Wet oxidation of maleic acid by copper(II) Schiff base catalysts prepared using cashew nut shell liquid templates
F. B.
Hamad
a,
E. B.
Mubofu
*b and
Y. M. M.
Makame
b
aDepartment of Inorganic and Physical Chemistry, Laboratory of Organometallic Chemistry and Catalysis, Ghent University, Krijgslaan 281 (S-3), 9000 Ghent, Belgium
bChemistry Department, University of Dar es Salaam, P.O. Box 35061, Dar es Salaam, Tanzania. E-mail: ebmubofu@gmail.com
First published on 19th April 2011
Abstract
Novel heterogeneous copper(II) Schiff base catalysts have been successfully prepared using a Cashew Nut Shell Liquid (CNSL) templating agent. The preparation of catalyst supports was via a one-pot route and afforded aminopropyl-functionalized Micelle Templated Silica (MTS-AMP). The MTS-AMP support was then condensed with pyridine-2-carboxyaldehyde, 2-hydroxybenzaldehyde, 2-hydroxynaphthaldehyde to produce the corresponding Schiff bases. In each case, the Schiff base was complexed with copper(II) acetate solution at room temperature. The supports and heterogeneous catalysts were porous with pore diameters of up to 25 nm and grain sizes of up to 1.0 μm as revealed by Nitrogen Physisorption Study and Scanning Electron Microscopy (SEM) respectively. The maximum copper loading was about 3% w/w for heterogeneous catalysts supported on CNSL based supports whereas the maximum loading of about 2% w/w was obtained for copper catalysts prepared using commercially available dodecylamine and hexadecylamine templates. The catalysts were tested on the oxidation of maleic acid at room temperature using H2O2 as an oxidant and the catalysts had an excellent catalytic efficiency with a yield of up to 90% and turn over number of about 1000 in ten minutes. The performance of catalysts depended on the type of ligands, the template used to prepare the catalyst support and on the method of catalyst preparation. Catalysts prepared using CNSL templates were more efficient than those prepared using the commercially available templates. The catalysts prepared by a stepwise approach were robust and gave good results while those prepared by an imprinting approach leached out after one reaction cycle.
Introduction
Mesoporous silicas have gained a particular interest as catalyst supports due to their high specific surface areas and ordered porous structures of equal pores with tunable size, shape and connectivity.1–3 Moreover, these materials are easy to prepare in an easily controllable manner obtaining well-defined pore sizes. The discovery of Mobil Composite Material (MCM) by Beck and co-workers in 19924,5 has led to this rapid development of regular mesoporous materials that are now promising in various applications.6–17 Huo et al.19 for instance, reported the synthesis of mesoporous silica under acidic conditions whereas Tanev and Pinnavaia20 proposed a neutral templating synthetic route based on hydrogen bonding between primary amines and neutral inorganic species. The neutral templating synthetic approach has been successfully employed to prepare palladium–Schiff base heterogeneous catalysts that showed excellent activity on the Mizoroki–Heck8 and Miyaura–Suzuki9 reactions. A number of MCM synthetic procedures have been developed for controlling the morphologies of MCM-41 materials,10,11 leading to mesoporous silica nanospheres (MSNs) that have been utilized in a variety of applications including catalysis12,13 and drug delivery.14,15Mesoporous silicas with large pores have previously been prepared using a readily available cashew nut shell liquid (CNSL) surfactant.18 Cashew nut shell liquid is an agricultural by-product neutral surfactant obtained from cashew processing factories. The components of CNSL (Fig. 1) are phenolic compounds, anacardic acid (1) and cardanol (2), having a long chain alkyl substituent meta to the phenolic group.21–23
 | ||
| Fig. 1 Major components of cashew nut shell liquid (CNSL). | ||
MTS materials prepared with CNSL templates possess more desirable properties than those prepared using commercially available templates like dodecylamine (DDA) and hexadecylamine (HDA).
