Synergistic effects between copper and tungsten on the structural and acidic properties of CuOx/WOx–ZrO2 catalyst
Received
15th December 2010
, Accepted 1st February 2011
First published on 11th March 2011
Abstract
Different amounts of tungsten oxide were introduced to zirconia to form WOx–ZrO2 solid solutions by a coprecipitation method, followed by impregnation with copper nitrate. The received catalysts were characterized by X-ray diffraction, Brunauer–Emmett–Teller surface area, X-ray photoelectron spectroscopy, Raman spectroscopy, diffuse reflectance UV-vis spectroscopy, H2 temperature-programmed reduction and in situFTIR of NH3/NO+O2 adsorption. Higher dispersions of CuOx are obtained on the tungsten-containing catalysts due to the increased surface area of the support. Highly dispersed WOx clusters in tetrahedral coordination are generated when tungsten exceeds its solubility in the solid solutions, and polymerized WOx clusters are formed for further increasing the loading amount of tungsten. The formation of Cun+–O2−–Wn+ bonds at the interface between CuOx and WOx clusters leads to an improved capacity to form nitrite and an enhanced Brønsted acidity, which enhance the activity of the catalyst for NH3-SCR reaction.
1. Introduction
Interactions between metal oxides and oxide supports have attracted much attention because of the wide application of supported metal oxide systems. WO3–TiO2 mixed oxides, which act as the support of commercial V2O5–WO3–TiO2 catalyst, are known to enhance the acidity of the catalyst and improve the dispersion of vanadium oxides.1 Furthermore, the electronic interactions between V2O5 and WO3–TiO2 support lead to enhanced redox properties of V2O5 species in the catalyst, which are proved to be responsible for the higher deNOx activity in ternary V2O5–WO3–TiO2 catalyst than in binary V2O5–TiO2 and WO3–TiO2 catalysts at low temperatures.2,3
Copper based catalysts are considered as promising candidates for deNOx catalysts due to their non-toxicity and affordability. Copper oxides supported on zeolite,4,5zirconia,6–9alumina,10titanium11,12 and ceria,13 which show good low-temperature SCR activities with ammonia/CO/hydrocarbon as reductants, have received much attention. According to the studies reported,4–13 the activity and selectivity of copper oxide based catalysts are determined to a great extent by the texture and dispersion of CuOx, which are significantly influenced by supports. However, few papers have demonstrated the effects of strong interactions between copper and support components on the SCR reaction performance. For example, a possible explanation for the high activity of copper exchanged zeolite is the capability of the zeolite to disperse active metals at the atomic level.4,5 Komova et al.11 reported that the interaction between Cu2+ ions and TiO2 support led to chain structure of CuO on the supports, which acted as more active copper sites for NH3–SCR. Chen et al.13 found that the formation of CuOx–CeO2 solid solutions resulted in highly dispersed CuOx and thereby promoted the CO-SCR activity of the catalyst. In the present study, the synergistic effects between copper oxide and WOx–ZrO2 support on the adsorption behavior and the redox properties of catalysts may help to further interpret the possible mechanisms.
The adsorption of the reactants (NO and NH3) on copper based catalysts has been widely studied. It has been noted that copper oxides supported on zirconia are more active than those on titanium, silica and alumina due to the faster NH3 dissociative chemisorption on the former catalyst.6 Arising from the inhibition effect on ammonia oxidation, sulfates have been adopted to improve the acidity of CuO/ZrO2 catalysts for NO reduction with ammonia.7,8 Unfortunately, the maximal NO conversion rate temperature for CuO/ZrO2 catalysts shifts to higher temperatures due to the lowered reducibility of Cu2+ caused by sulfate modification. WO3 is added to zirconia for isomerization and alkylation of hydrocarbons due to the large surface area and high acidity of WO3–ZrO2 solid solutions.14,15 Therefore, an improvement on NO and NH3 adsorption properties of CuOx/ZrO2 catalysts can be expected by introducing tungsten into zirconia. To the best of our knowledge, little is known about the effects of interaction between copper and tungsten on zirconia, which plays an important role in improving dispersion of copper and enhancing NO/NH3 adsorptive properties of catalysts.
In our previous report,16 the CuOx/WOx–ZrO2 catalysts achieved high NH3-SCR activity and near 100% N2 selectivity at 200–300 °C, and the loading amount of CuO was optimized at 10 wt%. However, the structural and electronic interactions between copper and tungsten species were not clearly presented. In the present work, the tungsten content in the catalyst was adjusted to investigate the interaction between copper and tungsten at a fixed copper content, to obtain a more clear reaction mechanism for the NH3-SCR reaction with this catalyst.
2. Experimental
The WOx–ZrO2 solid solutions were synthesized by a coprecipitation method. ZrO(NO3)2·5H2O (99%, Rongruida), 5(NH4)2O·12WO3·5H2O (99%, Juneng) and polyethylene glycol (PEG) (99%, Juneng) were dissolved in deionized water at a mass ratio of H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :(WO3+ZrO2):PEG = 100:1:1. Excess ammonia was added to keep the pH value of the mixed solution at 10. The mixed solution was magnetically stirred at 100 °C for 4 h. The received precipitates were dried at 110 °C overnight and calcined at 500 °C for 3 h in static air in a muffle. A similar method was used for synthesis of ZrO2 support.
