Catalytic synthesis of homoallyloxyalcohols and 1,2-bis(homoallyloxy)ethanes through ring-opening allylation of cyclic acetals with allylsilanes over solid acids†
Received
2nd February 2011
, Accepted 22nd March 2011
First published on 20th April 2011
Abstract
Ring-opening allylation of cyclic acetals with allylsilanes afforded silyl ethers using a silica–alumina catalyst. For example, the reaction of 2-phenyl-1,3-dioxolane with allyltrimethylsilane gave the corresponding silyl ether in 83% yield. After the allylation, addition of water to the reaction mixture allowed hydrolysis of the silyl ether to afford the homoallyloxyalcohol in 77% yield based on the acetal used. The reaction pathway of the silica–alumina-catalyzed allylation was examined. Solid-state 13C and 29Si NMR analyses revealed the generation of SiMe3 groups on the silica–alumina surface. The part of the (surface)–O–SiMe3 groups was found to be an active site for the allylation reaction. During the silica–alumina-catalyzed allylation, a small amount of 1,2-bis(homoallyloxy)ethane formed as a co-product. The selectivity toward the 1,2-bis(homoallyloxy)ethane increased from 7% to 60% using H+-montmorillonite instead of silica–alumina. Detailed reaction pathways for the syntheses of 1,2-bis(homoallyloxy)ethanes were also investigated.
Introduction
Allylation of acetals is promoted by acid reagents.1,2 However, a stoichiometric amount of Lewis acid to the acetal is necessary for ring-opening allylation of cyclic acetals.2 To obtain the homoallyloxyalcohol by the Lewis acid-promoted ring-opening allylation, water or an alcohol can be added to decompose the “Lewis acid-allylation product complex (alkoxide)” (Scheme 1, path A).2 It was reported that a catalytic amount of a Lewis acid did not show good performance for the ring-opening allylation of cyclic acetals.3 The reason for this seems to be that the Lewis acidic species is not regenerated after the allylation. To regenerate a catalytically active species, acid anhydrides were used as additives to afford ester products instead of the homoallyloxyalcohols (Scheme 1, path A (dotted line)).3 It is believed that the acid anhydride captures the “Lewis acid-allylation product complex”.
 |
| | Scheme 1 Lewis acid-promoted ring-opening allylation of cyclic acetal (path A) and catalytic synthesis of homoallyloxyalcoholvia introduction of a leaving group (LG) to ring-opened acetal (path B). | |
If a leaving group (LG) of the allylating reagent is introduced to the allylation product, regeneration of the promoter connected to the ring-opened acetal should be unnecessary (Scheme 1, path B). It means the catalytic ring-opening allylation of the cyclic acetal can proceed. It can be expected that an interaction between the cyclic acetal and the leaving group during the allylation creates the possibility of introducing the leaving group to the allylation product.
It is well known that Si species acts as Lewis acid sites for several nucleophilic addition reactions, including allylation.4 These active Si species can be generated by the reaction between an allylsilane and a Brønsted acid.5 These facts encouraged us to use allylsilane as an allylating reagent and a Brønsted acid as an initiator of the Si species for the ring-opening allylation of cyclic acetals. Herein, we report the catalytic ring-opening allylation of cyclic acetals with allylsilanes using silica–alumina as a solid acid. The ring-opening allylation afforded silyl ethers that can be hydrolyzed to homoallyloxyalcohols in the presence of the silica–alumina (eqn (1)).6 In addition, 1,2-bis(homoallyloxy)ethane, a potentially useful compound with many functional groups, formed as a co-product during the allylation. The selectivity toward the 1,2-bis(homoallyloxy)ethane increased with an H+-montmorillonite catalyst (eqn (2)). Detailed reaction pathways of the catalytic ring-opening allylation and 1,2-bis(homoallyloxy)ethane synthesis were also investigated.
