On the rate-determining step and the ligand electronic effects in rhodium catalysed hydrogenation of enamines and the hydroaminomethylation of alkenes†
José A.
Fuentes
,
Piotr
Wawrzyniak
,
Geoffrey J.
Roff
,
Michael
Bühl
* and
Matthew L.
Clarke
*
School of Chemistry, University of St Andrews, St Andrews, Fife, KY16 9ST, UK. E-mail: mc28@st-andrews.ac.uk; Fax: +44 (0) 1334 463808
First published on 1st April 2011
Abstract
In the course of studies on the tandem hydroformylation–reductive amination (hydroaminomethylation), fluorinated mono-phosphines were found to be more active than their more electron-donating counterparts in the enamine hydrogenation step of the reaction; this is in contrast to the widely held view that alkene hydrogenation activity increases with ligand donor strength. DFT calculations comparing the reaction pathways for a simple alkene and a representative enamine show that the rate-determining step changes from the first insertion into a Rh–H bond for but-2-ene to the final reductive elimination step from the Rh–hydride–alkyl species in the enamine hydrogenations.
Introduction
The hydrogenation of alkenes is one of the key reactions in synthetic chemistry.1 Despite this being a well-established technology and in many cases an easy reaction to perform even with high enantioselectivity, there are some examples of substrates that do not perform well under normal alkene hydrogenation conditions. For example, asymmetric enamine (not enamide!) hydrogenation2 is an under-developed technology that has also been highlighted as one of the more important aspirational green transformations for the pharmaceutical industry.3 A greater understanding of how the more challenging alkenes differ from those that react so smoothly thus seems desirable. In addition, there has been increasing research on tandem hydroformylation processes where the aldehydes are directly converted into another functionality, with the most important of these domino reactions being hydroaminomethylation (tandem hydroformylation–reductive amination).4 Efficient asymmetric hydroaminomethylation, as a domino process or even as a two-stage process has not yet been achieved, and is a long desired reaction for tertiary amine synthesis. One approach to a net asymmetric hydroaminomethylation process is hydroaminovinylation of an alkene to selectively form enamine products, followed by asymmetric hydrogenation of the enamine produced. However, this represents quite a stiff challenge; even though controlled formation of enamines can be achieved through a delicate balance of reaction conditions,5 asymmetric enamine hydrogenation is challenging. There is little known about the enamine hydrogenation step of this reaction and our own preliminary studies were hampered by poor reactivity and low enantioselectivity. In order to make progress in this area, we chose to examine ligand electronic effects on the hydroaminomethylation and enamine hydrogenation processes, since, to the best of our knowledge, this has not been evaluated. In this paper, we report surprising ligand electronic effects on hydroaminomethylation and more specifically enamine hydrogenation. In contrast to standard alkene substrates, the enamines produced are hydrogenated much more readily using Rh complexes of electron deficient ligands. We also report DFT calculations that provide an answer as to the origin of this effect.Results and discussion
Hydroformylation generally benefits from an electron poor phosphine,6 while it is generally accepted (and taught) that one of the most clear-cut cases of electronic effects in catalysis is that of alkene hydrogenation being accelerated by electron rich ligands.7 Electron donating phosphines accelerate the oxidative addition of hydrogen and what is more likely the rate-determining step, the insertion of the alkene into the Rh–hydride bond. The hydride has been calculated to act as a nucleophile in this process.8 Literature precedent would therefore suggest that the ligand requirements of hydroformylation and hydrogenation are at odds with each other in tandem hydroformylation–reduction process. None-the-less, active and regioselective hydroaminomethylation catalysts derived from electron-deficient diphosphines4d have been reported and led us to consider ligand effects on enamine hydrogenation in more detail.We initially investigated Rh catalysed reactions of styrene, syngas and morpholine using ligands of contrasting electronic properties (Scheme 1). The donor properties of the ligands can be ordered as Cy3P > Bun3P > L2 > Ph3P > L3 > L1 by consideration of their carbonyl stretching frequencies in the complexes of type trans-[RhCl(CO)L2].10a Hydroaminomethylations are generally carried out above 100 °C, but lower temperatures were used in this study in order to analyse the reactions before complete conversion is reached.