:(WO3+ZrO2):PEG = 100:1:1. Excess ammonia was added to keep the pH value of the mixed solution at 10. The mixed solution was magnetically stirred at 100 °C for 4 h. The received precipitates were dried at 110 °C overnight and calcined at 500 °C for 3 h in static air in a muffle. A similar method was used for synthesis of ZrO2 support.
The copper loaded catalysts were prepared through a wet incipient impregnation method. The water absorption capacities of ZrO2 and WOx–ZrO2 powders were measured. Then, the required amount of Cu(NO3)2 solution (10 mol L−1) was mixed with these powders. The mixture was dried at 100 °C for 4 h and calcined at 500 °C for 1 h in static air in a muffle. The nominal compositions of the catalysts are listed in Table 1.
Table 1 Nominal compositions of the catalysts
| Sample |
CuO (wt%) |
WO3 (wt%) |
ZrO2 (wt%) |
|
C10Z
|
10 |
— |
90 |
|
C10W5Z
|
10 |
5 |
85 |
|
C10W10Z
|
10 |
10 |
80 |
|
C10W20Z
|
10 |
20 |
70 |
2.2 Characterization
The X-ray diffraction (XRD) experiments were determined on a Japan science D/mas-RB diffractometer employing Cu-Kα radiation (λ = 0.15418 nm). The X-ray tube was operated at 40 kV and 120 mA. The XRD spectra were recorded at 0.02° intervals in range of 20° < 2θ < 80°. The identification of the phases was made with the help of JCPDS cards (Joint Committee on Powder Diffraction Standards). The mean crystallite sizes of tetragonal ZrO2 and CuO were calculated by the Scherrer equation.
N2 adsorption isotherms at −196 °C by Brunauer–Emmett–Teller (BET) method for the determination of specific surface areas were carried out on Quantachrome NOVA instrument. The samples were evacuated at 200 °C for 2 h before measurements.
The X-ray photoelectron spectroscopy (XPS) experiments were carried out on a PHI-Quantera SXM system equipped with a monochromatic Al Kα X-rays under UHV (6.7 × 10−8 Pa). Sample charging during the measurement was compensated by an electron flood gun. The electron takeoff angle was 45° with respect to the sample surface. The XPS data from the regions related to the Cu 2p and W 4f core levels were recorded for each sample. The binding energy (BE) was calibrated internally by the carbon deposit C 1s BE at 284.8 eV.
The Raman spectra were obtained with a Renishaw RM2000 Confocal Raman spectrometer at room temperature (RT) and atmospheric pressure. 514.5 nm was used as the exciting source from an argon ion laser. The laser beam was focused onto an area 0.1 × 0.1 mm2 in size of the sample surface. The wavenumber values of the Raman spectra are accurate to 1 cm−1.
Diffuse reflectance UV-vis spectroscopy (DR UV-vis) was measured by using a Hitachi U-3010 UV-vis spectrophotometer equipped with an integrating sphere. A BaSO4 pellet was used as a reference. The spectra were recorded at RT in the spectral range 200–800 nm.
H2 temperature-programmed reduction (H2-TPR) was performed in a fixed-bed reactor with the effluent gases monitored using a quadrupole mass spectrometer (MS) (Omnistar 200). Prior to the H2-TPR experiment, 50 mg sample was treated with O2 (2 vol%)/He with a total flow rate of 50 ml min−1 at 500 °C for 30 min, then cooled down to RT in the same atmosphere, and subsequently flushed by He (50 ml min−1) for 30 min to remove the physically adsorbed molecules. Finally, the reactor temperature was raised to 900 °C at a constant heating rate of 10 °C min−1 in H2 (5 vol%)/He with a flow rate of 50 ml min−1. H2 consumption during the experiment was monitored by MS.
Using a thermo Nicolet 6700 Fourier Transform Infrared (FTIR) spectrometer equipped with a high-temperature environmental cell fitted with KBr window, In situFTIR spectra of adsorbed species, which arise from NH3/NO+O2 adsorption, were recorded in the range of 4000–650 cm−1. Prior to the adsorption, the sample was placed in a crucible located in a high-temperature cell and heated up to 500 °C in a 20% (v/v) O2/N2 flow mixture with a total flow of 100 ml for 30 min to remove traces of organic residues. After that, the sample was cooled down to 180 °C and was flushed by 100 ml min−1 N2 for 30 min to remove the physisorbed molecules for background collection. Then, a gas mixture containing 1000 ppm NH3 in N2 with a total flow rate of 100 ml min−1 passed through the sample for 60 min, and the FTIR spectra of ammonia adsorbed species were collected at the same time. After purging the weakly adsorbed or gaseous NH3 molecules by N2 flow for 30 min, a gas mixture containing 1000 ppm NO in N2 with a total flow rate of 100 ml min−1 passed through the sample, and the FTIR spectra of catalysts were collected simultaneously.