Results and discussion
Ring-opening allylation over Brønsted acids
Ring-opening allylations of 2-phenyl-1,3-dioxolane (1a) with 1.2 equivalents of allyltrimethylsilane (2a) in the presence of heterogeneous inorganic acids were carried out at 60 °C. The results are summarized in Table 1. An 81% yield of the ring-opened allylation product (3aa) could be achieved with silica–alumina (entry 1). 1,2-Bis(homoallyloxy)ethane (4aa) formed as a by-product in 7% yield. H+-beta also showed catalytic activity to afford 43% yield of 3aa (entry 2). Other zeolites used, such as H+-mordenite and H+-USY, showed much lower performances under the reaction conditions (entries 3–6). These results suggest that the higher accessibility of substrates to the active site on amorphous silica–aluminaversus other zeolites induces the higher performance of silica–alumina.
| Entry |
Acid (SiO2/Al2O3) |
Conv. of 1ab (%) |
Conv. of 2ab (%) |
Yieldb (%) |
|
3aa
c
|
4aa
d
|
|
Reaction conditions:
1a (3.7 mmol), 2a (4.4 mmol), acid (0.05 g), toluene (3.0 mL), 60 °C, Ar atmosphere, 27 h.
Determined by GC using an internal standard.
Yield based on acetal 1a.
Yield = [4aa (mmol) × 2/1a (mmol)] × 100.
0.15 g of zeolite was used.
|
| 1 |
Silica–alumina (6.3) |
95 |
94 |
81 |
7 |
| 2 |
H+-Beta (25) |
75 |
86 |
43 |
3 |
| 3e |
H+-Mordenite (18) |
42 |
56 |
8 |
<1 |
| 4e |
H+-USY (5.9) |
24 |
52 |
1 |
1 |
| 5e |
H+-ZSM-5 (23.8) |
24 |
33 |
<1 |
<1 |
| 6 |
FSM-16
|
25 |
45 |
18 |
2 |
| 7 |
Amorphous SiO2 |
4 |
37 |
<1 |
<1 |
| 8 |
Na–silica–alumina |
11 |
40 |
1 |
<1 |
An 18% yield of 3aa was obtained with mesoporous silica FSM-16 (entry 6),7 however, amorphous SiO2 did not show catalytic activity (entry 7). It can be said that the Si–OH group did not act as an active site for the allylation. To confirm the role of the Brønsted acid site of silica–alumina, the allylation using silica–alumina treated with NaOH/NaCl solution (Na–silica–alumina) was examined. The reaction afforded only 1% yield of 3aa (entry 8). These results indicate that the acid site on the silica–alumina acts as a catalytically active site for the allylation, because the amount of acid site on the silica–alumina is estimated to be 0.154 mmol g−1 by NH3-TPD analysis.
Reaction time and solvent were optimized in the silica–alumina-catalyzed allylation of 1a with 2a, as shown in Table 2. It was expected that strong electron donation from solvent molecules would decrease the activity of the acid site. To determine the effect of solvent polarity on the catalytic performance, the dielectric values ε of the solvents as a criterion of polarity are also listed. An 83% yield of 3aa was obtained after 8 h with toluene (entry 3). n-Heptane and 1,4-dioxane also acted as good solvents (entries 5 and 6), whereas acetonitrile, 2-propanol, and DMF were substantially less active (entries 7–9). These results suggest that nonpolar solvents with low dielectric constants are good media for the ring-opening allylation. The active Brønsted acid site on silica–alumina can be deactivated by the electron donating effect of highly polar solvents.8
| Entry |
Solvent [dielectric constant (ε)] |
Time/h |
Conversion of 1ab (%) |
Yieldb (%) |
|
3aa
c
|
4aa
d
|
|
Reaction conditions:
1a (3.7 mmol), 2a (4.4 mmol), silica–alumina (0.05 g), solvent (3.0 mL), 60 °C, Ar atmosphere.
Determined by GC using an internal standard.