Maleic acid as a central intermediate in phenol oxidation
Maleic acid plays a central role in the reaction pathway of phenol oxidation as it appears almost in all reaction conditions.24Catalytic wet air oxidations of maleic acid, oxalic acid and formic acid have been studied in a batch reactor operated at atmospheric pressure using Pt/Al2O3 and the sulfonated poly(styrene-co-divinylbenzene) resin as catalysts.25Catalytic wet air oxidation of maleic acid has also been studied by using a Pt/graphite catalyst in a three phase slurry26 and it was found that between 10% and 90% of maleic acid is degraded at a 120 to 157 °C temperature range. The allylic and benzylic alcohol oxidation into respective ketones by tert-butyl hydroperoxide (TBHP) in the presence of copper salts under phase-transfer catalysis conditions is also documented.27Numerous examples of copper catalysts supported on Micelle Templated Silica (MTS) are available. For instance, a copper–Schiff base complex of salicylaldehyde covalently anchored into the organic-modified MCM-41 has displayed excellent catalytic efficiency in epoxidation reactions with various olefinic compounds using t-BuOOH as an oxidant.28 Also, the copper metal impregnated on MCM-41 has shown high activity in wet oxidation of phenol.29 Generally the number of copper catalysts supported on the micelle templated silica is on the increase because copper is an important catalyst in many reactions such as ligand-free Sonogashira cross-coupling,30 liquid-phase oxidation reactions, and it is a cheap and abundant metal on the earth's crust.
Despite the great number of papers related to silica supported copper on modified mesoporous silicas, only a few reports exist in the literature on copper(II) Schiff base catalysts supported on MTS prepared from CNSL templated silica. In this study, we have assessed the suitability of CNSL based MTS materials for copper(II) Schiff base catalyst immobilization. The immobilized copper(II) Schiff base catalysts have been tested in wet maleic acid oxidation using hydrogen peroxide as an oxidant. For comparison, the commercially available dodecylamine (DDA) and hexadecylamine (HDA) templates were also employed.
Results and discussion
Adsorption–desorption isotherms of the supports and supported catalysts
Adsorption–desorption isotherms of the aminopropyl modified MTS prepared using different surfactants are shown in Fig. 2. | ||
Fig. 2 Adsorption–desorption isotherms of the MTS-AMP (DDA) 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶9, MTS-AMP (HDA) 1∶9 and MTS-AMP (CNSL) 1∶9. ∶9, MTS-AMP (HDA) 1∶9 and MTS-AMP (CNSL) 1∶9. | ||
In accordance with the IUPAC classification, the DDA template based MTS-AMP exhibits type IVc adsorption–desorption behaviour. This behaviour indicates that the material is mesoporous silica, even though the presence of microporosity is detected at low relative pressure. The isotherm shows three steps on the rapid nitrogen uptake with the first step at low relative pressure (<0.1). The first uptake is ascribed to a gradual microporous filling by the nitrogen and the formation of a monolayer of nitrogen on the walls of the mesopores. The second nitrogen uptake at a relative pressure of about 0.3 is due to the formation of a multilayer of the nitrogen adsorbate, causing capillary condensation in the pores.31 The final increase at a relative pressure of about 0.9 is due to multilayer adsorption on the outer surface of the particles that causes capillary condensation in the inter-particle pores.32 There are no pronounced hysteresis loops in the isotherms although there is a slight up-turn in nitrogen adsorption at very high relative pressure. These behaviours imply that the majority of pores are framework confined and this is indeed proved in the pore size distribution curves shown in Fig. 3.
 | ||
| Fig. 3 Pore size distribution of the MTS-AMP (DDA), MTS-AMP (HDA) and MTS-AMP (CNSL). | ||
The isotherms obtained when HDA is employed as a template were similar to those obtained for DDA. However, the isotherms of HDA based aminopropyl functionalized MTS had a steeper inflection point at a relative pressure of about 0.3 with a clear hysteresis loop. This observable difference implies that the pore distributions in these materials are less uniform relative to those obtained from DDA template based materials. CNSL template based materials produced isotherms of mesoporous characteristics. The isotherms of these materials form a plateau followed by a gradual rise in nitrogen uptake at a relative pressure of about 0.6. A sharp rise in nitrogen uptake at a relative pressure of about 0.8 (Fig. 2) is clearly noted. The sharp rise in nitrogen uptake at a relative pressure above 0.8 is associated with the formation of a multilayer of adsorbate. The formation of an adsorbate multilayer causes capillary condensation in the inter-particle pores. The isotherms of CNSL based materials showed hysteresis loops at a relative pressure above 0.8. The hysteresis loop is an indirect evidence of mesoporous characteristics of the MTS materials and may also be due to the wider inter-particle pore size distribution of the MTS.