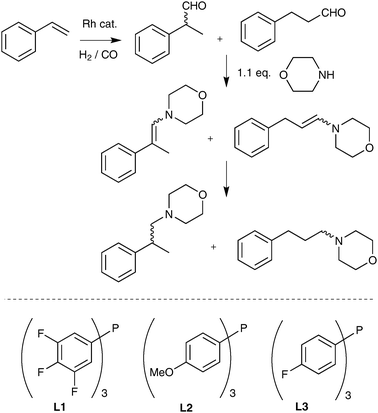 | ||
| Scheme 1 Products formed from a hydroaminomethylation of styrene and morpholine, and some of the ligands used in this study. | ||
A reaction time of 20 hours at 70 °C proved to be suitable conditions to differentiate the various catalysts; after 20 hours, the reactions were stopped and analysed using 1H NMR and GCMS (authentic samples of possible reaction products were prepared to confirm the identities of intermediates and products). The results are shown in Table 1.
| Entrya | Ligand | Aldehyde (%) | Enamine (%) | Amine (L) | Amine (B) |
|---|---|---|---|---|---|
a Conditions: 0.2% [RhCl(COD)]2 as pre-catalyst, 60 bar syngas (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1), toluene as solvent. Conversion of styrene >99% in all cases. Product composition determined using 1H NMR. Enamine and aldehydes yields are mixtures of isomers.
b Temperature = 90 °C.
c 92% isolated yield of amine as a 3.5∶1 mix of regio-isomers.
d Time = 2 hours.
e 0.2% [Rh(acac)(CO)2] used as a catalyst precursor. ∶1), toluene as solvent. Conversion of styrene >99% in all cases. Product composition determined using 1H NMR. Enamine and aldehydes yields are mixtures of isomers.
b Temperature = 90 °C.
c 92% isolated yield of amine as a 3.5∶1 mix of regio-isomers.
d Time = 2 hours.
e 0.2% [Rh(acac)(CO)2] used as a catalyst precursor.
|
|||||
| 1 | 0.4% L1b | 3 | 0 | 21 | 76c |
| 2 | 0.8% L1d | 13 | 71 | 0 | 16 |
| 3 | 0.8% L1 | 0 | 92e | 0 | 0 |
| 4 | 0.8% L1 | 0 | 0 | 8 | 92 |
| 5 | 0.8% L2 | 32 | 66 | 0 | 2 |
| 6 | 0.8% Ph3P | 21 | 72 | 0 | 7 |
| 7 | 0.8% Cy3P | 26 | 61 | 0 | 13 |
| 8 | 0.8% Bun3P | 23 | 71 | 0 | 6 |
| 9 | 0.4% L1 | <1 | 0 | 12 | 88 |
| 10 | 0.7% L1/0.1% L2 | 5 | 16 | 8 | 71 |
| 11 | 0.4% L1/0.4% L2 | 16 | 80 | 0 | 4 |
A number of useful conclusions can be made: under the conditions of hydroaminomethylation, even the poor hydroformylation catalysts derived from tris-(4-methoxyphenyl)phosphine, L2 completely convert styrene into aldehydes or subsequent products. The use of [Rh(acac)(CO)2] and (3,4,5-F3C6H2)3P, L1 results in essentially exclusive formation of the enamine, suggesting that the mono-hydrides formed from reaction of [Rh(acac)L] complexes with hydrogen and/or the resting states in hydroformylation [Rh(H)(PR3)n(CO)3−n] are not catalysts for enamine reduction. This also suggests that the use of [RhCl(COD)]2 as a pre-catalyst can form sufficient amounts of both type of catalysts necessary for hydroformylation and hydrogenation. Reduction of the enamines does not take place using the catalysts derived from more electron-donating ligands, PPh3, (4-MeO–C6H4)3P, (Bun)3P and Cy3P (entries 5–8). In addition, it is striking that using mixtures of (3,4,5-F3C6H2)3P and (4-MeO–C6H4)3P results in an inhibition of the enamine hydrogenation step of the reaction (Table 1, entries 10 and 11). A combination of (3,4,5-F3C6H2)3P, L1 and [Rh(COD)Cl]2 allows complete conversion to the desired amine with reasonable regioselectivity and high yield (entry 4). There are two main mechanistic scenarios for the observed reactivity: (i) ligand electronic effects on alkene hydrogenation using syngas as a hydrogen source are different to those using pure hydrogen, or (ii) the high activity of Rh complexes of electron rich phosphines normally employed in the hydrogenation of alkenes does not extend to enamine substrates that are rarely studied in alkene hydrogenation. This then provided us with impetus to investigate the rates of enamine hydrogenation as a function of the ligands.