The catalytic activity measurement for reduction of NO by ammonia (NH3-SCR) with excess oxygen was carried out in a fixed bed reactor made of stainless steel with 0.5 g catalysts (diluted to 2 ml by silica) inside. The reaction gas mixture simulating diesel engine exhaust gases consisted of 500 ppm NO, 500 ppm NH3, 5% O2, and N2 in balance. The NOx conversion was measured from room temperature to 500 °C at a heating rate of 10 °C min−1. The total flow of the gas mixture was 1 L min−1 at a gas hourly space velocity (GHSV) of 30000 h−1. The concentrations of nitrogen oxides and ammonia were measured at 120 °C by a Thermo Nicolet 380 FTIR spectrometer equipped with 2 m path-length sample cell (250 ml volume). The gas path from the reactor to FTIR spectrometer was maintained a constant temperature at 120 °C to avoid NH4NO2/NH4NO3 deposition. The NOx conversions were calculated as follows:
| |  | (1) |
3. Results
3.1
XRD and BET
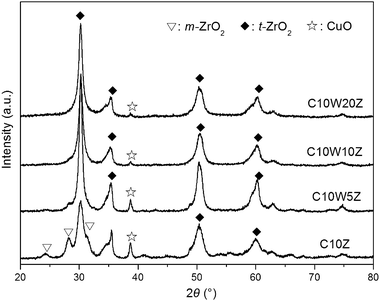
The XRD patterns of the samples are shown in Fig. 1. C10Z presents a two-phase mixture of tetragonal (t-ZrO2) and monoclinic zirconia (m-ZrO2), while only the characteristic peaks of t-ZrO2 are found on the tungsten-containing catalysts. No diffraction peaks of tungsten oxides are observed. These results are consistent with our previous works and other reports that the phase transformation from t- to m-ZrO2 is inhibited by the formation of WOx–ZrO2 solid solutions.16–18 The peak at 2θ = 38°, corresponding to CuO (111), decreases in intensity with the increase of the tungsten content, suggesting the high dispersion of copper oxides on the high-tungsten-content WOx–ZrO2 supports. The possible presence of low-valent copper and tungsten species will be validated by XPS in section 3.2.
The BET surface areas and structural properties of catalysts are summarized in Table 2. The smaller crystallite sizes of t-ZrO2 and higher BET surface area of the high-tungsten-content samples suggest the inhibition effect of tungsten modification on the sintering of zirconia-based crystallites. This effect is limited when the tungsten content exceeds 10 wt%.
Table 2 Textural and structural properties of the catalysts
| Samples |
BET surface area (m2 g−1) |
Crystallite size of t-ZrO2 (nm) |
|
C10Z
|
19 |
14.4 |
|
C10W5Z
|
37 |
11.0 |
|
C10W10Z
|
70 |
8.6 |
|
C10W20Z
|
85 |
8.0 |
3.2
XPS studies
The XPS results of catalysts are shown in Fig. 2. The fittings of Cu 2p peaks were performed by two components with the B.E. at 933.9 and 932.9 eV corresponding to the surface Cu2+ and Cu+/Cu0 species, respectively.19 The W 4f doublets at 37.9 ± 0.2 and 35.8 ± 0.2 eV are attributed to W6+ atoms, and the lower ones at 37.1 ± 0.2 and 35.1 ± 0.2 eV correspond to W5+ atoms.20 The relative percentages of different Cu/W species were calculated by the area ratios of the corresponding characteristic peaks and the results are listed in Table 3. More Cu2+ cations are reduced to Cu+/Cu0 on the WOx–ZrO2 supports compared with C10Z, while the ratio of W6+/W5+ increases with the tungsten content. This opposite trend implies a possible electronic interaction occurring between these two elements.
Table 3
Oxidation states of surface Cu and W species derived from XPS results
| Samples |
Cu2+/(Cu++Cu0) |
W6+/W5+ |
|
Cu10ZrO2 |
6.6 |
— |
|
Cu10W5ZrO2 |
2.3 |
0.8 |
|
Cu10W10ZrO2 |
1.3 |
1.0 |
|
Cu10W20ZrO2 |
1.4 |
1.1 |
It has been shown that no crystalline WOx is detected by XRD. However, the possibility of the presence of WOx clusters cannot be excluded. Raman spectroscopy is very sensitive to the microstructure of tungsten oxides.18,19 Thus, the Raman spectra of the catalysts were determined and the results are shown in Fig. 3. The Raman spectra of C10W10Z and C10W20Z exhibit a distinct band at 933–935 cm−1. This band is attributed to the stretching vibration of the W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O mono-oxo species, indicating the presence of highly dispersed WOx clusters in tetrahedrally coordinated state on the surface of these two catalysts.18,20 The broad band in the region 980–800 cm−1 on C10W20Z with a shoulder at 850 cm−1 suggests that the multi-states of tungsten species containing highly dispersed WOx clusters and polymerized WOx are obtained. The band at 850 cm−1 assigned to polymerized WOx is not obvious for the other catalysts. The band at 311–323 cm−1 is ascribed to W–O–W shifts towards lower frequencies with increasing tungsten content, which is also related to the polymerization of WOx clusters.