No significant difference was observed between isotherms of modified supports (MTS-AMP) and that of MTS-supported copper catalysts. For both modified supports prepared using different templates, the only slight observable difference between them is in the amount of nitrogen adsorbed at the same relative pressure (Fig. 4). The maintenance of isotherm shapes is an indirect proof that there was no destruction of the mesostructure during the immobilization processes.
 | ||
| Fig. 4 Adsorption–desorption isotherms of the MTS-AMP (CNSL) and catalyst 1 (CNSL). | ||
Surface areas and porosity of supports and heterogeneous catalysts
The BET surface areas and pore diameters of the prepared materials were determined and are summarized in Table 1.| Material | Surface area (BET)/m2 g−1 | Pore diameter (BJH)/nm | Pore volume (BJH)/cc nm−1 g−1 | Cu (% w/w) AAS | NH2/mmol g−1 |
|---|---|---|---|---|---|
| a The hot filtration method was used for surfactant extraction.n.d = not determined. n.a = not applicable because the material does not contain the group/metal. | |||||
| MTS (DDA) | 722 | 3.2 | 0.43 | n.a | n.a |
| MTS (DDA) | 1228 | 3.1 | 0.25 | n.a | n.a |
| MTS (HDA) | 741 | 4.2 | 0.92 | n.a | n.a |
| MTS-AMP (DDA) 1∶4 |
214 | 3.1 | 0.14 | n.a | 2.75 |
|
MTS-AMP-NAPH (DDA) 1∶4 |
103 | 3.0 | 0.12 | n.a | n.a |
| MTS-AMP (DDA) 1∶9 |
732 | 3.6 | 0.25 | n.a | 1.20 |
| MTS-AMP (DDA) 1∶9a |
83 | 3.2 | 0.35 | n.a | 2.80 |
| MTS-AMP (CNSL) 1∶4 |
187 | 25.1 | 4.07 | n.a | 3.00 |
| MTS-AMP (CNSL) 1∶9 |
258 | 17.5 | 1.03 | n.a | 2.10 |
| MTS-AMP (CNSL) 1∶9a |
6 | 4.2 | 0.13 | n.a | 3.32 |
| MTS-AMP (HDA) 1∶4 |
160 | 4.1 | 0.56 | n.a | 2.30 |
| MTS-AMP (HDA) 1∶9 |
380 | 6.5 | 0.20 | n.a | 2.00 |
| MTS-AMP (HDA) 1∶9a |
104 | 3.5 | 0.51 | n.a | n.d |
| MTS-AMP-NAPH-Cu (CNSL )-cat 1 | 82 | 3.0 | 0.10 | 3.18 | n.a |
| MTS-AMP-NAPH-Cu-(DDA)Water-cat 2 | 67 | 3.6 | 0.6 | 2.07 | n.a |
| MTS-AMP-NAPH-Cu-(DDA)methanol | n.d | n.d | n.d | 2.25 | n.a |
| MTS-AMP-NAPH-Cu-(DDA)toluene | n.d | n.d | n.d | 2.16 | n.a |
Generally, the type of template used in the preparation, the technique employed in the extraction of the template and the organosilane to silica precursor molar ratio affect the surface area and the pore diameter of the prepared material. CNSL templates produced materials with larger pore diameters and smaller surface areas than DDA and HDA templates. Because the packing parameter and hence the resulting MTS are dependent on the alkyl chain length and the head group area33 of the templating agent, the difference in pore diameters and surface areas may be due to a large phenolic head group in CNSL compared to an amine head group in DDA and HDA.
The surface areas of materials showed a slight decrease after each step of modification. For instance, the surface area of MTS-AMP (DDA) 1∶4 is 214 m2 g−1 and upon modification to MTS-AMP-NAPH (DDA), its surface area drops to 103 m2 g−1. This decrease in surface areas and pore diameters after each step of MTS modification implies that the anchored materials occupy some space of MTS pore volume and hence block some of the pores. The BET results also reveal that there is a significant difference between the surface areas of the materials depending on the technique employed for template extraction. The hot filtration method of surfactant extraction gave lower BET surface areas compared to the Soxhlet method for the same material. For instance, the hot filtration method gave materials with a surface area of 6 m2 g−1 for MTS-AMP (CNSL) 1∶9 while the Soxhlet method produced materials with a surface area of 258 m2 g−1 for the same material. This difference in surface areas shows that the extraction of templates by the Soxhlet method is more effective than hot filtration.
Fig. 3 shows the pore size distribution of MTS-AMP prepared using different templates. Apparent from the figure is that the CNSL based materials possess wider pore size distribution with the majority of pores ranging from 10 nm to 40 nm. In contrast, the pore size distribution of DDA and HDA based materials is narrow with the greater part of pore sizes being centered reasonably well on the average pore diameters of 3 nm. The effect of the organosilane to silica precursor ratio is clearly seen in Table 1. The organosilane to silica precursor molar ratio of 1 ∶9 resulted in higher surface areas compared to the 1∶4 molar ratio.