A sample of 4-(2-phenylprop-1-enyl)morpholine was prepared by refluxing purified branched aldehyde, 2-phenyl-propanal with morpholine, and studied in hydrogenation. The results shown in Table 2 indicate that (3,4,5-F3C6H2)3P, L1 is a far more active catalyst for this reduction, regardless of whether the reactions are carried out using syngas or pure hydrogen.
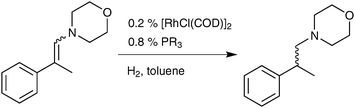
The results described in Table 2 strongly support the suggestion that electron-deficient ligands are preferred in this class of enamine hydrogenation. In order to confirm this was not some special effect confined only to this substrate, we varied the secondary amine in hydroaminomethylation of styrene (pyrrolidine and N-methyl-N-benzylamine) using catalysts derived from (3,4,5-F3C6H2)3P and (4-MeO–C6H4)3P ligands. These results (Table 3) are again striking and in complete agreement with the findings in Tables 1 and 2. (3,4,5-F3C6H2)3P provides a much better catalyst than {(4-MeO)–C6H4}3P and in particular smoothly converts the enamine into the tertiary amine product, whereas the latter ligand stalls at the enamine stage. Reaction of cis-stilbene and morpholine was also carried out, although this gave poor conversion using {(4-MeO)–C6H4}3P and reduced chemo-selectivity using (3,4,5-F3C6H2)3P. None-the-less it is clear that, using the electron rich ligand, some enamine is present and there is no amine detected, whereas for (3,4,5-F3C6H2)3P, almost no enamine remains and a significant amount of amine has been produced, entirely consistent with the previous results. We would therefore suggest that electron-poor phosphines are the best-suited monodentate ligands for a significant number of enamine hydrogenations.
| Entrya | Alkene | Amine | Ligand | T/°C | Conversionb % | Aldehyde % | Enamine d % | Amine % | Yieldc (%) |
|---|---|---|---|---|---|---|---|---|---|
|
a Conditions: 0.2% [RhCl(COD)]2, 0.8% P ligand, toluene, 1 equiv. alkene, 1.1. equiv. secondary amine, initial pressure 60 bar CO/H2 (1∶1), reaction time 20 hours unless stated otherwise.
b Conversion = alkene consumed against 2-methylnaphthalene internal standard using 1H NMR; the composition of the reaction mixture was calculated in the same way. <1 refers to barely detectable traces or undetectable amounts.
c Yield of purified amine after column chromatography or acid/base extraction.
d Mixture of E and Z isomers, and in the case of styrene branched and linear enamine.
e
b/l = 7∶1.
f
b∶l = 6.4∶1.
g
b∶l = 9∶1.
h
b/l = 7∶1.
|
|||||||||
| 1 | Styrene | Pyrrolidine | L1 | 70 | >99 | 1 | 15 | 59 | 53e |
| 2 | Styrene | Pyrrolidine | L2 | 70 | >99 | 6 | 82 | <1 | nd |
| 3 | Styrene | Bn(Me)NH | L1 | 90 | >99 | <1 | <1 | 95 | 87f |
| 4 | Styrene | Bn(Me)NH | L1 | 70 | >99 | 3 | 3 | 70 | 49g |
| 5 | Styrene | Bn(Me)NH | L2 | 70 | >99 | 31 | 65h | 2 | nd |
| 6 | cis-Stilbene | Morpholine | L1 | 80 | 72 | <1 | 3 | 40 | 38 |
| 7 | cis-Stilbene | Morpholine | L2 | 80 | 9 | <1 | 8 | <1 | nd |
These ligand electronic effects are quite contrasting with the accepted theory for alkene hydrogenation and are not easily reconciled with the early rate-determining step expected in such reactions. To gain further insights into mechanistic details of the hydrogenation step, some exploratory calculations were performed at a suitable level of density functional theory (DFT).9 The reaction between the widely studied and accepted active catalyst, Rh(PPh3)2Cl(H)2, and the (E)-4-(2-phenylprop-1-enyl)morpholine substrate was considered. To model the key intermediate resulting from the first Rh–H addition across the double bond a number of possible isomers was screened, in which Rh was attached either at the α or β position (with respect to the nitrogen atom in the morpholine group) and stabilised by secondary interactions with neighbouring groups. The most stable structure turned out to be an α-adduct with an additional Rh–N contact of 2.23 Å (1b in Fig. 1) with all other isomers at least 50 kJ mol−1 higher in energy.8b