O mono-oxo species, indicating the presence of highly dispersed WOx clusters in tetrahedrally coordinated state on the surface of these two catalysts.18,20 The broad band in the region 980–800 cm−1 on C10W20Z with a shoulder at 850 cm−1 suggests that the multi-states of tungsten species containing highly dispersed WOx clusters and polymerized WOx are obtained. The band at 850 cm−1 assigned to polymerized WOx is not obvious for the other catalysts. The band at 311–323 cm−1 is ascribed to W–O–W shifts towards lower frequencies with increasing tungsten content, which is also related to the polymerization of WOx clusters.
Characteristic bands for both t-ZrO2 (640 cm−1) and m-ZrO2 (470 and 190 cm−1) are found in the spectrum of C10Z,18,20 while the tungsten-containing samples only present typical bands assigned to t-ZrO2 (635–670 and 446–473 cm−1). This finding agrees well with the XRD results.
It has been reported that bulk CuO loaded on Al2O3 presents three bands at 290, 340 and 628 cm−1.21 Although the identification of these bands are disturbed to a great extent by bands of zirconia, the bands assigned to bulk CuO (290 and 628 cm−1) appear to be more intensive on C10Z than on the tungsten-containing catalysts, indicating that the dispersion of CuOx is improved by modification of the zirconia support with tungsten.
Compared with Raman and XRD techniques, DR UV-vis is a more useful technique to characterize different states of copper species. Furthermore, this technique can give some meaningful information of electronic interaction between nanosized oxide clusters. DR UV-vis spectra of the catalysts are shown in Fig. 4. All the catalysts present an absorption band at 250–350 nm attributed to the Cu2+ ← O2− ligand–metal charge transfer between surface oxygen and isolated Cu2+ ions.21–27 Again, the broader bands in this region for C10W10Z and C10W20Z indicate higher dispersion of CuOx clusters on the high-tungsten-content catalysts.20,21
Another typical absorption band appears in the region of 600–900 nm corresponding to the 2Eg → 2T2g spin-allowed transition of the Cu2+ ion in the octahedral symmetry.5,21–27 The absorption band shifts from 645 to 750 nm with the increase of the tungsten content, suggesting the transformation of copper oxide from bulk crystallites to highly dispersed clusters on the high-tungsten-content catalysts.5 According to the literature,5,23 the tetragonal distortion of the octahedral Oh configuration may lead to a shift of the Cu2+d-d transition absorption band towards lower wavelengths. The appearance of a similar phenomenon in this work demonstrates that the coordinating state of Cu2+ is significantly influenced by tungsten, which will be discussed later. Both highly dispersed CuOx clusters and bulk WO3 present absorption bands between 350–450 nm.21–29 However, C10W10Z and C10W20Z show absorption bands at 350–550 nm with absorption edge energy at ca. 2.3 eV, which is obviously lower than that of C10Z and C10W5Z (ca. 3.0 eV). These results suggest electronic interaction between highly dispersed copper and tungsten oxide clusters.
3.5
H2-TPR
The H2-TPR method is a useful technique to characterize different types of copper species and to determine the redox properties of catalysts. The H2-TPR profiles and the features of TPR peaks are given in Fig. 5 and Table 4, respectively. All the catalysts present distinct reduction peaks below 200 °C (α peak) ascribed to highly dispersed CuOx clusters or Cu2+ ions with an octahedral environment.30–38 The TPR curves for the tungsten-containing catalysts exhibit a bimodal shape of two overlapped peaks at low temperatures, α1 and α2 peaks, which are ascribed to the hydrogen uptake of nested oxygen ions related to highly dispersed CuOx, respectively.30–33 The H2 consumption corresponding to all peaks are listed in Table 4. It shows that more interfacial oxygens (nested oxygen) are generated on the copper loaded on high-tungsten-content WOx–ZrO2 support. The α1 peak temperature shifts towards low temperatures with an increase in the tungsten content, indicating that the reduction of interface oxygen is influenced by the interaction between copper and the support. The peak at 230–270 °C (β peak) is attributed to the reduction of polymerized CuOx clusters.34–36 The β peak area decreases with increasing tungsten content, indicating a decreased degree of polymerization of CuOx clusters. The γ peak on C10Z corresponds to bulk CuO on zirconia support.37,38 The TPR peaks for pure tungsten oxides and zirconia are all located at temperatures higher than 300 °C (not shown).