The identity of supported functional groups
Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS) was used in the qualitative determination of functional groups on the supports and supported catalysts. Infrared spectra on steps towards supported catalyst (DDA) are shown in Fig. 5. | ||
| Fig. 5 DRIFT spectra of (a) MTS-AMP (DDA), (b) MTS-AMP-Naph. (DDA), (c) catalyst 3 (DDA), (d) free copper acetate. | ||
The spectrum of MTS-AMP (DDA) (Fig. 5a) shows a weak C–H stretching vibration band in the region of 2950 cm−1 and the corresponding weak bending vibration bands for H–N–H and H–C–H in the regions 1650 cm−1 to 1550 cm−1 and 1550 cm−1 to 1300 cm−1, respectively, are observed. These vibration bands were absent in the spectrum of unmodified MTS and it is therefore clear that aminopropyl silane has been anchored into the MTS matrix. The MTS-AMP was functionalized with the ligand in order to prepare the Schiff base functionalized MTS. The success of this functionalization was confirmed by the variation of the absorption frequencies of the new spectrum with respect to the parent one (Fig. 5b). The new spectrum shows changes in the regions where bending vibrations due to the H–N–H and H–C–H occur. In addition, a new band at about 1640 cm−1 that is attributed to a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration is observed. Other new bands are observed at 1545 cm−1 and 1560 cm−1 and these correspond to the aromatic ring breathing mode. Upon complexation of copper into MTS-AMP-NAPH, the CN stretching vibration shifted to a lower frequency. The presence of the azomethine vibration band in the spectrum of the supported Schiff base and its shift to lower frequencies in the spectra of supported copper Schiff base catalysts confirm the coordination of azomethine nitrogen with copper (Fig. 5c). No significant difference was noted between the spectra of catalysts prepared using CNSL templates and those prepared using the commercially available templates, HDA and DDA.
N stretching vibration is observed. Other new bands are observed at 1545 cm−1 and 1560 cm−1 and these correspond to the aromatic ring breathing mode. Upon complexation of copper into MTS-AMP-NAPH, the CN stretching vibration shifted to a lower frequency. The presence of the azomethine vibration band in the spectrum of the supported Schiff base and its shift to lower frequencies in the spectra of supported copper Schiff base catalysts confirm the coordination of azomethine nitrogen with copper (Fig. 5c). No significant difference was noted between the spectra of catalysts prepared using CNSL templates and those prepared using the commercially available templates, HDA and DDA.
The morphology and grain size of the supports and supported catalysts
Scanning Electron Microscopy (SEM) was used to analyze the morphology and grain sizes of the materials. Some micrographs of MTS-AMP (DDA) and of MTS-AMP (CNSL) are presented in Fig. 6 and 7 respectively. | ||
| Fig. 6 Scanning electron micrograph of MTS-AMP (DDA). | ||
 | ||
| Fig. 7 Scanning electron micrograph of MTS-AMP (CNSL). | ||
The micrograph of MTS-AMP (DDA) comprises of aggregates of roughly spherical particles with a typical average size of about 0.5 μm while the particles from MTS-AMP (CNSL) are relatively more distinct and their surfaces are smoother with the average size of 1.0 μm. The large size of materials prepared by using CNSL templates may be due to the large head group of CNSL templates. A large surfactant head group leads to a large packing parameter, micelle size and hence leads to MTS with large pores.
The effect of immobilization process on the grain sizes and shapes is noticed. It is evident from Fig. 8 and 9 that relative to the parent MTS, as the average particle size decreases the grain size changes to a less distinct grain size after every step of catalyst preparation.
 | ||
| Fig. 8 Scanning electron micrograph of catalyst 1 (CNSL). | ||
 | ||
| Fig. 9 Scanning electron micrograph of catalyst 2 (DDA). | ||
This destruction of grain size is attributed to an uneven distribution of supported catalysts within the silica mesopores. The anchored materials are seen in the micrographs of supported catalysts as randomly distributed species. However, this effect is significant in the materials prepared using CNSL templates than those prepared by using commercial templates. This difference may be attributed to the large pore diameters for materials prepared using CNSL templates.