![Salient stationary points involved in the Rh-catalysed olefin hydrogenation; generic scheme with a Rh–dihydride fragment (top), optimised structures involving (phenylpropenyl)morpholine (middle) and 2-butene (bottom) with the Rh(PPh3)2Cl(H)2 model catalyst, together with relative energies [kJ mol−1] computed at the B97-D/ECP2 level of DFT. Peripheral H atoms and phenyl groups shown as wireframe for clarity; C, H, N, O, P and Cl atoms are rendered in black, white, blue, red, orange and green, respectively.](/image/article/2011/CY/c1cy00026h/c1cy00026h-f1.gif) | ||
| Fig. 1 Salient stationary points involved in the Rh-catalysed olefin hydrogenation; generic scheme with a Rh–dihydride fragment (top), optimised structures involving (phenylpropenyl)morpholine (middle) and 2-butene (bottom) with the Rh(PPh3)2Cl(H)2 model catalyst, together with relative energies [kJ mol−1] computed at the B97-D/ECP2 level of DFT. Peripheral H atoms and phenyl groups shown as wireframe for clarity; C, H, N, O, P and Cl atoms are rendered in black, white, blue, red, orange and green, respectively. | ||
The transition state for the reductive elimination of the saturated product from 1b was located at 111 kJ mol−1 (TS1b, Fig. 1). This is a surprisingly high barrier, given that it is usually the first H-transfer step, rather than this second one, which is believed to be rate-limiting in Rh-catalysed hydrogenation reactions.8a A transition state for the first H-transfer could be located (TS1a), which, however, connects to the separated reactants, i.e. the olefin and a coordinatively unsaturated Rh(PPh3)2Cl(H)2 fragment (not shown). Starting from these reactants, the resting state before this first H-transfer will be stabilised, e.g. through coordination of solvent or the olefin. The only such olefin complex that could be located is 1a (Fig. 1). Taking this isomer as representative for the intermediate leading to 1b, a barrier of ca. 52 kJ mol−1 can be estimated for the first H-addition step. Thus, even allowing for this being an informed estimation, the second H transfer (1b → TS1b) clearly has a much larger energy barrier and is indeed indicated to be rate-limiting with this substrate.
When the computations were repeated for 2-butene, a model substrate without the N functionality, the expected, “normal” profile was obtained (lower part in Fig. 1). That is, the first H-transfer from 2aviaTS2a has a higher barrier (ca. 75 kJ mol−1) than the second one from 2b to TS2b (56 kJ mol−1). These results therefore indicate that the mechanistic details of homogenous olefin hydrogenation can change notably with an enamine as substrate. Key to this mechanistic change is the stabilisation of the organometallic intermediate 1b through an interaction with the lone pair of the adjacent nitrogen atom to form a Rh–C–N ring. This intermediate is lowered so much in energy that the barrier for the subsequent reductive elimination can become rate-limiting. Stable Rh–alkyl–hydrides have been detected in enamide hydrogenation before,10 but the reductive elimination in these cases has been a lower energy process relative to their formation.8b It is well established that electron-withdrawing ligands accelerate reductive elimination processes,11 and this therefore can be concluded to be the origin of the ligand effects we have observed. Quantitatively, our computed numbers may be associated with a large uncertainty because only a very limited region of the vast conformational space could be explored. However, the final step proposed to be rate-determining is calculated as approximately 60 kJ mol−1 greater in energy than the first hydride insertion. Thus we would estimate with some confidence that any subtle differences in phosphine conformation are extremely unlikely to stabilise any transition states to an extent that could alter the rate-determining steps from those calculated here, giving considerable certainty on the qualitative conclusions.
