refinement were performed by using the SHELXL-97 package.19Crystal data and processing parameters for C2 and C3 is collected in Table 4.| C2 | C3 | |
|---|---|---|
| Empirical formula | C22H24Cl2CoN2 | C24H28Cl2CoN2 |
| Formula weight | 446.26 | 474.31 |
| Crystal color | Yellow | Blue |
| T/K | 173(2) | 173(2) |
| λ/Å | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P21/n |
| a/Å | 11.337(2) | 10.125(2) |
| b/Å | 15.712(3) | 16.885(3) |
| c/Å | 11.611(2) | 13.533(3) |
| β/° | 95.49(3) | 90.01(3) |
| V/Å3 | 2058.6(7) | 2313.7(8) |
| Z | 4 | 4 |
| D c/Mg m−3 | 1.440 | 1.362 |
| μ/mm−1 | 1.102 | 0.985 |
| F(000) | 924 | 988 |
| Crystal size/mm | 0.13 × 0.12 × 0.03 | 0.20 × 0.20 × 0.20 |
| θ range/° | 2.40–27.45 | 2.35–27.51 |
| Limiting indices | −13 ≤ h ≤ 14 | −13 ≤ h ≤ 13 |
| −20 ≤ k ≤ 17 | −21 ≤ k ≤ 15 | |
| −–15 ≤ l ≤ 15 | −16 ≤ l ≤ 17 | |
| Completeness to θ (%) | 99.7 (θ = 27.45) | 98.7 (θ = 27.51) |
| R int | 0.0500 | 0.0595 |
| Absorption correction | Empirical | Empirical |
| Data/restraints/parameters | 4682/82/272 | 5245/42/282 |
| Goodness-of-fit on F2 | 1.200 | 1.113 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0574, wR2 = 0.1313 | R 1 = 0.0537, wR2 = 0.1082 |
| R indices (all data) | R 1 = 0.0616, wR2 = 0.1336 | R 1 = 0.0595, wR2 = 0.1117 |
| Largest diffraction peak and hole/e Å−3 | 0.402 and −0.353 | 0.423 and −0.415 |
Acknowledgements
This work is supported by MOST 863 program no. 2009AA033601.References
- (a) B. L. Small, M. Brookhart and A. M. A. Bennett, J. Am. Chem. Soc., 1998, 120, 4049 CrossRef CAS; (b) G. J. P. Britovsek, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. J. McTavish, G. A. Solan, A. J. P. White and D. J. Williams, Chem. Commun., 1998, 849 RSC.
- (a) G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed., 1999, 38, 428 CrossRef CAS; (b) S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169 CrossRef CAS; (c) V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283 CrossRef CAS; (d) V. C. Gibson, C. Redshaw and G. A. Solan, Chem. Rev., 2007, 107, 1745 CrossRef CAS; (e) C. Bianchini, G. Giambastiani, I. G. Rios, G. Mantovani, A. Meli and A. M. Segarra, Coord. Chem. Rev., 2006, 250, 1391 CrossRef CAS; (f) C. Bianchini, G. Giambastiani, L. Luconi and A. Meli, Coord. Chem. Rev., 2010, 254, 431 CrossRef CAS; (g) W.-H. Sun, S. Zhang and W. Zuo, C. R. Chim., 2008, 11, 307 CrossRef CAS; (h) S. Jie, W.-H. Sun and T. Xiao, Chin. J. Polym. Sci., 2010, 28, 299 CAS.