|
Catalysts
|
Peak T/°C |
H2 consumption (μmol g−1) |
| α1 |
α2 |
β |
γ |
α1 |
α2 |
β |
γ |
|
C10Z
|
— |
176 |
— |
356 |
— |
20.0 |
— |
4.0 |
|
C10W5Z
|
155 |
169 |
227 |
— |
6.8 |
10.2 |
7.0 |
— |
|
C10W10Z
|
152 |
173 |
238 |
— |
8.2 |
8.1 |
6.1 |
— |
|
C10W20Z
|
149 |
172 |
238 |
— |
12.7 |
9.1 |
2.0 |
— |
3.6
In situ
FTIR
Fig. 6 shows the in situFTIR spectra of the adsorbed species on C10W10Z from contact with NH3 and then purging by NO+O2 at 180 °C. From Fig. 6(a), bands at 3400–3100, 1611 and 1216 cm−1, correspond to NH3 coordinated to Lewis acid sites.39 Bands at 1724 and 1483 cm−1 are attributed to ammonia coordinated to Brønsted acid sites. Two strong negative bands at 3643 and 997 cm−1 are observed with an increase in ammonia adsorption time, which arise from the formation of ammonium ions from the contact of ammonia and hydroxyls (Brønsted acid sites) and the adsorption of ammonia on WO groups (Lewis acid sites), respectively. The coordinatively unsaturated Wn+ sites may accept the electron pairs of ammonia, leading to a weakened interaction between zirconia and Wn+ sites and a shift of bands for WO to lower wavenumbers. Therefore, the corresponding positive band is observed at 954 cm−1. However, the weak intensity of this band with respect to the strong negative band at 997 cm−1 indicates that some reactions occur between WO and ammonia, which may be one of the incentives for the generation of Brønsted acid sites on catalysts (eqn (2)). No species derived from ammonia oxidation are observed at 180 °C. When the ammonia adsorbed samples were purged by NO + O2 (Fig. 6(b)), the bands of NH3 and NH4+ decreased quickly. The regeneration of Brønsted acid sites and Lewis acid sites can be observed with an increase in negative bands at 3643 and 997 cm−1. It is notable that the band at 1716 cm−1 changes little when purged with NO+O2 for 10–30 min. This result indicates that parts of NH3 move from Lewis sites to Brønsted acid sites, leading to a supplemental addition in NH4+. After purging with NO+O2 for 30 min, NO adsorbed on copper sites at 1888 cm−1 is observed, and multi-bands at 1600–1500 cm−1 appear simultaneously, which suggest the formation of nitrite species after the consumption of pre-adsorbed ammonia.40 These results indicate that SCR reactants over C10W10Z are NH3/NH4+ and NO2+NO, consistent with our previous report.16| | | –WO + NH3 → –W–O–NH4+ | (2) |
Fig. 7 shows the in situFTIR spectra of the adsorbed species on C10Z from contact of NH3 and then purged by NO+O2 at 180 °C. A comparison with Fig. 6 shows that bands of NH3 adsorbed on Lewis acid sites over C10Z locate at much lower frequencies (3357–3138 and 1195 cm−1), indicating a lower Lewis acidity of the un-modified catalyst. Meanwhile, no bands of ammonium on Brønsted acid sites are found in Fig. 7(a). Instead, ammonia-derived species, such as NH2 and N2H4, are present, with bands at 1410–1456 cm−1. The increase of the band at 3710 cm−1 with time is ascribed to the formation of hydroxyls from ammonia oxidation. A band of NH3 (1616 cm−1) disappears completely in Fig. 7(b) after purging by NO+O2 for 10 min. Specifically, an immediate increase of bands at 1202 cm−1 with the presence of bands at 1494 and 1006 cm−1 suggests the fast formation of ammonia nitrates on C10Z. Ammonia nitrate deposition on the surface of the catalyst may suppress the low temperature SCR activity of C10Z.
3.7
SCR activity
NH3-SCR performance for various catalysts was measured as a function of temperature, and the results are shown in Fig. 8. The NOx conversion increases rapidly with the increase of reaction temperature and reaches a maximum at about 250 °C. The onset temperature of NOx conversion decreases to 150 °C for the tungsten-containing catalysts compared with C10Z. Thus, it is plausible to suppose that tungsten oxide plays an important role in low-temperature NH3-SCR reaction. Among these catalysts, C10W10Z shows the highest NH3-SCR activity (90%) and the widest temperature window (200–300 °C). Comparatively, the maximal NOx conversion on C10Z only reaches 70%. These results demonstrate that WOx addition can remarkably enhance the NH3–SCR activity of CuOx/ZrO2 catalysts and broaden the temperature window for NOx conversion. It is notable that the C10Z catalyst, which is only Lewis acidic, also presents remarkable NH3–SCR activity, suggesting that Lewis acid sites are responsible for NH3–SCR, and that the existence of Brønsted acidity is not a prerequisite for the NH3-SCR reaction. However, Brønsted acidity helps to enhance the ammonia adsorption capacity and facilitate the inhibition of ammonia oxidation on catalysts (see results in section 3.6). As a result, the selectivity to N2 is improved, which has been primarily discussed in our previous study.16
 |
| | Fig. 8
NH3-SCR activities of CuOx/WOx–ZrO2 catalysts. | |
4. Discussion
Based on our previous report,16copper and tungsten provide two distinct adsorption sites on the CuOx/WOx–ZrO2 catalyst: NO is prone to adsorb on copper sites and ammonia tends to coordinate to tungsten sites. In this way, two NH3-SCR reaction routes, the “ammonia-NO2” route at low temperatures (<200 °C) and the “amine-NO/NO2” route at high temperatures (>250 °C), were established. In the above mechanisms, WOx–ZrO2 solid solutions as a support provide moderate Lewis acidity and abundant Brønsted acidity to achieve a balance between the NO conversion and ammonia oxidation over the CuOx/WOx–ZrO2 catalyst. According to the above experimental results, the structural and electronic interactions between copper and tungsten have been confirmed in this work, which can help us explain the alternations between surface acidity and the adsorptive properties of catalysts.