The amounts of anchored functional groups
Acid titration and atomic absorption spectroscopy were used to quantitatively determine the amounts of amino groups on the supports and the amount of copper on the supported catalysts respectively. The maximum loading of copper catalysts was 3.18% w/w. The results of some representative samples are shown in Table 1. It is obvious from the results that CNSL template based materials acquired higher amino group loading than those based on HDA and DDA templates. Consequently, CNSL based supports produced catalysts with higher copper loading. This catalyst loading difference is associated with the large pore diameter characteristic of a CNSL based support and thus allows more active sites to be easily available for coordination with a copper complex. The effect of solvent on copper loading is also observable in the table. Methanol produces material with higher copper loading followed by toluene, with water at the bottom.We see that heterogeneous copper(II) Schiff base catalysts can successfully be anchored on the Micelle Templated Silica (MTS). Both CNSL templates and commercially available templates produced good catalyst supports. Materials prepared using CNSL templates showed higher pore diameter than those prepared using DDA and HDA hence being suitable candidates for immobilization of biomolecules.
Catalytic activities and catalyst reuses on oxidation of maleic acid
The activities and reusability of the supported copper catalysts were tested on the oxidation of maleic acid at room temperature using H2O2 as an oxidant. It was anticipated that a copper complex coordinated by a Schiff base ligand will be a good catalyst because the imino moiety of the Schiff base is a π-electron acceptor that makes copper more electrophilic and thus accelerates the reactivity of copper with nucleophilic reactants.Oxidation of maleic acid
The MTS supported copper(II) Schiff base catalysed oxidation of maleic acid by H2O2 at room temperature to carbon dioxide and water was carried out and the progress of the reaction was monitored by using an HPLC technique. A calibration curve obtained using standard solutions was used to obtain amounts of maleic acid converted. The peak areas of the analyzed samples were then converted into concentration by using the calibration curve. The extent to which maleic acid is degraded by H2O2 at room temperature in the absence of a catalyst was also established (Fig. 10). It is evident from these results that while the reaction proceeds to some extent without a catalyst, the reaction is highly accelerated by the heterogeneous copper catalyst. The reaction reached about 90% yield after 10 minutes while in the absence of the catalyst, it was less than 5% yield after the same reaction time.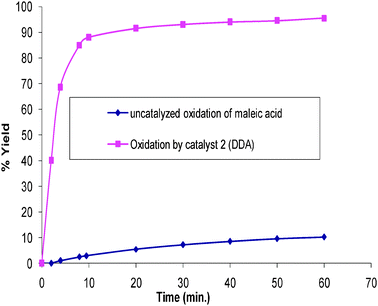 | ||
| Fig. 10 Wet oxidation of maleic acid at room temperature using hydrogen peroxide as an oxidant (maleic acid = 0.02 g; catalyst = 0.01 g; water = 100 ml; H2O2 = 20 ml). | ||
The influence of templates on the activity of catalysts was studied. For instance, catalysts prepared using CNSL were compared with those prepared using DDA. Fig. 11 shows the activity of catalyst 3, which was prepared using different templates. Under the same reaction conditions, it is clear that about 95% of maleic acid is degraded after about 40 minutes when catalyst 3 (CNSL) is employed while only 67% of maleic acid is degraded after one hour when catalyst 3 (DDA) is employed.
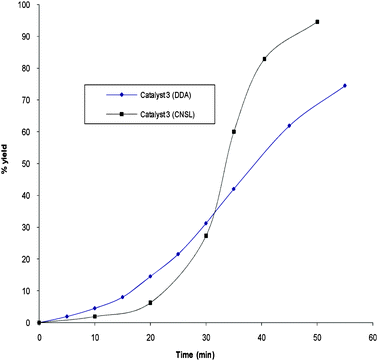 | ||
| Fig. 11 Oxidation of maleic acid by catalyst 3 prepared using a DDA or CNSL template at room temperature using hydrogen peroxide as an oxidant (maleic acid = 0.5 g; water = 100 ml; H2O2 = 40 ml; catalyst = 0.01 g). | ||
Furthermore, different catalysts prepared using different ligands were compared on the oxidation of maleic acid and the results are shown in Fig. 12. It can be seen that maleic acid degrades up to 95% with catalyst 3 (CNSL) after forty minutes and 87% with catalyst 1 (CNSL) after one hour while only 78% of maleic acid degrades with catalyst 2 (CNSL) after one hour.