Yield based on acetal 1a.
Yield = [4aa (mmol) × 2/1a (mmol)] × 100.
|
| 1 |
Toluene (2.4) |
1 |
40 |
38 |
4 |
| 2 |
Toluene (2.4) |
3 |
75 |
67 |
6 |
| 3 |
Toluene (2.4) |
8 |
95 |
83 |
7 |
| 4 |
Toluene (2.4) |
27 |
95 |
81 |
7 |
| 5 |
n-Heptane (1.9) |
27 |
96 |
76 |
12 |
| 6 |
1,4-Dioxane (2.2) |
27 |
90 |
76 |
13 |
| 7 |
Acetonitrile (37.5) |
27 |
15 |
3 |
1 |
| 8 |
2-Propanol (18.6) |
27 |
36 |
<1 |
<1 |
| 9 |
DMF (36.7) |
27 |
<1 |
<1 |
<1 |
Ring-opening allylation of 1a with other allylsilanes proceeded as shown in Scheme 2. The reactions with methallyltrimethylsilane, crotyltrimethylsilane, and allyltriethylsilane afforded the corresponding allylated products in 76, 52, and 50% yields, respectively.
Reaction pathways of ring-opening allylation
It is reported that reactions between the surface H+ site or Si–OH group and allylsilane give (surface)–O–SiR3 groups and propylene.5,9Scheme 3 shows possible reaction pathways for the allylation involving (surface)–O–SiR3 group formation [Scheme 3(A)]. Here, Si1 and Si2 are the SiR3 group on the silica–alumina surface and the SiR3 group in the allylsilane, respectively. As shown in Scheme 3(B), the surface Si species (Si1) might be introduced to the cyclic acetal, and then nucleophilic addition of the allyl group of the allylsilane occurs. The surface Si species (Si2) is regenerated by the Si group in the allylsilane. To confirm this reaction pathway, the following experiments were conducted. The reaction of an excess amount of allyltrimethylsilane (2a) with silica–alumina afforded propylene and SiMe3-functionalized silica–alumina (SiMe3–silica–alumina) (Scheme 4). The solid-state 29Si and 13C CP/MAS NMR spectra of the SiMe3–silica–alumina are shown in Fig. 1(A) and 2(A), respectively. Signals at around 14 ppm in the 29Si NMR spectrum and 0 ppm in the 13C NMR spectrum clearly indicate the presence of (surface)–O–SiMe3 groups on silica–alumina.10 Next, the reaction of acetal (1a) and allyltriethylsilane (2b) in the presence of the SiMe3–silica–alumina was carried out. The result is shown in Scheme 5. Formation of 1% yield of the ring-opened allylation product with a SiMe3 group was detected.11 After the reaction of 1a, 2b, and SiMe3–silica–alumina, the silica–alumina was recovered and solid-state NMR measurements were conducted [Fig. 1(B) and 2(B)]. As shown in Fig. 2(B), a new signal at around 5.4 ppm was detected in the 13C NMR spectrum. This indicates the presence of a (surface)–O–SiEt3 group on the silica–alumina after the allylation with 2b.12 These reaction and NMR results strongly support the allylation pathway involving Si exchange [Scheme 3(B)]. After the reaction with 2b, the 0 ppm signal still remained in the 13C NMR spectrum [Fig. 2(B)]. This result indicates the existence of an inactive (surface)–O–SiMe3 group on the silica–alumina. Because amorphous SiO2 had no catalytic activity (Table 1, entry 8), the inactive species might be the Si–O–SiMe3 generated from the simple Si–OH group and 2a, as shown in Scheme 6A. It is suggested that the catalytically active Si species on the silica–alumina was generated by the reaction between the Brønsted acid site due to the [Si–O(H+)–Al] moiety and 1a (Scheme 6B).
 |
| | Scheme 3 Formation of Si species on the silica–alumina surface (A) and possible reaction pathways of ring-opening allylation involving: (B) Si exchange reaction and (C) intramolecular addition of Si species via a 6-membered transition state. Si1 and Si2 are trialkylsilyl species such as SiMe3. | |
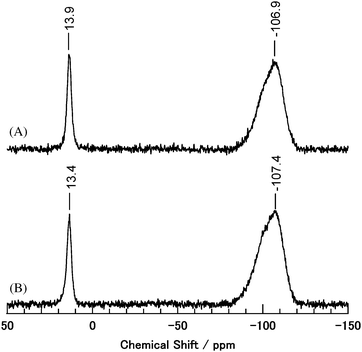 |