4.1 Structure of catalyst
As indicated by the XRD and BET results, the incorporation of tungsten into the zirconia support inhibits the transformation of t- to m-ZrO2 and prevents the sintering of the support oxide.17 As a result, more highly dispersed CuOx clusters are obtained on the high-surface-area supports with high tungsten content, which are verified by UV-vis and H2-TPR measurements.
When the tungsten content increases to 10 wt%, highly dispersed WOx clusters with tungsten cations at tetrahedral coordination are formed on the support in addition to those incorporated into the WOx–ZrO2 solid solutions.17,25,26 As the tungsten content further increases, octahedral coordinated W6+ ions appear on C10W20Z due to the polymerization of WOx clusters. These results are confirmed by the increase of W6+/W5+ ratio on surface of catalyst (the XPS results).
Based on the above description, the structural features of C10W10Z catalysts are shown in Scheme 1.
 |
| | Scheme 1 Structure of the catalysts containing different amounts of tungsten. | |
4.2 Interactions between copper and tungsten
The variation in dispersion of copper oxides and tungsten oxides leads to different structural and electronic interactions between these two components. As shown in Scheme 1, the structural interactions may lead to the formation of abundant Cun+–O2−–Wn+ interfaces, which is verified by the Raman shift. The band at 850 cm−1 for C10W20Z falls outside of the typical location of W–O–W vibrations (810 cm−1) because the W–O stretching model may shift to higher frequencies due to the lower mass of Cun+. CuWO3 is reported be prepared at 800 °C which is much higher than the temperatures used in this work for the synthesis of catalysts.41 Therefore, XRD and Raman measurements in this work do not detect the CuWO3 species, indicating that CuWO3 species are not largely formed. CuO/Cu2O and WO3 are known in the fields of photocatalysis and gas sensors as candidates for p- and n-type semiconductors with band-gaps at 1.2/2.0 and 2.7 eV, respectively.42–44 Generally, both pure WO3 and highly dispersed CuOx present an absorption edge at about 450 nm. The absorption edge of C10W10Z and C10W20Z extends to 550 nm, which strongly supports the formation of an interface containing Cun+–O2−–Wn+ bonds and with lower band gap. The interface may lead to a higher concentration of electrons and holes on its sides, resulting in an improved redox property and enhanced Brønsted acidity of catalysts (discussed later). Among the catalysts investigated, C10W10Z presents the lowest energy absorption edge, indicating that highly dispersed copper oxide and tungsten oxide clusters facilitate the formation of a Cun+–O2−–Wn+ interface.
From the discussion above, in addition to the improved dispersion of copper, the combination of zirconia and tungsten oxides in the form of solid solutions may also generate stronger acid sites compared with separate zirconia due to the induced charge imbalance. Furthermore, the superior catalytic performance of this catalyst may also be ascribed to strong interactions between copper and tungsten oxides.
The interface containing a Cun+–O2−–Wn+ bond (acting as p-n heterojunctions generated from the interaction of highly dispersed CuOx and WOx) may result in a high electron density of CuOx (p-type semiconductor), leading to the feasibility of oxygen activation at the CuOx sites (eqn (3)), which is verified by the H2-TPR results (leading to the lowered reduction temperature of copper oxide). This effect also facilitates the preferential adsorption of NO in the form of nitrite on copper sites (eqn (4)) because NO is more easily captured by active oxygen. However, the further oxidation of nitrite to nitrate (eqn (5)) is inhibited on the p-type copper sites because this reaction is accompanied by a release of electrons. Without the electronic interaction between copper and tungsten over C10Z, the deep oxidation of nitrite to nitrate (eqn (5)) occurs on the C10Z catalyst, leading to ammonia nitrate deposition. Ammonia nitrate deposition is considered as the main cause for the low NH3-SCR activity of catalysts at low temperature. However, ammonia nitrite may decompose easily even at temperatures lower than 100 °C. Furthermore, nitrite species may enhance the low temperature (<200 °C) SCR activity of catalystsvia “fast SCR routes” (eqn (6)), because reaction rate via “fast SCR routes” is ten times faster than the standard SCR route (eqn (7)).
| | | NO2− + O− (ads) → NO3− + e | (5) |
| | | 2NH3 + NO2 + NO → 2N2 + 3H2O | (6) |
| | | 4NH3 + 4NO + O2 → 6N2 + 6H2O | (7) |
The results from
Fig. 6 reveal that not only higher Lewis but also Brønsted acid strengths are obtained on C10W10Z compared with C10Z. First of all,
tungsten oxides can easily induce the formation of Brønsted acid sites due to the interaction between the –W
O bond and
water or
ammonia. Furthermore, the interface containing Cu
n+–O
2−–W
n+ may lead to a high concentration of holes on the n-type WO
x. The Brønsted acidity of the
catalyst is thereby enhanced from contacts between
H2O/
NH3 and holes (with strong
oxidation potential
44) (
eqn (8)–(10)). These mechanisms also explain the non-Brønsted acidity of C10Z, which does not contain
tungsten oxides. The high Brønsted acidity endows the
catalyst with the inhibition of ammonia
oxidation beside the enhanced ammonia
adsorption, which has been verified by ammonia
adsorption. Ammonia
oxidation is considered as the main reason for the lowered NH
3-SCR activity at higher temperatures (>250 °C). Therefore, the high-temperature SCR activity of CuO
x–ZrO
2 catalyst may be improved by the addition of tungsten oxides.
| | | H2O + h+ → OH* (ads) + H+ | (8) |
| | | NH3 + h+ → NH2* (ads) + H+ | (9) |
| | | NH3 + H+ → NH4+ (ads) | (10) |
Overall, tungsten and copper may play different roles in the NH
3-SCR reaction over C10W10Z
catalyst (shown in
Scheme 2): highly dispersed WO
x act as main sites for ammonia
adsorption, and Brønsted acid sites arising from WO
x lead to the enhanced ammonia
adsorption and inhibited ammonia
oxidation, which is crucial for high
NH3–SCR activity of
catalysts (especially at higher temperatures (>250 °C)). Copper sites are responsible for NO
adsorption and
oxidation to
nitrite species, which facilitate a high NH
3-SCR reaction rate
via “fast SCR routes”. Compared with C10Z, the CuO
x–WO
x interface helps to enhance the ammonia adsorption and facilitates the formation of ammonia nitrite rather than ammonia nitrate, which may be a crucial factor for obtaining a high SCR activity of the CuO
x/WO
x–ZrO
2 catalyst.
 |
| | Scheme 2
NH3-SCR reaction over C10W10Z catalyst. | |
5. Conclusions
CuOx/WOx–ZrO2 catalyst was prepared by impregnating copper nitrate on WOx–ZrO2 supports with different tungsten content. Excess tungsten (10 wt%) for its solubility in WOx–ZrO2 solid solutions results in the formation of highly dispersed WOx clusters in addition to a high-surface-area solid solution support. Highly dispersed CuOx clusters are obtained on the tungsten-containing catalysts. Highly dispersed (CuOx and WOx) oxides facilitate the higher Lewis acidity of catalysts, which enhances the ammonia adsorption on catalysts. On the other hand, strong structural and electronic interactions between highly dispersed CuOx and WOx clusters lead to the formation of an interface containing Cun+–O2−–Wn+ bonds that facilitate the formation of nitrites rather than nitrates over catalysts at low temperatures (<200 °C). Furthermore, the Brønsted acidity of catalysts is enhanced simultaneously, facilitating the inhibition of ammonia oxidation on catalysts. These effects endow the CuOx/WOx–ZrO2 catalyst with an improved NH3-SCR activity compared with CuOx–ZrO2 catalyst.
Acknowledgements
We would like to acknowledge the Ministry of Science and Technology, PR China for financial support of Project 2009AA06Z304 and 2009AA064801. We would also thank the State Key Lab of New Ceramics and Fine Processing in Tsinghua University.
References
- G. Busca, L. Lietti, G. Ramis and F. Berti, Appl. Catal., B, 1998, 18, 1 CrossRef CAS.
- P. Forzatti, Appl. Catal., A, 2001, 222, 221 CrossRef CAS.
- M. L. Paganini, L. Dall’Acqua, E. Giamello, L. Lietti, P. Forzatti and G. Busca, J. Catal., 1997, 195 Search PubMed.
- C. C. Liu and H. Teng, Appl. Catal., B, 2005, 58, 69 CrossRef CAS.
- H. Praliaud, S. Mikhailenko, Z. Chajar and M. Primet, Appl. Catal., B, 1998, 16, 359 CrossRef CAS.
- G. Centi and S. Perathoner, Appl. Catal., A, 1995, 132, 179 CrossRef CAS.
- D. Pietrogiacomi, D. Sannino, A. Magliano, P. Ciambelli, S. Tuti and V. Indovina, Appl. Catal., B, 2002, 36, 217 CrossRef CAS.
- D. Pietrogiacomi, A. Magliano, D. Sannino, M. C. Campa, P. Ciambelli and V. Indovina, Appl. Catal., B, 2005, 60, 83 CrossRef CAS.
- A. L. Kustov, S. B. Rasmussen, R. Fehrmann and P. Simonsen, Appl. Catal., B, 2007, 76, 9 CrossRef CAS.
- S. Suárez, J. A Martín, M. Yates, P. Avila and J. Blanco, J. Catal., 2005, 229, 227 CrossRef CAS.
- O. V. Komova, A. V. Simakov, V. A. Rogov, D. I. Kochubei, G. V. Odegova, V. V. Kriventsov, E. A. Paukshtis, V. A. Ushakov, N. N. Sazonova and T. A. Nikoro, J. Mol. Catal. A: Chem., 2000, 161, 191 CrossRef CAS.
- X. Jiang, G. Ding, L. Lou, Y. Chen and X. Zheng, J. Mol. Catal. A: Chem., 2004, 218, 187 CrossRef CAS.
- J. Chen, J. Zhu, Y. Zhan, X. Lin, G. Cai, K. Wei and Q. Zheng, Appl. Catal., A, 2009, 363, 208 CrossRef CAS.
- T. Onfroy, G. Clet and M. Houalla, J. Phys. Chem. B, 2005, 109, 3345 CrossRef CAS.
- A. Martínez, G. Prieto, M. A. Arribas, P. Concepción and J. F. Sánchez-Royo, J. Catal., 2007, 248, 288 CrossRef CAS.
- Z. Si, D. Weng, X. Wu, J. Li and G. Li, J. Catal., 2010, 271, 43 CrossRef CAS.
- M. A. Cortés-Jácome, C. Angeles-Chavez, E. López-Salinas, J. Navarrete, P. Toribio and J. A. Toledo, Appl. Catal., A, 2007, 318, 178 CrossRef CAS.
- M. A. Cortés-Jácome, C. Angeles-Chavez, X. Bokhimi and J. A. Toledo-Antonio, J. Solid State Chem., 2006, 179, 2663 CrossRef CAS.
- T. Venkov, M. Dimitrov and K. Hadjiivanov, J. Mol. Catal. A: Chem., 2006, 243, 8 CrossRef CAS.
- J. R. Sohn and M. Y. Park, Langmuir, 1998, 14, 6140 CrossRef.
- V. R. K. Chary, G. V. Sagar, D. Naresh, K. K. Seela and B. Sridhar, J. Phys. Chem. B, 2005, 109, 9437 CrossRef.
- V. Indovina, D. Pietrogiacomi and M. C. Campa, Appl. Catal., B, 2002, 39, 115 CrossRef CAS.
- H. Praliaud, S. Mikhailenko, Z. Chajar and M. Primet, Appl. Catal., B, 1998, 16, 359 CrossRef CAS.
- G. Águila, F. Gracia, J. Cortés and P. Araya, Appl. Catal., B, 2008, 77, 325 CrossRef CAS.
- M. A. Cortés-Jácome, J. A. Toledo, C. Angeles-Chavez, M. Aguilar and J. A. Wang, J. Phys. Chem. B, 2005, 109, 22730 CrossRef CAS.
- E. I. Ross-Medgaarden and I. E. Wachs, J. Phys. Chem. C, 2007, 111, 15089 CrossRef.
- M. Scheithauer, R. K. Grasselli and H. Knzinger, Langmuir, 1998, 14, 3019 CrossRef CAS.
- C. Lu, L. Qi, J. Yang, X. Wang, D. Zhang, J. Xie and J. Ma, Adv. Mater., 2005, 17, 2562 CrossRef CAS.
- J. Macht, C. D. Baertsch, M. May-Lozano, S. L. Soled, Y. Wang and E. Iglesia, J. Catal., 2004, 227, 479 CrossRef CAS.
- R. Zhou, T. Yu, X. Jiang, F. Chen and X. Zheng, Appl. Surf. Sci., 1999, 148, 263 CrossRef CAS.
- W. P. Dow, Y. P. Wang and T. J. Huang, J. Catal., 1996, 160, 171 CrossRef CAS.
- W. P. Dow, Y. P. Wang and T. J. Huang, J. Catal., 1996, 160, 155 CrossRef CAS.
- S. Bennici and A. Gervasini, Appl. Catal., B, 2006, 62, 336 CrossRef CAS.
- G. Águila, F. Gracia, J. Cortés and P. Araya, Appl. Catal., B, 2008, 77, 325 CrossRef CAS.
- M. D. Rhodes and A. T. Bell, J. Catal., 2005, 233, 198 CrossRef CAS.
- Y. Fu, Y. Tian and P. Lin, J. Catal., 1991, 132, 85 CrossRef CAS.
- H. Praliaud, S. Mikhailenko, Z. Chajar and M. Primet, Appl. Catal., B, 1998, 16, 359 CrossRef CAS.
- M. Manzoli, R. D. Monte, F. Boccuzzi, S. Coluccia and J. Kašpar, Appl. Catal., B, 2005, 61, 192 CrossRef CAS.
- G. Ramis, L. Yi, G. Busca, M. Turco, E. Kotur and R. J. Willey, J. Catal., 1995, 157, 523 CrossRef CAS.
- K. I. Hadjiivanov, Catal. Rev. Sci. Eng., 2000, 42, 71 CrossRef CAS.
- O. Yu. Khyzhun, V. L. Bekenev and Yu. M. Solonin, J. Alloys Compd., 2009, 480, 184 CrossRef.
- H. Yu, J. Yu, S. Liu and S. Mann, Chem. Mater., 2007, 19, 4327 CrossRef CAS.
- W. Siripala, A. Ivanovskaya, T. F. Jaramillo, S. H. Baeck and E. W. McFarland, Sol. Energy Mater. Sol. Cells, 2003, 77, 229 CrossRef CAS.
- T. Arai, M. Yanagida, Y. Konishi, Y. Iwasaki, H. Sugihara and K. Sayama, J. Phys. Chem. C, 2007, 111, 7574 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.