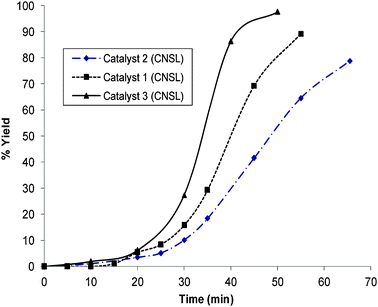 | ||
| Fig. 12 Reaction profile of the oxidation of maleic acid by different catalysts prepared using different ligands at room temperature using hydrogen peroxide as an oxidant (maleic acid = 0.5 g; water = 100 ml; H2O2 = 40 ml; catalyst = 0.01 g). | ||
In addition to the effect of ligand and templates, the effect of the solvent used in the preparation of the catalysts on the performance of the catalyst was studied. No significant difference was noted in the performance of catalyst 3 (DDA) prepared using different solvents (Table 2).
Catalyst reuse on wet oxidation of maleic acid at room temperature
The catalysts were subjected to reuse studies where the used catalyst was filtered from the reaction mixture at the end of each reaction. The catalyst was then washed with dichloromethane to remove any surface adsorbed products. The catalyst was then reused in a fresh batch and the results show that within the experimental limits, the product yield remains the same (Fig. 13). | ||
| Fig. 13 Catalyst 2 (DDA) (∼0.01 g) reuse on the oxidation of maleic acid (0.02 g). | ||
Generally, we have seen that the catalysts were very active at room temperature compared to those used in other work that performed the same reaction in the temperature range of 120 °C to 157 °C.34 It is obvious that the efficiency of these catalysts is dependent on the type of surfactant, method of catalysts preparation and ligand used during catalyst preparation. For instance, catalysts prepared using CNSL templates showed higher activity than those prepared using DDA templates. The efficacy of the CNSL template based catalysts is attributed to their larger pores implying that more active sites are accessed by the reactant. Catalysts prepared using 2-hydroxynaphthaldehyde are more effective on the oxidation of maleic acid followed by catalysts prepared using pyridine-2-carboxyaldehyde while catalysts prepared using 2-hydroxybenzaldehyde were the least active. This variation in activities is associated with differences in the electronic and steric environments of the ligands used in the preparation of the catalysts. This also serves as an indirect proof that the active catalysts are heterogeneous as otherwise they would have the same catalytic activity. Moreover, the supported catalysts prepared by one pot were found to be robust and reusable. On the other hand, catalysts prepared by grafting leached after only one run. The catalyst leaching for imprinted catalysts suggests that the coordination of the active metal to homogeneous imine was not efficient during catalyst preparation and hence copper was only physisorbed.
Experimental
Materials and reagents
3-Aminopropyltrimethoxysilane 97% (Aldrich), pyridine-2-carboxyaldehyde >98% (Fluka), 2-hydroxybenzaldehyde 99% (Aldrich), 2-hydroxynaphthaldehyde 98% (Sigma-Aldrich), and 2-thiophene carboxaldehyde 98% (Aldrich), tetraethoxysilane 98% (Fluka), dodecylamine 98% (Fluka), NaOH pellets 98% (Fluka), copper(II) acetate 99% (BDH Limited, Poole England), HCl 32% (Fluka), norbornene >95.0% (Fluka), maleic acid 99% (May & Baker), ethanol 96% (Sigma-Aldrich), methanol 99.9% (Rochelle Chemicals), acetonitrile 99.8% (Sigma-Aldrich), toluene 99.8% (Rochelle Chemicals), nitric acid 96% (Codex) and distilled water (University of Dar es salaam, Chemistry Department) were used as received. The technical CNSL was collected from the cashew nut processing factory in Dar es Salaam, Tanzania.Catalyst preparation
The catalysts were prepared as reported elsewhere35 with little modification (Scheme 1). Three types of catalysts were prepared by varying the ligands where pyridine-2-carboxyaldehyde produced cat 1, 2-hydroxybenzaldehyde produced cat 2 and 2-hydroxynaphthaldehyde (4) shown in Scheme 1 produced cat 3.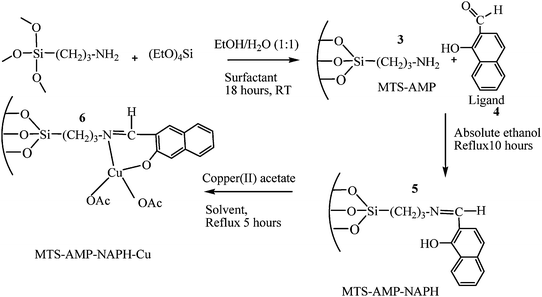 | ||
| Scheme 1 Stepwise synthesis of MTS-AMP-NAPH-Cu catalyst (cat 3). | ||
Preparation of MTS-AMP (CNSL) (3)