These two main mechanistic scenarios share a dependence on catalyst, substrate and hydrogen to deliver the species that undergoes rate-determining reactions. However, it was felt useful to collect several other pieces of data regarding the kinetic behaviour of these reactions, and these are archived in the ESI.† Measurements of turnover frequency under essentially identical conditions for L1 and L2 give initial average TOF of 515 mol mol−1 h−1 and 4 mol mol−1 h−1 for L1 and L2, respectively, and increased rates in going from 10 bar to 50 bar pressure of hydrogen. The reaction is slightly unusual in being promoted further by increasing phosphine/Rh ratios. The comparative studies described in the previous pages are done using low ratios, even more pronounced differences would be seen if using a significant excess of phosphine. This can be reconciled by either, (i) strong enamine binding and/or improved kinetics for phosphine loss from a saturated Rh species, or (ii) weak non-competitive product inhibition. In any case, these effects do not alter the likelihood that the rate-determining step changes when one moves from a simple alkene to an enamine.
Summary
In conclusion, we have found that there is a pronounced electronic effect from the phosphine ligands employed in Rh-catalysed enamine hydrogenation, and during Rh catalysed alkene hydroaminomethylation using secondary amines. (3,4,5-F3C6H2)3P emerges as an excellent ligand for these hydrogenations, and seems to be around two orders of magnitude more reactive than (4-MeOC6H4)3P. The origin of this surprising ligand-effect is a change in the rate-determining step for these reactions from the first insertion into the rhodium dihydride species for simple alkenes to the reductive elimination of product for the enamine examined using DFT calculations. This altered reaction profile can be ascribed to a rather stable Rh–alkyl that takes the form of a 3 membered Rh–C–N chelate ring. It remains to be seen the degree to which the rate-determining step in alkene hydrogenation is subject to change as the substrate structure is altered (and inverted ligand electronic effects are observed). It is known that despite many alkene hydrogenations being very easy processes, there are quite a range of substrates known to be difficult when examined with the common toolbox of (generally) electron-donating phosphine ligands. It is possible that a different choice of ligand may render some of these processes feasible, and this work shows once again the value of screening ligands to search for reactivity trends rather than 'hits', and combining such studies with calculations. Enantioselective processes using enamine hydrogenations enabled by these findings will be investigated and will be reported in due course.Experimental
Full experimental and computational details are available in the ESI.†General procedure for the reaction of styrene, syngas and morpholine
[Rh(COD)Cl]2 (3.8 mg, 0.0077 mmol, 0.2 mol%) and the corresponding phosphine PR3 were added into a Biotage 5 ml microwave vial. A stirring bar was added and the vial was sealed with a crimp cap and put under an inert atmosphere. Toluene (2 ml) was added and the mixture stirred for one minute, followed by the addition of styrene (443 μl, 3.88 mmol) and morpholine (373 μl, 4.27 mmol) using a syringe. Two needles were pierced into the vial and this was introduced into the autoclave, which had been previously purged with three vacuum/argon cycles. The autoclave was then purged three times with syngas (1∶1 H2/CO), pressurised to 60 bar and immersed into an oil bath preheated at the required temperature. After the desired reaction time, the autoclave was cooled down to room temperature, the pressure slowly released and opened. A small sample was taken and analysed by 1H NMR to calculate the conversion. Authentic samples of the possible reaction products were prepared as detailed in the ESI† to assist the assignment of the NMR spectra. In some cases, the hydroaminomethylation products were isolated as follows: the reaction mixture was concentrated and partitioned between 1 M HCl and diethyl ether. The HCl layer was made basic with 1 M NaOH and extracted three times into ether. The combined organic phases were dried with MgSO4 and the final solution concentrated under vacuum to give the corresponding mix of branched and linear amines.
Acknowledgements
MLC wishes to thank the Royal Society of Edinburgh for a Research Support Lectureship, the Leverhulme Trust, and the EPSRC for supporting this project. MB wishes to thank EaStCHEM for support and access to the EaStCHEM Research Computing Facility, maintained by Dr H. Früchtl. We thank all the technical staff in the School of Chemistry for assistance.References
- The comprehensive handbook of homogeneous hydrogenation, ed. C. J. Elsevier and J. N. H. DeVries, Wiley-VCH, Weinheim, 2006 Search PubMed.