- (a) B. L. Small and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 7143 CrossRef CAS; (b) G. J. P. Britovsek, M. Bruce, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. Mastroianni, S. J. McTavish, C. Redshaw, G. A. Solan, S. Strömberg, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1999, 121, 8728 CrossRef CAS; (c) G. J. P. Britovsek, S. Mastroianni, G. A. Solan, S. P. D. Baugh, C. Redshaw, V. C. Gibson, A. J. P. White, D. J. Williams and M. R. J. Elsegood, Chem.–Eur. J., 2000, 6, 2221 CrossRef CAS; (d) E. P. Talsi, D. E. Babushkin, N. V. Semikolenova, V. N. Zudin, V. N. Panchenko and V. A. Zakharov, Macromol. Chem. Phys., 2001, 202, 2046 CrossRef CAS; (e) Z. Ma, W.-H. Sun, Z. L. Li, C. X. Shao, Y. L. Hu and X. H. Li, Polym. Int., 2002, 51, 994 CrossRef CAS; (f) Y. Chen, C. Qian and J. Sun, Organometallics, 2003, 22, 1231 CrossRef CAS; (g) I. S. Paulino and U. Schuchardt, J. Mol. Catal. A: Chem., 2004, 211, 55 CrossRef CAS; (h) Z. Zhang, S. Chen, X. Zhang, H. Li, Y. Ke, Y. Lu and Y. Hu, J. Mol. Catal. A: Chem., 2005, 230, 1 CrossRef CAS; (i) J.-Y. Liu, Y. Zheng, Y.-G. Li, L. Pan, Y.-S. Li and N.-H. Hu, J. Organomet. Chem., 2005, 690, 1233 CrossRef CAS.
- (a) V. C. Gibson, M. J. Humphries, K. P. Tellmann, D. F. Wass, A. J. P. White and D. J. Williams, Chem. Commun., 2001, 2252 RSC; (b) G. J. P. Britovsek, G. K. B. Clentsmith, V. C. Gibson, D. M. L. Goodgame, S. J. McTavish and Q. A. Pankhurst, Catal. Commun., 2002, 3, 207 CrossRef CAS; (c) K. P. Bryliakov, N. V. Semikolenova, V. A. Zakharov and E. P. Talsi, Organometallics, 2004, 23, 5375 CrossRef CAS; (d) K. P. Bryliakov, N. V. Semikolenova, V. N. Zudin, V. A. Zakharov and E. P. Talsi, Catal. Commun., 2004, 5, 45 CrossRef CAS; (e) J. Cámpora, A. M. Naz, P. Palma, E. Álvarez and M. L. Reyes, Organometallics, 2005, 24, 4878 CrossRef CAS; (f) M. W. Bouwkamp, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2005, 127, 9660 CrossRef CAS; (g) S. C. Bart, K. Chlopek, E. Bill, M. W. Bouwkamp, E. Lobkovsky, F. Neese, K. Wieghardt and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 13901 CrossRef CAS; (h) C. Wallenhorst, G. Kehr, H. Luftmann, R. Fröhlich and G. Erker, Organometallics, 2008, 27, 6547 CrossRef CAS; (i) R. J. Trovitch, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2008, 130, 11631 CrossRef CAS; (j) V. L. Cruz, J. Ramos, J. Martínez-Salazar, S. Gutiérrez-Oliva and A. Toro-Labbé, Organometallics, 2009, 28, 5889 CrossRef CAS; (k) R. Raucoules, T. Bruin, P. Raybaud and C. Adamo, Organometallics, 2009, 28, 5358 CrossRef CAS; (l) K. P. Bryliakov, E. P. Talsi, N. V. Semikolenova and V. A. Zakharov, Organometallics, 2009, 28, 3225 CrossRef CAS; (m) A. C. Bowman, C. Milsmann, C. C. H. Atienza, E. Lobkovsky, K. Wieghardt and P. J. Chirik, J. Am. Chem. Soc., 2010, 132, 1676 CrossRef CAS; (n) A. C. Bowman, C. Milsmann, E. Bill, E. Lobkovsky, T. Weyhermüller, K. Wieghardt and P. J. Chirik, Inorg. Chem., 2010, 49, 6110 CrossRef CAS; (o) A. M. Tondreau, C. Milsmann, A. D. Patrick, H. M. Hoyt, E. Lobkovsky, K. Wieghardt and P. J. Chirik, J. Am. Chem. Soc., 2010, 132, 15046 CrossRef CAS; (p) C. C. H. Atienza, A. C. Bowman, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2010, 132, 16343 CrossRef.
- (a) R. Cowdell, C. J. Davies, S. J. Hilton, J.-D. Maréchal, G. A. Solan, O. Thomas and J. Fawcett, Dalton Trans., 2004, 3231 RSC; (b) A. R. Karama, E. L. Cataría, F. López-Linaresa, G. Agrifoglioa, C. L. Albanoa, A. Díaz-Barriosa, T. E. Lehmanna, S. V. Pekerara, L. A. Albornoza, R. Atencioa, T. Gonzáleza, H. B. Ortegab and P. Joskowicsb, Appl. Catal., A, 2005, 280, 165 CrossRef CAS; (c) A. Karama, R. Tenia, M. Martínez, F. López-Linares, C. Albano, A. Diaz-Barrios, Y. Sánchez, E. Catarí, E. Casas, S. Pekerar and A. Albornoz, J. Mol. Catal. A: Chem., 2007, 265, 127 CrossRef; (d) B. L. Small, R. Rios, E. R. Fernandez and M. J. Carney, Organometallics, 2007, 26, 1744 CrossRef CAS; (e) K. Tenza, M. J. Hanton and A. M. Z. Slawin, Organometallics, 2009, 28, 4852 CAS.
- (a) W.-H. Sun, P. Hao, S. Zhang, Q. Shi, W. Zuo, X. Tang and X. Lu, Organometallics, 2007, 26, 2720 CrossRef CAS; (b) Y. Chen, P. Hao, W. Zuo, K. Gao and W.-H. Sun, J. Organomet. Chem., 2008, 693, 1829 CrossRef CAS; (c) L. Xiao, R. Gao, M. Zhang, Y. Li, X. Cao and W.-H. Sun, Organometallics, 2009, 28, 2225 CrossRef CAS.
- W.-H. Sun, P. Hao, G. Li, S. Zhang, W. Wang, J. Yi, M. Asma and N. Tang, J. Organomet. Chem., 2007, 692, 4506 CrossRef CAS.
- (a) W.-H. Sun, S. Jie, S. Zhang, W. Zhang, Y. Song and H. Ma, Organometallics, 2006, 25, 666 CrossRef CAS; (b) S. Jie, S. Zhang, W.-H. Sun, X. Kuang, T. Liu and J. Guo, J. Mol. Catal. A: Chem., 2007, 269, 85 CrossRef CAS; (c) S. Jie, S. Zhang, K. Wedeking, W. Zhang, H. Ma, X. Lu, Y. Deng and W.-H. Sun, C. R. Chim., 2006, 9, 1500 CrossRef CAS; (d) S. Jie, S. Zhang and W.-H. Sun, Eur. J. Inorg. Chem., 2007, 5584 CrossRef CAS; (e) M. Zhang, W. Zhang, T. Xiao, J.-F. Xiang, X. Hao and W.-H. Sun, J. Mol. Catal. A: Chem., 2010, 320, 92 CrossRef CAS.
- (a) M. Zhang, P. Hao, W. Zuo, S. Jie and W.-H. Sun, J. Organomet. Chem., 2008, 693, 483 CrossRef CAS; (b) M. Zhang, R. Gao, X. Hao and W.-H. Sun, J. Organomet. Chem., 2008, 693, 3867 CrossRef CAS.
- (a) A. Köppl and H. G. Alt, J. Mol. Catal. A: Chem., 2000, 154, 45 CrossRef CAS; (b) G. J. P. Britovsek, S. P. D. Baugh, O. Hoarau, V. C. Gibson, D. Wass, A. J. P. White and D. J. Williams, Inorg. Chim. Acta, 2003, 345, 279 CrossRef CAS; (c) V. C. Gibson, C. M. Halliwell, N. J. Long, P. J. Oxford, A. M. Smith, A. J. P. White and D. J. Williams, Dalton Trans., 2003, 918 RSC; (d) K. Wang, K. Wedeking, W. Zuo, D. Zhang and W.-H. Sun, J. Organomet. Chem., 2008, 693, 1073 CAS.
- (a) S. Zhang, I. Vystorop, Z. Tang and W.-H. Sun, Organometallics, 2007, 26, 2456 CrossRef CAS; (b) S. Zhang, W.-H. Sun, X. Kuang, I. Vystorop and J. Yi, J. Organomet. Chem., 2007, 692, 5307 CrossRef CAS.
- (a) R. Gao, K. Wang, Y. Li, F. Wang, W.-H. Sun, C. Redshaw and M. Bochmann, J. Mol. Catal. A: Chem., 2009, 309, 166 CrossRef CAS; (b) R. Gao, Y. Li, F. Wang, W.-H. Sun and M. Bochmann, Eur. J. Inorg. Chem., 2009, 4149 CrossRef CAS.
- S. Zhang, W.-H. Sun, T. Xiao and X. Hao, Organometallics, 2010, 29, 1168 CrossRef CAS.
- (a) T. V. Laine, K. Lappalainen, J. Liimatta, E. Aitolal, B. Löfgren and M. Leskelä, Macromol. Rapid Commun., 1999, 20, 487 CrossRef CAS; (b) L. Wang, W.-H. Sun, Z. Li, L. Han, Y. Hu, C. He and C. Yang, J. Organomet. Chem., 2002, 650, 59 CrossRef CAS; (c) C. Bianchini, G. Mantovani, A. Meli and F. Migliacci, Organometallics, 2003, 22, 2545 CrossRef CAS; (d) C. Bianchini, G. Giambastiani, G. Mantovani, A. Meli and D. Mimeau, J. Organomet. Chem., 2004, 689, 1356 CrossRef CAS; (e) W.-H. Sun, X. Tang, T. Gao, B. Wu, W. Zhang and H. Ma, Organometallics, 2004, 23, 5037 CAS; (f) C. Bianchini, D. Gatteschi, G. Giambastiani, I. G. Rios, A. Ienco, F. Laschi, C. Mealli, A. Meli, L. Sorace, A. Toti and F. Vizza, Organometallics, 2007, 26, 726 CrossRef CAS; (g) M. Tanabiki, Y. Sunada and H. Nagashima, Organometallics, 2007, 26, 6055 CrossRef CAS; (h) V. Rosa, S. A. Carabineiro, T. Avilés, P. T. Gomes, R. Welter, J. M. Campos and M. R. Ribeiro, J. Organomet. Chem., 2008, 693, 769 CrossRef CAS.
- (a) R. J. Butcher and E. Sinn, Inorg. Chem., 1977, 16, 2334 CrossRef CAS; (b) T. V. Laine, M. Klinga and M. Leskelä, Eur. J. Inorg. Chem., 1999, 959 CrossRef CAS; (c) F. Speiser, P. Braunstein, L. Saussine and R. Welter, Inorg. Chem., 2004, 43, 1649 CrossRef CAS; (d) P. Chavez, I. Rios, A. Kermagoret, R. Pattacini, A. Meli, C. Bianchini, G. Giambastiani and P. Braunstein, Organometallics, 2009, 28, 1776 CrossRef CAS; (e) W.-H. Sun, W. Zhang, T. Gao, X. Tang, L. Chen, Y. Li and X. Jin, J. Organomet. Chem., 2004, 689, 917 CrossRef CAS.
- (a) D. Guo, L. Han, T. Zhang, W.-H. Sun, T. Li and X. Yang, Macromol. Theory Simul., 2002, 11, 1006 CrossRef CAS; (b) T. Zhang, D. Guo, S. Jie, W.-H. Sun, T. Li and X. Yang, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4765 CrossRef CAS.
- M. Atiqullah, H. Hammawa and H. Hamid, Eur. Polym. J., 1998, 34, 1511 CrossRef CAS.
- (a) E. Y. -X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391 CrossRef CAS; (b) X. Tang, D. Zhang, S. Jie, W.-H. Sun and J. Chen, J. Organomet. Chem., 2005, 690, 3918 CrossRef CAS; (c) W. Zhang, W.-H. Sun, S. Zhang, J. Hou, K. Wedeking, S. Schultz, R. Fröhlich and H. Song, Organometallics, 2006, 25, 1961 CrossRef CAS; (d) W. Zhang, W.-H. Sun, X. Tang, T. Gao, S. Zhang, P. Hao and J. Chen, J. Mol. Catal. A: Chem., 2007, 265, 159 CrossRef CAS.
- Sheldrick, G. M. SHELXTL-97, Program for the Refinement of Crystal Structures; University of Göttingen: Germany, 1997.
Footnote |
| † Electronic Supplementary Information (ESI) available: CCDC reference numbers 805826 and 805827 for crystallographic data of complexes C2 and C3. See http://dx.doi.org/10.1039/c1cy00028d |
| This journal is © The Royal Society of Chemistry 2011 |