To a stirred solution of CNSL (5 g) in aqueous ethanol (104 ml of absolute ethanol and 106 ml of distilled water) at room temperature, tetraethoxysilane (TEOS, 36.67 g, 156.84 mmol) and 3-aminopropyltriethoxysilane (AMPS, 8.7 g, 39.3 mmol) were added. TEOS and AMPS were separately, but rapidly and simultaneously added to the stirred mixture. The mixture was initially clear, but increasingly became cloudy after 10 min and later turned into a thick brown paste. The thick paste was filtered and was washed with excess ethanol after 18 h. The template on the brown powder obtained during filtration was removed by the Soxhlet extraction using ethanol as solvent. The final powder was dried overnight at 100 °C in an oven and kept for further characterization and functionalisations. This procedure was adopted for dodecylamine (DDA) and hexadecylamine templates except that 10 g of these templates were used instead of 5 g used for CNSL templates. Because hot filtration as a method for template extraction did not produce porous materials, the Soxhlet extraction using ethanol solvent was the preferred method for template extraction in all prepared materials.Preparation of the MTS supported Schiff base (5) and subsequent catalyst (6)
To a stirred solution of 2-hydroxynaphthaldehyde (4) in absolute ethanol (50 cm3ethanol and 5.46 mmol ligand), 2 g of the dried MTS-AMP (3) was added slowly and the mixture was refluxed and vigorously stirred for 10 h. After this time, the reflux was stopped and the Schiff base modified silica (MTS-AMP-NAPH) (5) was filtered under reduced pressure and washed with excess ethanol and dried overnight in an oven at 100 °C.The MTS supported copper(II) catalyst (6) was achieved by the reaction of the modified Schiff base support (5) with a 5.46 mmol solution of copper(II) acetate in water, methanol or toluene. In all cases, the mixture was refluxed for 5 h and the supported catalyst was filtered under reduced pressure and washed thoroughly with the solvent used for the preparation until the washings were colourless. The resulting catalyst was then left over-night in air at room temperature to dry. The resulting yellowish green supported catalyst was later conditioned for a total of 6 h by refluxing it in ethanol and acetone so as to remove any surface physisorbed copper acetate. The conditioned catalyst was dried over-night in an oven at 100 °C and characterized.
Catalysts characterization
Nitrogen physisorption was used to determine the porosity behavior of catalysts. The surface area was analyzed by the BET method while pore size distributions were determined by the BJH method. Acid titration and atomic absorption spectroscopy were used for quantitative determination of amino groups on the supports and the copper content on the supported catalysts respectively. Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFT) was used for qualitative analysis of the prepared supports and supported catalysts. The grain size and morphology of supports and supported catalysts were analyzed using Scanning Electron Microscopy and atomic force microscopy.Oxidation of maleic acid
Oxidation of maleic acid was performed in a 250 ml round bottomed flask. The initial mixture containing 0.01 g catalyst, 100 ml water, and 0.02 g maleic acid was stirred at room temperature for about 1 min, and then 10 ml of 30% hydrogen peroxide was added. The amount of H2O2 was in excess with respect to the substrate since in the reaction mixture it partially undergoes self-decomposition into water and atomic oxygen. Small samples (ca. 0.1 ml) of the reaction mixture were taken after various time intervals and analysed on a Merch-Hitachi HPLC instrument equipped with a L-6200A intelligent pump, AS-2000A Auto sampler, Merck T-6300 Column heater, L-4250 UV-VIS Detector and D-2500 Chromator-Intergrator. The separation of components was achieved by a 300 × 8 ID mm Shodex, Rspak KC-811 Column. The eluent used was a mixture of distilled water (99%) and phosphoric acid (1%). After integration of the chromatographic peaks, conversion of the substrates and the yield of the products were calculated.Acknowledgements
Financial support from Sida/SAREC under Material Science and Solar energy project of Faculty of Science, University of Dar es Salaam, is gratefully acknowledged.Notes and references
- A. Taguchi and F. Schuth, Microporous Mesoporous Mater., 2005, 77, 1 CrossRef CAS.
- T. Tsoncheva, M. Jarn, D. Paneva, M. Dimitrov and I. Mitov, Microporous Mesoporous Mater., 2011, 137, 56 CrossRef CAS.
- S. M. Seo, E. J. Cho, S. J. Lee, K. C. Nam, S.-H. Park and J. H. Jung, Microporous Mesoporous Mater., 2008, 114, 448 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. Chu, W. D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834 CrossRef CAS.
- J. Dhainaut, J.-P. Dacquin, A. F. Lee and K. Wilson, Green Chem., 2010, 12, 296 RSC.