| | Fig. 1 Solid-state 29Si CP/MAS NMR for (A) SiMe3–silica–alumina, and (B) recovered SiMe3–silica–alumina after the reaction with 1a and 2b. The signals at around −107 ppm are assignable to bulk Si in silica–alumina. | |
 |
| | Fig. 2 Solid-state 13C CP/MAS NMR for (A) SiMe3–silica–alumina, and (B) recovered SiMe3–silica–alumina after the reaction with 1a and 2b. | |
 |
| | Scheme 6 Proposed reaction pathway involving formation of both active and inactive SiMe3 species on the silica–alumina surface. | |
If the allylation proceeds via the reaction pathway involving the Si exchange reaction [Scheme 3(B)], four types of allylation products should form by cross-allylation of 1a in the presence of both methallyltrimethylsilane (methallyl–SiMe3) and allyltriethylsilane (allyl–SiEt3). The cross-allylation was carried out, as shown in Scheme 7. According to our expectations, four types of allylation products including allyl–SiMe3 groups and methallyl–SiEt3 groups were obtained. This result supports the reaction pathway including Si transfer [Scheme 3(B)]. However, these results cannot completely exclude a concerted mechanism through a 6-membered ring transition state [Scheme 3(C)], which was reported in the allylation of carbonyl compounds using allylsilanes.4,13 The presence of both reaction pathways is possible in the ring-opening allylation using silica–alumina.
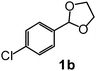
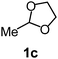
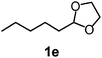
After the allylation of acetals (1) with 2a using silica–alumina, water was added to the reaction mixture. Hydrolysis of the silyl ethers (3) occurred to afford homoallyloxyalcohols (5). These results are summarized in Table 3. In the case of acetal 1a, the corresponding homoallyloxyalcohol (5aa) was obtained in 77% yield after 23 h (entry 3). The recovered silica–alumina was calcined at 500 °C, and then reused for the allylation–hydrolysis sequence. A 70% yield of 5aa was obtained after a prolonged reaction time for the allylation (entry 4). The hydrolysis reaction scarcely proceeded in the absence of the silica–alumina (entry 5). This result suggests that hydrolysis of 3 was promoted by the silica–alumina. The silica–alumina catalyst was also applicable toward other ethylene acetals, such as 2-methyl-1,3-dioxolane (1c) and simple 1,3-dioxolane (1d), affording 51–77% yields of homoallyloxyalcohols (entries 6–11). In the case of acetals with small sizes, the product selectivity in the allylation step was moderate due to the formation of the bis(homoallyloxy)ethane (4) as a by-product. Reaction of 2-phenyl-4-methyl-1,3-dioxolane (1h) afforded both primary and secondary alcohols (entry 12). 1,3-Dioxane (1i) also reacted with 2a to give the corresponding homoallyloxyalcohol (entry 13). Allylation of ethylene ketal, such as 2,2-dimethyl-1,3-dioxolane, hardly proceeded in the reaction conditions.
| Entry |
Acetal (1) |
Time/h |
Product (5) |
Yield of 5b (%) |
|
Allylation
|
Hydrolysis
|
Reaction conditions: [allylation] acetal 1 (3.7 mmol), 2a (4.4 mmol), silica–alumina (0.05 g), toluene (3.0 mL), 60 °C, Ar atmosphere. [hydrolysis] water (11 mmol), acetone (3.0 mL), 25 °C.
Determined by GC using an internal standard. Yield based on acetal 1.
Reuse experiment.
Result of hydrolysis reaction of allylation product 3aa in the absence of silica–alumina.
Silica–alumina (0.10 g) was used.
Isolate yield.
A mixture of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 diastereoisomers was obtained. Product structures and ratio were determined by 1H and 13C NMR, H–H COSY, C–H COSY, and H–H NOESY. ∶1 diastereoisomers was obtained. Product structures and ratio were determined by 1H and 13C NMR, H–H COSY, C–H COSY, and H–H NOESY.
|
| 1 |

|
8 |
3 |

|
45 |
| 2 |
1a
|
8 |
6 |
5aa
|
63 |
| 3 |
1a
|
8 |
23 |
5aa
|
77 |
| 4 |
1a
|
20 |
23 |
5aa
|
70c |
| 5 |
1a
|
— |
3 |
5aa
|
3d |
| 6 |
|
8 |
8 |

|
77 |
| 7 |
|
27 |
27 |

|
60 |
| 8 |

|
27 |
27 |

|
68 |
| 9 |
|
27 |
3 |

|
51 |
| 10 |

|
14 |
20 |

|
51ef |
| 11 |

|
14 |
20 |

|
58e,f |
| 12 |

|
20 |
6 |

|
59e,f,g |

|
14e,f.g |
| 13 |

|
22 |
6 |

|
53f |
1,2-Bis(homoallyloxy)ethane synthesis and reaction pathway
During the above ring-opening allylation of cyclic acetals with allylsilanes, we detected the formation of small amounts of 1,2-bis(homoallyloxy)ethane (4aa). The 1,2-bis(homoallyloxy)ethane structure might have potentially utility for organic synthesis and polymer chemistry because of its two terminal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and ether functions. Only one example was reported for the synthesis of 4aa from a diphenyl dioxane using an electrochemical procedure.14 Our solid acid-catalyzed approach toward 4aa has the following advantages: (i) readily available cyclic acetals can be used as substrates, (ii) various functional groups can be introduced by choosing appropriate acetal structures. These facts encouraged us to investigate efficient procedures for 1,2-bis(homoallyloxy)ethane synthesis.
C bonds and ether functions. Only one example was reported for the synthesis of 4aa from a diphenyl dioxane using an electrochemical procedure.14 Our solid acid-catalyzed approach toward 4aa has the following advantages: (i) readily available cyclic acetals can be used as substrates, (ii) various functional groups can be introduced by choosing appropriate acetal structures. These facts encouraged us to investigate efficient procedures for 1,2-bis(homoallyloxy)ethane synthesis.
Because of the larger size of 4aa than 3aa, homogeneous acids and heterogeneous acids, which can be used for transformation of large substrates,15 are thought to be effective to increase the yield of 4aa. Among these acid catalysts, montmorillonite was reported by Kaneda and co-workers as a highly efficient heterogeneous acid catalyst for reactions of large molecules, such as BHEPF, 1,3-diphenyl-2-propanone, and steroid.15a–c These catalysts were employed in the reaction of 1a with 2a. The results are summarized in Table 4. The reaction using H+-montmorillonite as a solid acid afforded 4aa in up to 60% yield (entry 2). Nafion and Amberlyst also showed higher selectivity toward 4aa than 3aa (entries 3 and 4), however, the yield of 4aa was lower than with the H+-montmorillonite. A 35% yield of 4aa was obtained with homogeneous trifluoromethane sulfonic acid (CF3SO3H) (entry 6). The reaction hardly proceeded with other homogeneous acids used, such as p-toluenesulfonic acid and H2SO4.
| Entry |
Acid |
t/h |
Conversion of 1ab (%) |
Yieldb (%) |
|
3aa
c
|
4aa
d
|
|
Reaction conditions:
1a (3.7 mmol), 2a (4.4 mmol), acid (0.05 g), toluene (3.0 mL), 60 °C, Ar atmosphere.
Determined by GC using an internal standard.
Yield based on acetal 1a.
Yield = [4aa (mmol) × 2/1a (mmol)] × 100.
The reaction of 1a (3.7 mmol) and 2a (3.7 mmol) was carried out for 8 h. Then, 2a (0.7 mmol) was added, and the reaction was conducted for an additional 2 h.
CF3SO3H (0.18 mmol).
|
| 1 |
H+-Montmorillonite |
4 |
>95 |
19 |
56 |
| 2e |
H+-Montmorillonite |
10 |
>99 |
23 |
60 |
| 3 |
Nafion
|
28 |
90 |
31 |
48 |
| 4 |
Amberlyst |
24 |
64 |
25 |
42 |
| 5 |
SO4/ZrO2 |
6 |
41 |
32 |
17 |
| 6 |
CF3SO4H
f |
1 |
79 |
8 |
35 |
1,2-Bis(homoallyloxy)ethane synthesis from 2-methyl-1,3-dioxolane (1c) and 4-methyl-2-phenyl-1,3-dioxolane using H+-montmorillonite were carried out, as shown in Scheme 8. A 54% yield of the corresponding product formed from 2-methyl-1,3-dioxolane (1c). In the reaction of the 4-methyl-2-phenyl-1,3-dioxolane, the presence of a methyl group in the product clearly indicates that the 1,2-bis(homoallyloxy)ethane was constructed with one protecting group of the acetal.
 |
| | Scheme 8 Reaction of 2-methyl-1,3-dioxolane (1c) and 4-methyl-2-phenyl-1,3-dioxolane with allyltrimethylsilane (2a) using H+-montmorillonite. | |
To reveal the reaction pathway for the 1,2-bis(homoallyloxy)ethane formation, silyl ether (3aa) was treated with H+-montmorillonite at 60 °C. Only 4% yield of 4aa was obtained (Scheme 9). This result indicates that the dimerization of 3aa is not a main pathway toward 4aa. Next, the conversion of 3aa using H+-montmorillonite in the presence of 1c and 2a was performed, as shown in Scheme 10. 1,2-Bis(homoallyloxy)ethane having both phenyl and methyl groups (4ca) was obtained in 38% yield along with 1% yield of 4aa. From this result, the main reaction pathway toward 1,2-bis(homoallyloxy)ethane from acetal and allylsilane is proposed as follows: (i) acetal exchange reaction16 of a silyl ether 3 with an acetal 1, (ii) nucleophilic addition of allylsilane 2 to the acetal intermediate, (iii) formation of 1,2-bis(homoallyloxy)ethane 4 along with bis(trimethylsiloxy)ethane (Scheme 11). The formation of bis(trimethylsiloxy)ethane during the H+-montmorillonite-catalyzed reaction supports this reaction pathway.
 |
| | Scheme 9 Reaction of 3aa using H+-montmorillonite. | |
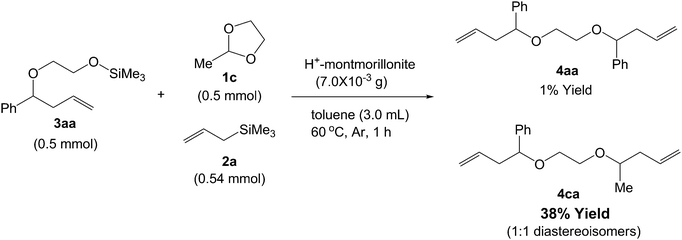 |
| | Scheme 10 Reaction of ring-opening allylation product 3aa in the presence of acetal 1c and allyltrimethylsilane 2a using H+-montmorillonite. | |
 |
| | Scheme 11 Proposed reaction pathway for 4. | |
Conclusion
We have found that silica–alumina acts as an efficient heterogeneous catalyst for the ring-opening allylation of cyclic acetals with allylsilanes. The corresponding homoallyloxyalcohols formed after silica–alumina-catalyzed allylation/hydrolysis sequences. An active (surface)–O–SiR3 group was generated on silica–alumina by the reaction between the Brønsted acid site [Si–O(H+)–Al] and the allylsilane. The catalytic allylation pathway involving the formation of a (surface)–O–SiR3 group on silica–alumina followed by Si transfer has been proposed for the present ring-opening allylation. In addition, good yields of 1,2-bis(homoallyloxy)ethanes could be obtained from cyclic acetals and allylsilane using H+-montmorillonite instead of silica–alumina.
Experimental section
Proton (1H) and carbon (13C) nuclear magnetic resonance (NMR) spectra were recorded in CDCl3 with a JNM-AL300 spectrometer operating at 300 and 75 MHz, respectively. Analytical GLC was measured using a Shimadzu GC-8A equipped with a Silicon SE-30 and OV-17 column and a thermal conductivity (TCD) detector. A Shimadzu QP5000 equipped with DB-1 column was used as GC-MS. The products were confirmed by comparison with reported NMR and MS data. Solid-state 13C and 29Si CP/MAS NMR spectra (MAS rate = 5 kHz) were recorded with an AVANCE III spectrometer operating at 100.6 and 79.5 MHz, respectively. Cross-polarization (CP) contact times of 13C and 29Si CP/MAS NMR measurements were 0.5 and 10.0 ms, respectively. The accumulation number and delay time were about 100000 and 1.0 s (13C), and 2500 and 4.0 s (29Si), respectively. Adamantane (δ 38.52 and 29.47 ppm) and hexamethylcyclotrisiloxane (δ −9.66 ppm) were used as external standards for the calibration of chemical shifts.
Silica–alumina was available from Nikki Chemical Co. as N633L (SiO2, 80.6, Al2O3, 12.8 wt%, 408 m2 g−1). H+-montmorillonite was prepared from Na+-montmorillonite (Knipia F) using an ion exchange procedure with aqueous hydrogen chloride.17H+-beta (SiO2/Al2O3 = 25), H+-mordenite (SiO2/Al2O3 = 18.0), and H+-USY (SiO2/Al2O3 = 5.9) were purchased from Nikki Chemical Co. H+-ZSM-5 (SiO2/Al2O3 = 23.8) was obtained from Toso Co. FSM-16 was obtained from Fuji Silysia Chemical Co. Aerosil 300 was used as amorphous SiO2. SO4/ZrO2 was purchased from Japan Energy Co. (S content: 3.2 wt%). These inorganic solid acids, except for H+-montmorillonite, were treated at 500 °C under air and 120 °C under vacuum before use. Amberlyst was purchased from Organo Co. as Amberlyst® 15DRY. Nafion was purchased from Aldrich Inc. as Nafion® NR50. Na–silica–alumina (Na content: 4.7 wt%) was prepared from silica–alumina using an ion exchange procedure with 1 M NaCl solution at 80 °C for 3 h. The pH of the solution was adjusted to 9 by addition of aqueous NaOH solution. After the treatment, the solid was filtered, washed with deionized water, then dried at 110 °C. Unless otherwise noted, other materials were purchased from Wako Pure Chemicals, Tokyo Kasei Co., Kanto Kagaku Co. and Aldrich Inc.
Into a glass reactor were placed silica–alumina (0.05 g), toluene (3.0 mL), 1a (3.7 mmol), and 2a (4.4 mmol) under a dry Ar atmosphere. The resulting mixture was vigorously stirred at 60 °C. After 27 h, the catalyst was separated by filtration and GC analysis of the filtrate showed an 81% yield of the allylated product 3aa and 7% yield of 4aa. The filtrate was evaporated and the crude product was purified by column chromatography using silica (n-hexane/ether = 9/1) to afford pure products 3aa and 4aa.
After the ring-opening allylation using silica–alumina, water (0.2 mL, 11 mmol) and acetone (3.0 mL) were added to the same flask. The resulting mixture was vigorously stirred at 25 °C for 3 h. The catalyst was separated by filtration and GC analysis of the filtrate showed a 77% yield of homoallyloxyalcohol 5aa. The filtrate was evaporated and the crude product was purified by column chromatography using silica (n-hexane/ether = 9/1 to 1/1) to afford pure product 5aa.
Acknowledgements
This work was supported by a Grant-in-Aid for Young Scientists (B) (Grant No. 21760626) and The Noguchi Institute (NJ200909). This study was also supported by the Cooperative Research Program of Catalysis Research Center (CRC), Hokkaido University. (Grant No. 10B2006). K.M. thanks Prof. Kenji Hara (CRC, Hokkaido University) and Prof. Atsushi Fukuoka (CRC, Hokkaido University). The authors thank Center for Advanced Materials Analysis (Suzukakedai), Technical Department, Tokyo Institute of Technology, for NMR and elemental analysis.
References
-
Allylation of acyclic acetals:
(a) A. Hosomi, M. Endo and H. Sakurai, Chem. Lett., 1976, 941 CAS;
(b) A. Hosomi, M. Endo and H. Sakurai, Chem. Lett., 1978, 499 CrossRef CAS;
(c) H. Sakurai, K. Sasaki, J. Hayashi and A. Hosomi, J. Org. Chem., 1984, 49, 2808 CrossRef CAS;
(d) T. Mukaiyama, H. Nagaoka, M. Murakami and M. Ohshima, Chem. Lett., 1985, 977 CAS;
(e) M. Kawai, M. Onaka and Y. Izumi, Chem. Lett., 1986, 381 CrossRef CAS;
(f) N. Komatsu, M. Uda, H. Suzuki, T. Toshikazu, T. Domae and M. Wada, Tetrahedron Lett., 1997, 38, 7215 CrossRef CAS;
(g) L. C. Wieland, H. M. Zerth and R. S. Mohan, Tetrahedron Lett., 2002, 43, 4597 CrossRef CAS;
(h) H. M. Zerth, N. M. Leonard and R. S. Mohan, Org. Lett., 2003, 5, 55 CrossRef CAS.
-
Allylation of cyclic acetals:
(a) P. A. Bartlett, W. S. Johnson and J. D. Elliott, J. Am. Chem. Soc., 1983, 105, 2088 CrossRef CAS;
(b) W. S. Johnson, P. H. Crackett, J. D. Elliott, J. J. Jagodzinski, S. D. Lindell and S. Natarajan, Tetrahedron Lett., 1984, 25, 3951 CrossRef CAS;
(c) Y. Yamamoto, S. Nishii and J.-I. Yamada, J. Am. Chem. Soc., 1986, 108, 7116 CrossRef CAS;
(d) S. E. Denmark and N. G. Almstead, J. Am. Chem. Soc., 1991, 113, 8089 CrossRef CAS;
(e) S. E. Denmark and N. G. Almstead, J. Org. Chem., 1991, 56, 6458 CrossRef CAS;
(f) T. Sammakia and R. S. Smith, J. Am. Chem. Soc., 1992, 114, 10998 CrossRef;
(g) T. Sammakia and R. S. Smith, J. Org. Chem., 1992, 57, 2997 CrossRef;
(h) S. Jiang, G. E. Agoston, T. Chen, M.-P. Cabal and E. Turos, Organometallics, 1995, 14, 4697 CrossRef CAS;
(i) Y. Egami, M. Takayanagi, K. Tanino and I. Kuwajima, Heterocycles, 2000, 52, 583 CrossRef CAS;
(j) S. Bogaczyk, M.-R. Brescia, Y. C. Shimshock and P. DeShong, J. Org. Chem., 2001, 66, 4352 CrossRef CAS;
(k) F. Carrel, S. Giraud, O. Spertini and P. Vogel, Helv. Chim. Acta, 2004, 87, 1048 CrossRef CAS;
(l) G. K. Friestad and H. J. Lee, Org. Lett., 2009, 11, 3958 CrossRef CAS.
- M. J. Spafford, J. E. Christensen, M. G. Huddle, J. R. Lacey and R. S. Mohan, Aust. J. Chem., 2008, 61, 419 CrossRef CAS.
-
(a) Y. Yamamoto and N. Asao, Chem. Rev., 1993, 93, 2207 CrossRef CAS;
(b) J. Beignet, P. J. Jervis and L. R. Cox, J. Org. Chem., 2008, 73, 5462 CrossRef CAS.
-
(a) A. P. Davis and M. Jaspars, J. Chem. Soc., Chem. Commun., 1990, 1176 RSC;
(b) K. Higuchi, M. Onaka and Y. Izumi, Bull. Chem. Soc. Jpn., 1993, 66, 2016 CAS.
- K. Motokura, H. Yoneda, A. Miyaji and T. Baba, Green Chem., 2010, 12, 1373 RSC.
- Effects of mesoporous structures on acid-catalyzed reactions have been reported by Iwamoto and co-workers:
(a) M. Iwamoto, Y. Tanaka, N. Sawamura and S. Namba, J. Am. Chem. Soc., 2003, 125, 13032 CrossRef CAS;
(b) H. Ishitani, H. Naito and M. Iwamoto, Catal. Lett., 2008, 120, 14 CrossRef CAS.
- K. Motokura, M. Tomita, M. Tada and Y. Iwasawa, Chem.–Eur. J., 2008, 14, 4017 CrossRef CAS.
-
(a) T. Shimada, K. Aoki, Y. Shinoda, T. Nakamura, N. Tokunaga, S. Inagaki and T. Hayashi, J. Am. Chem. Soc., 2003, 125, 4688 CrossRef CAS;
(b) K. Aoki, T. Shimada and T. Hayashi, Tetrahedron: Asymmetry, 2004, 15, 1771 CrossRef CAS.
- D. Derouet and C. N. Ha Thuc, J. Appl. Polym. Sci., 2008, 109, 2113 CrossRef CAS.
- The amount of acid sites in 0.20 g silica–alumina is estimated to be 3.1 × 10−2 mmol from NH3-TPD results. This value is larger than that for the SiMe3-containing allylation product (4.0 × 10−3 mmol). The amount of active H+ sites for the allylation might be lower than the acid sites determined by NH3-TPD.
- The 13C NMR chemical shifts of Et3Si–OH are δ 5.81 and 6.60 ppm (CDCl3), see: T. Tokuyasu, S. Kunikawa, A. Masuyama and M. Nojima, Org. Lett., 2002, 4, 3595 Search PubMed.
- A. Hosomi, S. Kohara, K. Ogata, T. Yanagi and Y. Tominaga, J. Org. Chem., 1990, 55, 2415 CrossRef CAS.
- M. Okajima, S. Suga, K. Itami and J.-I. Yoshida, J. Am. Chem. Soc., 2005, 127, 6930 CrossRef CAS.
- Montmorillonite-catalyzed transformations of large molecules:
(a) K. Ebitani, T. Kawabata, K. Nagashima, T. Mizugaki and K. Kaneda, Green Chem., 2000, 2, 157 RSC;
(b) T. Kawabata, T. Mizugaki, K. Ebitani and K. Kaneda, Tetrahedron Lett., 2001, 42, 8329 CrossRef CAS;
(c) T. Kawabata, M. Kato, T. Mizugaki, K. Ebitani and K. Kaneda, Chem. Lett., 2003, 32, 648 CrossRef CAS. Other catalysts:
(d) A. Kumar, M. Dixit, S. P. Singh, R. Raghunandan, P. R. Maulik and A. Goel, Tetrahedron Lett., 2009, 50, 4335 CrossRef CAS;
(e) S. G. Rha and S.-K. Chang, J. Org. Chem., 1998, 63, 2357 CrossRef CAS.
-
Acetal exchange reactions between cyclic acetals and silyl ethers have been reported: T. Suzuki and T. Oriyama, Synth. Commun., 1999, 29, 1263 Search PubMed.
- K. Motokura, N. Fujita, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, Angew. Chem., Int. Ed., 2006, 45, 2605 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cy00040c |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here. 