- Recently, a few examples of selected enamines have been hydrogenated with high selectivity, although even these highly successful studies display much lower reactivity than is normally encountered in alkene hydrogenations, and a significant effort will be needed to realise the potential of these reactions: (a) G.-H. Hou, J.-H. Xie, L. X. Wang and Q. L. Zhou, J. Am. Chem. Soc., 2007, 128, 11774; (b) A. Baeza and A. Pfaltz, Chem.–Eur. J., 2009, 15, 2266 CrossRef CAS; (c) C. I. Tararov, R. Kadyrov, T. H. Riermeier, J. Holz and A. Borner, Tetrahedron Lett., 2000, 41, 2351 CrossRef CAS; (d) G.-H. Hou, J.-H. Xie, P.-C. Yan and Q. L. Zhou, J. Am. Chem. Soc., 2009, 131, 1366 CrossRef CAS; (e) G.-H. Hou, J.-H. Xie, L. X. Wang and Q. L. Zhou, J. Am. Chem. Soc., 2006, 128, 11774 CrossRef CAS.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, Jr., R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC.
- (a) For a review on hydroaminomethylation and related tandem processes, see: P. Eilbracht, L. Barfacker, C. Buss, C. Hollmann, B. E. Kitsos-Rzychon, C. L. Kranemann, T. Rische, R. Roggenbuck and A. Schmidt, Chem. Rev., 1999, 99, 3329 Search PubMed; (b) P. Eilbracht and A. Schmidt, Top. Organomet. Chem., 2006, 18, 65 CAS; (c) M. Ahmed, A. M. Seayad, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2003, 125, 10311 CrossRef CAS; (d) A. Seayad, M. Ahmed, H. Klein, R. Jackstell, T. Gross and M. Beller, Science, 2002, 297, 1676 CrossRef CAS; (e) P. S. Bäuerlein, I. A. Gonzalez, J. J. M. Weemers, M. Lutz, A. L. Spek, D. Vogt and C. Müller, Chem. Commun., 2009, 4944 RSC; (f) S. Chercheja, T. Rothenbücher and P. Eilbracht, Adv. Synth. Catal., 2009, 351, 339 CrossRef CAS; (g) O. Abillard and B. Breit, Adv. Synth. Catal., 2007, 349, 1891 CrossRef CAS; (h) K. Takahashi, M. Yamahita, T. Ichihara, K. Nakano and K. Nozaki, Angew. Chem., Int. Ed., 2010, 49, 4488 CrossRef CAS; (i) I. I. F. Boogaerts, D. F. S. White and D. J. Cole-Hamilton, Chem. Commun., 2010, 46, 2194 RSC; (j) J. J. R. Briggs, J. Klosin and G. T. Whiteker, Org. Lett., 2005, 7, 4795 CrossRef CAS; (k) A. M. Schmidt and P. Eilbracht, Org. Biomol. Chem., 2005, 3, 2333 RSC; (l) L. Routaboul, C. Buch, H. Klein, R. Jackstell and M. Beller, Tetrahedron Lett., 2005, 46, 7401 CrossRef CAS; (m) T. Rische, K.-S. Müller and P. Eilbracht, Tetrahedron, 1999, 55, 9801 CrossRef CAS; (n) M. Ahmed, R. P. J. Bronger, R. Jackstell, P. C. J. Kamer, P. W. N. M. Van Leeuwen and M. Beller, Chem.–Eur. J., 2006, 12, 8979 CrossRef CAS; (o) B. Hamers, P. S. Bäuerlein, C. Müller and D. Vogt, Adv. Synth. Catal., 2008, 350, 332 CrossRef CAS; (p) B. Hamers, E. Morizet-Kosciusko, C. Müller and D. Vogt, ChemCatChem, 2009, 1, 103 Search PubMed; (q) T. O. Viera and H. Alper, Chem. Commun., 2007, 2710 RSC.
- (a) M. L. Clarke and G. J. Roff, Green Chem., 2007, 9, 792 RSC; (b) Y.-S. Lin, B. E. Ali and H. Alper, J. Am. Chem. Soc., 2001, 123, 7719 CrossRef CAS; (c) M. Ahmed, A. Majeedseayad, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2003, 42, 5615 CrossRef CAS.
- (a) P. W. N. M. van Leeuwen, C. Claver, Rhodium catalysed hydroformylation, Kluwer, Dordrecht, 2000 Search PubMed; (b) M. L. Clarke, D. Ellis, K. L. Mason, A. G. Orpen, P. G. Pringle, R. L. Wingad, D. A. Zaher and R. T. Baker, Dalton Trans., 2005, 1294 RSC; (c) R. A. Baber, M. L. Clarke, K. Heslop, A. Marr, A. G. Orpen, P. G. Pringle, A. M. Ward and D. A. Zambrano-Williams, Dalton Trans., 2005, 1079 RSC; (d) B. Breit, R. Winde, T. Mackewitz, R. Paciello and K. Harms, Chem.–Eur. J., 2001, 7, 3106 CrossRef CAS; (e) A. van Rooy, E. N. Orji, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 1995, 14, 34 CrossRef CAS; (f) C. P. Casey, E. L. Paulsen, E. W. Beuttenmueller, B. R. Proft, L. M. Petrovich, B. A. Matter and D. R. Powell, J. Am. Chem. Soc., 1997, 119, 11817 CrossRef CAS; (g) M. L. Clarke, Curr. Org. Chem., 2005, 9, 701 CrossRef CAS; (h) A. M. Trzeciak, T. Glowiak, R. Grzybek and J. J. Ziolowski, Dalton Trans., 1997, 1831 RSC; (i) S. C. van der slot, P. C. J. Kamer, P. W. N. M. van Leeuwen, J. Fraanje, K. Goubitz, M. Lutz and A. L. Spek, Organometallics, 2000, 19, 2504 CrossRef CAS.
- (a) S. Montelatici, A. van der Ent, J. A. Osborn and G. Wilkinson, J. Chem. Soc. A, 1968, 1054 RSC; (b) A. S. C. Chan and J. Halpern, J. Am. Chem. Soc., 1980, 102, 838 CrossRef CAS; (c) J. Halpern and T. Okamoto, Inorg. Chim. Acta, 1984, 89, L53 CrossRef CAS; (d) P. D. Zgoliczm, M. I. Cabrera and R. J. Grau, React. Kinet. Catal. Lett., 2008, 93, 165 CrossRef; (e) J. Halpern, Inorg. Chim. Acta, 1981, 50, 11 CrossRef CAS; (f) T. V. RajanBabu, T. A. Ayers and A. L. Casalnuovo, J. Am. Chem. Soc., 1994, 116, 4101 CrossRef CAS; (g) Z. Herseczki, I. Gergely, C. Hegedüs, Á. Szöllosy and J. Bakos, Tetrahedron: Asymmetry, 2004, 15, 1763 CrossRef.
- (a) D. J. Nelson, R. Li and C. Brammer, J. Org. Chem., 2005, 70, 761 CrossRef CAS; (b) C. R. Landis, P. Hilfenhaus and S. Feldgus, J. Am. Chem. Soc., 1999, 121, 8741 CrossRef CAS; (c) We did consider a reversed polarity of the Rh–hydride and formation of beta adducts, but did not found any evidence for this in this case, see: J. Yu and J. B. Spencer, J. Am. Chem. Soc., 1997, 119, 5257 Search PubMed; (d) H.-C. Wu, S. A. Hamid, J.-Q. Yu and J. B. Spencer, J. Am. Chem. Soc., 2009, 131, 9604 CrossRef CAS.
- Computations were performed at a dispersion-corrected DFT level, B97-D, validated for bulky phosphine complexes, cf. N. Sieffert and M. Bühl, Inorg. Chem., 2009, 48, 4622 Search PubMed ; see ESI† for full details and references.
- For examples, see: (a) I. D. Gridnev, C. Fan and P. G. Pringle, Chem. Commun., 2007, 1319 RSC; (b) I. D. Gridnev, M. Yasutake, T. Imamoto and I. P. Beletskaya, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5385 CrossRef CAS.
- (a) M. L. Clarke and J. J. R. Frew, Specialist Periodical Reports: Organometallic Chemistry, ed. I. J. S. Fairlamb and J. Lynam, Royal Society of Chemistry, 2009, 35, 19 Search PubMed; (b) F. Ernst and D. M. Roddick, Organometallics, 1992, 11, 2972 CrossRef CAS; (c) G. Mann, Q. Shelby, A. H. Roy and J. F. Hartwig, Organometallics, 2003, 22, 2775 CrossRef CAS; (d) T. Korenaga, K. Abe, A. Ko, R. Maenishi and T. Sakai, Organometallics, 2010, 29, 4025 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cy00026h |
| This journal is © The Royal Society of Chemistry 2011 |