- R. Mokaya, J. Mater. Chem., 2002, 12, 3027 RSC.
- E. B. Mubofu, J. H. Clark and D. J. Macquarrie, Green Chem., 2001, 3, 23 RSC.
- J. H. Clark, D. J. Macquarrie and E. B. Mubofu, Green Chem., 2000, 2, 53 RSC.
- M. Grun, K. K. Unger, A. Matsumoto and K. Tsutsumi, Microporous Mesoporous Mater., 1999, 27, 207 CrossRef CAS.
- Q. Cai, Z.-S. Luo, W.-Q. Pang, Y.-W. Fan, X.-H. Chen and F.-Z. Cui, Chem. Mater., 2001, 13, 258 CrossRef CAS.
- H.-T. Chen, S. Huh, J. W. Wiench, M. Pruski and V. S.-Y. Lin, J. Am. Chem. Soc., 2005, 127, 13305 CrossRef CAS.
- A. Salis, M. S. Bhattacharyya and M. Monduzzi, J. Phys. Chem. B, 2010, 114, 7996 CrossRef CAS.
- I. Slowing, B. G. Trewyn and V. S.-Y. Lin, J. Am. Chem. Soc., 2006, 128, 14792 CrossRef CAS.
- I. Slowing, B. G. Trewyn and V. S.-Y. Lin, J. Am. Chem. Soc., 2007, 129, 8845 CrossRef CAS.
- Y.-Y. Li, J. Yang, W.-B. Wu, X.-Z. Zhang and R.-X. Zhuo, Langmuir, 2009, 25, 1923 CrossRef CAS.
- E. Otsuka, K. Kurumada, A. Suzuki, S. Matsuzawa and K. Takeuchi, J. Sol-Gel Sci. Technol., 2008, 46, 71 CrossRef CAS.
- A. Hilonga, Synthesis of Polyamine–Silica Hybrid by Non-electrostatic Surfactant Assembly, MSc thesis, University of Dar es Salaam, Tanzania, 2006 Search PubMed.
- Q. Huo, D. I. Margolese, U. Ciesla, P. Feng, T. E. Gier, P. Sieger, R. Leon, P. M. Petroff, F. Schuth and G. D. Stucky, Nature, 1994, 368, 317 CrossRef CAS.
- P. T. Tanev and T. J. Pinnavaia, Science, 1995, 267, 865 CrossRef CAS.
- J. H. P. Tyman, Chem. Soc. Rev., 1979, 8, 499 RSC.
- Cardolite Corporation, Inc., Test Plan for Cashew Nut Shell Liquid, CAS No. 8007-24-7 2002 at www.cardolite.com.
- P. Oghome and A. J. Kehinde, Afr. J. Sci. Technol., Sci. Eng. Ser., 2004, 5, 92 Search PubMed.
- H. R. Devlin and I. J. Harris, Ind. Eng. Chem. Fundam., 1984, 23, 387 CrossRef CAS.
- D. K. Lee, D. S. Kim, Catalysis in the Asia Pacific Region, 2000, vol. 63, p. 249 Search PubMed.
- E. A. J. Mwakilobolwa, Catalytic Wet Oxidation of Wastewater Pollutants: Modelling of In situ Remediation of the Platinum Catalyst During Wet Oxidation of Phenol, MSc thesis, UDSM, 2005 Search PubMed.
- G. Rothenberg, L. Feldberg, H. Wiener and Y. Sasson, J. Chem. Soc., Perkin Trans. 2, 1998, 2429 RSC.
- J. Sreyashi, D. Buddhadeb, B. Rajesh and K. Subratanath, Langmuir, 2007, 23, 2492 CrossRef CAS.
- Q. Wu, X. Hu, P. L. Yue, X. S. Zhao and G. Q. Lu, Appl. Catal., B, 2001, 32, 151 CrossRef CAS.
- M. B. Thathagar, J. Beckers and G. Rothenberg, Green Chem., 2004, 6, 215 RSC.
- S. J. Gregg and K. S. W. Sing, Adsorption, Surface Area and Porosity, Academic PressLondon, 1982 Search PubMed.
- Z. Luan, H. He, W. Zhou and J. Klinowski, J. Chem. Soc., Faraday Trans., 1995, 91, 2955 RSC.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525 RSC.
- R. A. Sheldon, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, VCH, Weinheim, 1996, vol. 1, p. 411 Search PubMed.
- E. B. Mubofu, in Novel Environmentally Benign Supported Palladium Catalysts, PhD thesis, University of York, UK, 2001 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |