Rapid synthesis of nanostructured Cu–TiO2–SiO2 composites for CO2 photoreduction by evaporation driven self-assembly†
Wei-Ning
Wang
,
Jinho
Park
and
Pratim
Biswas
*
Aerosol and Air Quality Research Laboratory, Department of Energy, Environmental and Chemical Engineering, Washington University in St. Louis, St. Louis, MO 63130, USA. E-mail: pratim.biswas@wustl.edu; Fax: +1-314-935-5464; Tel: +1-314-935-5548
First published on 4th April 2011
Abstract
Nanostructured copper doped titania–silica (Cu–TiO2–SiO2) photocatalyst composite particles were directly formed in a rapid manner by evaporation driven self-assembly of nanocolloids in a furnace aerosol reactor (FuAR). Aqueous suspensions of nanosized TiO2 and SiO2 colloids and copper nitrate solution were used as precursors. The size, composition, and porosity of the composite particles were tailored by manipulating the precursor concentration, stoichiometric ratio, and synthesis temperature, respectively. The as-prepared composite particles were characterized by means of SEM, TEM, XRD, UV-VIS, and nitrogen physisorption measurements, to determine particle diameter, morphology, crystallinity, absorption band, surface area, and pore size. CO2 photoreduction was conducted inside a quartz reactor under illumination of UV light followed by GC analysis. The results revealed that the composite particles were submicrometre-sized mesoporous spheres with average pore sizes of 20 to 30 nm, having optimal molar percentages of TiO2 and Cu to the whole particle of 2% and 0.01%, respectively, achieving a relatively high CO2 conversion efficiency, i.e. a CO yield of approximately 20 μmol/g TiO2/h.
1. Introduction
There are mounting concerns over the emission of greenhouse gases (GHG), in particular carbon dioxide (CO2), which is one of the major contributors to global climate change.1 A great deal of effort has been expended to reduce CO2 emissions. For instance, the development of technologies to generate renewable energies is the most desirable way with regard to the long-term view.2 However, in their current state, these technologies cannot fully replace the existing fossil fuel-based power generation. Thus, power generation viafossil fuel combustion with effective CO2 capture appears to become a key contributor to energy supply in the foreseeable future, which can be realized by pre-, post-, or oxy-fuel combustion processes.3 The carbon dioxide capture and sequestration (CCS) processes, however, are energy-intensive and of high-cost.2Recent innovations have made the photocatalysis technology a potentially promising alternative.2,4–6 The process can be applied for converting CO2 to carbon-contained energy bearing compounds, such as carbon monoxide (CO), formic acid (HCOOH), methane (CH4), methanol (CH3OH), and ethanol (CH3CH2OH). Another potential feature of the photocatalysis technology is that it can consume less energy in comparison to conventional methods by harnessing solar energy, which is abundant, cheap, and ecologically clean and safe.2 In addition, a variety of inexpensive metal oxides, such as titanium oxide (TiO2), are readily available as catalysts.
In spite of its economic and environmental benefits, the photocatalytic pathway suffers from low efficiency. A typical semiconductor-based photocatalyst needs to absorb light energy, generate electron–hole pairs, spatially separate them, transfer them to redox active species across the interface and minimize electron–hole recombination.6 The decrease of the electron–hole recombination rate is a key factor in terms of enhancement of the photocatalytic activity. For this purpose, several methods have been proposed, including synthesis of catalysts with porous structures.7–11 It is believed that the porous structure can effectively distribute the electron transfer, slow down the recombination rate, and hence enhance the photocatalytic performance. The other method is doping metals, such as copper (Cu) and iron (Fe), on the photocatalyst surface.12,13 The doping metal can absorb excited electrons, and thus separate electron–hole pairs to decrease the recombination rate.
The current photocatalysts, in particular, TiO2-based inorganic catalysts with porous structures are intensively investigated, due to their unique morphology, high thermal stability, and improved photocatalytic efficiency. They are typically prepared by batch processes, such as hydrothermal, sol–gel, precipitation, impregnation or ion exchange methods.7,8,10,12,14–18 The current photocatalysts for CO2 reduction, however, do not perform as effectively as current solar hydrogen generation catalysts. The maximum CO2 conversion rate was reported to be 25 μmol/g TiO2/h.6 Our recent results showed that the photoreduction efficiency could be improved by distributing Cu–TiO2 nanocrystals inside a mesoporous silica (SiO2) matrix prepared by a sol–gel method.19 The catalysts prepared by these liquid methods, however, typically have an inhomogeneous phase distribution due to macroscopic reactions, leading to non-optimal performance. Further, these liquid methods generally need a very long time for both reaction and post-treatments, such as surfactant/solvent removal and calcinations. Thus they are difficult to apply to commercial scale production because of their high energy consumption and significant batch-to-batch variation. Therefore, it is essential to develop a process for the rapid preparation of Ti-based CO2 photoreduction catalysts.
In this work, a rapid and direct synthesis of nanostructured copper-doped titania–silica (Cu–TiO2–SiO2) composite particles for CO2 photoreduction using a furnace aerosol reactor (FuAR) method20,21 is demonstrated for the first time. The FuAR method is simple yet effective, with an extremely short processing time of several seconds.22,23Catalyst particles are produced from micrometre-sized droplets, in which homogeneous phase distribution can be ensured by microscopic reactions inside the droplets. In addition, the FuAR method is applied in a continuous manner, avoiding batch-to-batch variations. The photocatalyst particles were prepared from aqueous suspensions of nanosized TiO2 and SiO2 colloids and copper nitrate solution, which are cheap and safe, and are readily available for mass production. The particle size, composition, structure, and hence the catalytic performance were tailored by manipulating precursor properties as well as process parameters. TiO2 nanocrystals as the effective photocatalysts were efficiently utilized by selectively distributing them on the surface of a silica mesoporous matrix by evaporation driven self-assembly24–26 of these nanocolloids inside the micrometre-sized droplets, resulting in high CO2 reduction efficiency.
2. Experimental section
Catalyst synthesis
The nanostructured Cu–TiO2–SiO2 composite particles were synthesized by the FuAR method, which is schematically shown in Fig. 1. The FuAR consisted of a 6-jet Collison nebulizer (BGI Instruments, Waltham, MA) as the atomizer, an electric furnace, a tubular alumina reactor, a microfiber filter, an air pump, and cooling and gas feeding systems. The schematic diagram of Cu–TiO2–SiO2 composite particle formation inside the FuAR is shown in Fig. 2a. Aqueous suspensions of colloidal silica (Snowtex® OL, Nissan Chemical Industries, Ltd., Chiba, Japan) and titania (AERODISP® W740X, Evonik Degussa Corporation, Parsippany, NJ), and copper nitrate trihydrate (Cu(NO3)2·3H2O, 99.5% purity, Strem Chemicals, Newburyport, MA) aqueous solution were used as precursors. The precursors were atomized into micrometre-sized droplets by means of the atomizer, and the mist was delivered by air into the tubular alumina reactor (1 m in length and 25 mm in inner diameter) maintained at predetermined temperatures (from 400 to 1000 °C), followed by heating for several seconds. During the process, the micrometre-sized droplets underwent solvent evaporation, evaporation driven self-assembly of TiO2 and SiO2 nanocolloids, and further drying and pyrolysis of Cu(NO3)2 to form the final composite particles. These composite particles were collected downstream of the reactor using the glass microfiber filter (EPM 2000, Whatman Inc., Florham Park, NJ) for particle characterization and CO2 photocatalytic reduction analysis.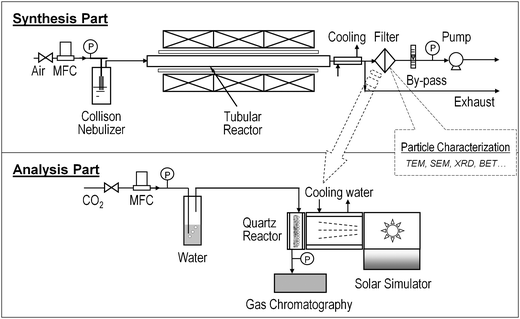 | ||
| Fig. 1 Schematic diagram of experimental setup for catalyst synthesis and analysis. | ||
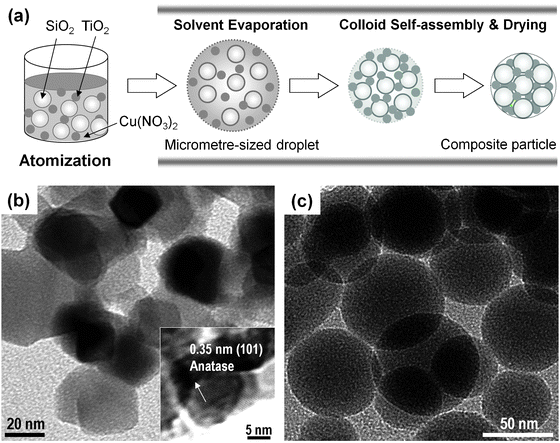 | ||
| Fig. 2 Cu–TiO2–SiO2 composite particle formation. (a) Possible formation mechanism, and TEM images of (b) TiO2 and (c) SiO2 nanocolloids. | ||
The TiO2 and SiO2 nanocolloids were characterized by transmission electron microscopy (TEM) to determine size and morphology. From Fig. 2b, the TiO2 nanocrystals, having an average diameter of 20 nm with an irregular shape, were clearly identified in the anatase phase (see the inset in Fig. 2b); while the SiO2 nanoparticles have an average diameter of 45 nm, which are amorphous with spherical morphology (Fig. 2c). The molar percentages of TiO2 and Cu(NO3)2 to the total components were controlled from 0–100% and 0–2%, respectively. The total precursor concentration was varied from 0.001 to 0.1 M. All chemicals were used as received without further purification. Air was used as the carrier gas with a flow rate of 8 l min−1 (litre per minute at STP = 1.33 × 10−4 m3s−1). The Reynolds number (Re)27 at 800 °C and 8 l min−1, as an example, was calculated to be 183, indicating that the flow pattern inside the reactor was laminar (Re < 2000), without any random eddies, vortices and other flow instabilities. The residence time (τr) in this process is generally very short, and was estimated from the following equation assuming that the temperature distribution inside the reactor is uniform:28
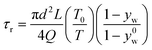 | (1) |
Composite particle characterization
The morphology and size of the composite particles were examined using field emission scanning electron microscopy (FESEM) (JSM-7001FLV, JEOL, Tokyo, Japan). The average diameters were determined by randomly sampling more than 300 particles from FESEM images. The inner structure was analyzed by TEM (JEM-2100F, JEOL, Tokyo, Japan). Before TEM analysis, the as-prepared particles were dried in a vacuum oven for 24 h to eliminate any organic residues inside the mesoporous structure, which were then suspended in ethanol and followed by sonication for 30 min prior to being dropped onto a TEM grid with a lacey support film. The crystal phase was determined by X-ray diffraction (XRD) (Geigerflex D-MAX/A, Rigaku Denki, Tokyo, Japan) using CuKα radiation (λ = 1.548 Å) at 30 kV and 30 mA, with a scan step of 0.02° in 2θ and a scan speed of 4° min−1. The surface area and pore size distribution were analyzed using the nitrogen physisorption method (Autosorb-1, Quantachrome Instruments, Boynton Beach, FL). In total, 79 adsorption and desorption points were analyzed. The surface area was calculated using the Brunauer–Emmett–Teller (BET) method and the pore size distribution was obtained from the desorption isotherm, which is more appropriate than the adsorption isotherm for evaluating the pore size distribution of an adsorbent because for the same volume of gas, the desorption isotherm exhibits a lower relative pressure, resulting in a lower free energy state, i.e., closer to true thermodynamic stability.29 The ultraviolet-visible (UV-VIS) spectra analysis was also performed (Cary 100, Varian, Inc., Palo Alto, CA), to check catalyst particles with different copper doping concentrations.CO2 photoreduction analysis
The photoreduction analysis system is shown in Fig. 1, which includes a home-made continuous flow reactor, a Xe arc lamp (Oriel 66021, Newport Co., Irvine CA), and a gas chromatograph (GC) (6895N, Agilent Technologies, Inc., Santa Clara, CA). Compressed CO2 (Instrument Gr. 4, Airgas, Inc., St. Louis, MO) was used as the source gas and the flow rate was controlled by a mass flow controller (MKS Instruments, Wilmington, MA). It passed through a water bubbler to generate a mixture of CO2 and water vapor. The mixture was then introduced into the reactor, which was cylindrically built with a stainless steel wall and a quartz window vertically facing the solar lighter. The inner cavity was 60 mm in diameter and 25 mm in depth. Glass wool was placed in the reactor as the support for the glass fiber filter loaded with the catalyst particles. The glass wool supporter was moisturized with 3.0 g deionized water to maintain saturated water vapor in the reactor. The catalyst particles (approximately 20 mg) were illuminated for 8 h by the Xe arc lamp which has an average light intensity of 2.4 mW cm−2 for 250 nm < λ < 400 nm measured by a spectroradiometer (ILT-900R, Polytec GmbH, Waldbronn, Germany). The concentrations of effluent gases (e.g.CO and CO2) from the reactor were continuously measured by the GC through an automated gas valve, using helium (He) as the carrier gas. The GC was equipped with a 30 m × 0.32 mm PLOT capillary column (Supelco Carboxen-1010) and a thermal conductivity detector (TCD). Before each test, the reactor loaded with catalysts was first purged with CO2 and water vapor at 100 ml min−1 for 1 h and then the flow rate was reduced and maintained at 3.0 ml min−1 during the whole analysis process. After 30 min when the flow was stabilized, the Xe lamp was turned on and the concentrations of effluent gases as a function of irradiation time were recorded. The maximum concentration of CO gas was used as the CO yield.3. Results and discussion
The effect of synthesis temperature, an important parameter in the FuAR process determining the particle formation,23 was first investigated. The composite particles were synthesized at various temperatures, i.e. from 400 to 1000 °C, by using the same precursor, that is, nanocolloidal TiO2 and SiO2 aqueous suspensions, with a TiO2 molar percentage of 10% and a total precursor concentration of 0.05 M. As shown in the FESEM images in Fig. 3, the catalyst particles obtained at all temperatures had a spherical morphology, which was originated from the droplets whose spherical shape was maintained because of a very low Bond number (much smaller than 1, see details in ESI†).30 | ||
| Fig. 3 FESEM images of TiO2–SiO2 composite particles prepared at different temperatures: (a) 400, (b) 600, (c) 800, and (d) 1000 °C, with a fixed TiO2 molar percentage of 10% and an air flow rate of 8 l min−1. | ||
The average diameters of the particles at each condition are almost similar, about 450 nm, since they were formed from the precursor with the same concentration. According to the typical one droplet to one particle (ODOP) principle, the estimated droplet size was calculated to be around 4 μm using the following equation:31
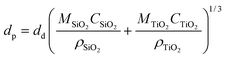 | (2) |
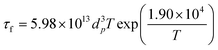 | (3) |
| Synthesis temperature/°C | Residence time/s | Sintering time/s | Specific surface area/m2 g−1 | Pore diameter/nm | Evaporation rate/g s−1 | CO yield (μmol/g TiO2/h) |
|---|---|---|---|---|---|---|
| 400 | 1.63 | 6.65 × 106 | 87.03 | 33.7 | 6.79 × 10−8 | 3.66 |
| 600 | 1.26 | 1.34 × 104 | 82.31 | 32.2 | 1.04 × 10−7 | 4.89 |
| 800 | 1.02 | 2.86 × 102 | 72.38 | 28.3 | 1.50 × 10−7 | 8.24 |
| 1000 | 0.86 | 2.10 × 101 | 70.35 | 23.8 | 1.77 × 10−7 | 6.82 |
Fig. 4 shows the representative TEM images of the particles obtained at 400 °C (Fig. 4a) and 800 °C (Fig. 4b). In Fig. 4a, one can find that the TiO2 nanocrystals are evenly distributed inside the composite particle. Taking a close inspection on the particle surface, for example, in the spots of a1 and a2, it was observed that most particles are spherical SiO2 nanoparticles. Some TiO2 nanocrystals were observed but not in large amounts. The composite is shown as the inset in the right corner of Fig. 4a. However, it is not the case for the particles obtained at 800 °C. In this case, more TiO2 nanocrystals moved to the surface of the composite particle, which were clearly indicated by the magnified TEM images of b1 and b2 spots in Fig. 4b.
 | ||
| Fig. 4 TEM and HR-TEM images of TiO2–SiO2 composite particles prepared at 400 (a) and 800 °C (b) shown in Fig. 3. The insets are schematic diagrams of the composites, in which the white balls represent SiO2 and the black spots indicate TiO2. | ||
The phenomenon was attributed to the droplet evaporation at high temperatures.24,33 In a FuAR route, it has been demonstrated that for a droplet containing colloids with different sizes, colloids with smaller size can move towards the surface of the droplet faster than the larger ones, since smaller particles have higher mobility.33 For example, the TiO2 nanocrystals in this work have a smaller size than that of the SiO2 nanocolloids, which can be expected to move faster than the SiO2 colloids to the droplet surface. The driving force of the movement is the local temperature gradient at the surface of the micrometre-sized droplet created by the solvent evaporation due to the high temperature heating. In general, as a droplet evaporates, a non-uniform temperature distribution, i.e. a temperature drop is established by heat loss due to the phase change from liquid to vapor at the evaporating surface.26,34 For common fluids, such as water, the surface tension increases with a decreasing temperature. As a result of the surface tension gradient as well as the thermophoretic force, the liquid interface is “pulled” toward the colder regions, i.e. the droplet surface. Viscous drag moves the fluid adjacent to the surface, and consequently, a surface-tension-driven Marangoni flow is initiated.35 The solvent evaporation rate can be enhanced at a higher temperature for droplets evaporated at different temperatures, creating a stronger evaporating cooling effect,34 moving more TiO2 nanocrystals to the composite particle surface. Evaporation rates under different synthesis temperatures are shown in Table 1 and are detailed in ESI.†
The above composite particles were also subjected to the physisorption measurement to determine the surface area, pore size, and its distribution. Fig. 5 shows the adsorption and desorption isotherms of the composite particles prepared at 800 °C and 8 l min−1, which can be assigned to be Type V, a typical type for a mesoporous structure. The isotherms also show A type hysteresis, indicating their cylindrical pore structure.29 The corresponding pore size distribution is shown as the inset, which reveals that the average pore size of the composite particles is approximately 28 nm. It should be noted that the mesoporous structure was directly formed in the FuAR process without using any templates avoiding contamination and reducing costs. The surface area and average pore size as a function of synthesis temperature are summarized in Table 1. It is apparent that both surface area and average pore size decrease with increasing synthesis temperature. For example, the surface area and average pore size at 400 °C were 87.03 m2 g−1 and 33.7 nm, respectively; while the corresponding data for 1000 °C were decreased to 70.35 m2 g−1 and 23.8 nm, respectively. The phenomenon was attributed to the shrinkage at high temperatures as explained before.
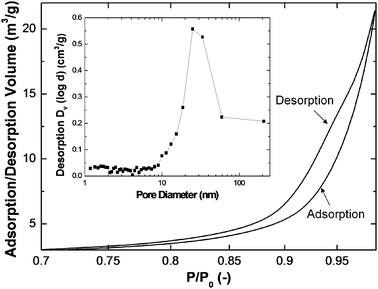 | ||
| Fig. 5 Representative nitrogen adsorption/desorption isotherm of TiO2–SiO2 particles synthesized at 800 °C with a fixed TiO2 molar percentage of 2%. The inset shows the pore size distribution calculated from the desorption isotherm. | ||
The photoreduction analysis was then carried out for these composite particles. The possible pathways for the photoreduction of CO2 by the mesoporous Cu–TiO2–SiO2 particles are schematically shown in Fig. 6. As explained in the Experimental section, the photoreduction of CO2 was conducted in a reactor where a mixture of CO2 and water vapor was presented. The mixture first diffused into the porous structure of the composite particle and then adsorbed on the TiO2 nanocrystal surface. Upon illumination by the UV light, the TiO2 got activated to generate electron–hole pairs for the reaction. The electron–hole pairs were then spatially separated and distributed inside the porous structure to slow down the electron–hole recombination. In addition, Cu doped on the TiO2 surface can trap electrons to separate the pairs and hence enhance the photoreduction efficiency. The photoreduction of CO2 happened in the third step, in which the electrons were transferred to the TiO2 nanocrystal surface to react with CO2 and water vapor to form various products, such as CO, CH4, HCOOH, CH3OH, and even CH3CH2OH. The mechanism of photoreduction of CO2 is quite complex. The reduction of CO2 by one electron to form CO−2 is highly unfavorable, having a reduction potential of −2.14 V vs.SCE.36 Most of the researchers now agree that this process is based on the proton-assisted multi-electron transfer (MET) mechanism (see ESI† for details).36,37 Based on the MET mechanism, although the formation of methane, methanol, and ethanol is thermodynamically favorable, it requires more electrons and protons. These products are more likely produced in CO2 photoreduction in aqueous solutions, since more protons can be supplied by water.12,18 Since this study focused on CO2 reduction on a gas–solid interface, CO was found as the main product in our flow reactor system as it needs a minimum number of electrons and protons. CO also has significant fuel value (ΔcH° = −283.0 kJ mol−1) and can be readily converted into methanol for use as a liquid fuel.38 The CO yield was investigated systematically as a function of synthesis temperature, TiO2 molar percentage, Cu molar percentage, as well as precursor concentration.
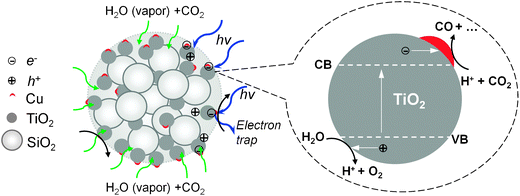 | ||
| Fig. 6 Proposed photoreduction pathways of mesoporous Cu–TiO2–SiO2 composite particles. | ||
Table 1 shows the synthesis temperature effect on the CO yield of TiO2–SiO2 particles with a TiO2 molar percentage of 2%. From the table, it is observed that the CO yield first increased from 3.66 μmol/g TiO2/h at 400 °C to 4.89 μmol/g TiO2/h at 600 °C. After reaching the maximum, i.e. 8.24 μmol/g TiO2/h at 800 °C, it slightly decreased to 6.82 μmol/g TiO2/h at 1000 °C. The increase of CO yield from 400 to 800 °C was due to the increased quantity of TiO2 nanocrystals on the mesoporous SiO2 matrix surface, which was clearly evidenced by the TEM images and explained as shown before. The slightly decreased CO yield at 1000 °C was due to the decrease in the surface area and pore size, as indicated in Table 1. It is believed that the decreased surface area and pore size of the composite catalyst particles would limit the adsorption of the CO2 and water vapor mixture on the catalyst particle surface and hence resulted in a relatively low CO yield. The corresponding XRD patterns of these composite particles were also obtained, from which no significant change in the crystal phase due to the variation of synthesis temperature was observed, implying that the crystal phase had negligible influence on the CO yield (see ESI†, Fig. S1, for details).
The effect of TiO2 molar percentage was investigated as well. Fig. 7a indicates that the CO yield first increased with increasing TiO2 molar percentage. After it reached a maximum value of 8.24 μmol/g TiO2/h at 2%, it decreased significantly. Only 1.51 μmol/g TiO2/h could be achieved at a TiO2 molar percentage of 10%, and an even lower value of 0.1 μmol/g TiO2/h was obtained when using pure TiO2. This was caused by agglomeration of the primary TiO2 nanocrystals during the synthesis. In general, the agglomeration rate increases with increasing particle number concentration.39 For instance, in the case of using 100% TiO2 nanocrystals, the final composite particles are actually agglomerated spheres of individual TiO2 nanocrystals with the average diameter of several hundred nanometres. In the agglomerates, the probability of electron–hole recombination would increase on the interfaces of individual TiO2 nanocrystals. However, in the case of low TiO2 molar percentages, less agglomeration would happen and isolated individual TiO2 nanocrystals would be mainly distributed on the surface of the porous SiO2 matrix, eliminating the surface and volume electron–hole recombination,2 thus enhancing the photoreduction efficiency.
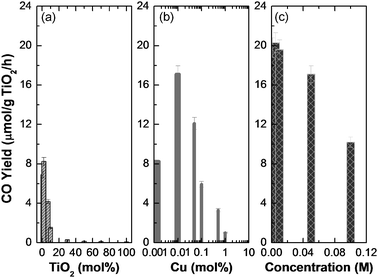 | ||
| Fig. 7 CO yield as a function of various parameters. (a) TiO2 molar percentage (no Cu loading), (b) Cu molar percentage, and (c) total precursor concentration (0.01% Cu loading). The total precursor concentration was maintained at 0.5 M for (a) and (b) and the TiO2 molar percentage was 2% for (b) and (c). The synthesis temperature and air flow rate were fixed at 800 °C and 8 l min−1, respectively. | ||
As demonstrated above, a maximum CO yield, i.e. 8.24 μmol/g TiO2/h was obtained by using an optimal TiO2 molar percentage of 2%. The value, however, is still very low in comparison to the previous research. In order to improve the reduction efficiency, copper doping was carried out to further decrease the electron–hole recombination rate. To check the doping effect, the TiO2 molar percentage and temperature were fixed at 2% and 800 °C. Fig. 7b shows that similar to that of the TiO2 molar percentage effect, there is an optimal Cu molar percentage at 0.01%, in which a high CO yield of 17.1 μmol/g TiO2/h was achieved. After that, the CO yield, however, decreased with increasing Cu molar percentage. It was probably due to the formation of CuO which covered the porous silica surface as well as TiO2, preventing the direct contact of CO2 and TiO2.
To further explore the reduction efficiency, the effect of total precursor concentration was also investigated as shown in Fig. 7c. Based on eqn (3), one can easily know that the particle size increases with increasing precursor concentration, assuming that the droplet size is the same, which was proved by previous research.40 The particle size as a function of total precursor concentration is presented in Table 2. It monotonically increased from 123 nm at 0.001 M to 571 nm at 0.10 M. It is also apparent from Table 2 that the number of individual TiO2 nanocrystals per composite particle increases with increasing total precursor concentration, indicating the increased probability of agglomeration of these individual nanocrystals, which is considered as one of the reasons for deteriorated photoreduction efficiency. The highest CO yield achieved was as high as 20.3 μmol/g TiO2/h at 0.005 M. Besides the agglomeration reason explained above, the specific surface area effect is also another major reason, since the specific surface area of the particles generally increases with decreasing particle size.
| Precursor concentration/M | Particle diameter/nm | N TiO2 a (#/particle) | Specific surface area/m2 g−1 | CO yield (μmol/g TiO2/h) |
|---|---|---|---|---|
| a The numbers of TiO2 colloids in one particle were roughly estimated based on the mass and volume conservation law by neglecting porosity of the particle. The TiO2 molar percentage was fixed at 2% for all above experiments. | ||||
| 0.001 | 123 | 3 | 92.54 | 19.2 |
| 0.005 | 210 | 15 | 88.32 | 20.3 |
| 0.01 | 265 | 31 | 81.71 | 19.6 |
| 0.05 | 453 | 155 | 72.38 | 17.1 |
| 0.1 | 571 | 310 | 65.46 | 10.2 |
To explain the effect of copper doping, UV-VIS spectra were also analyzed for the catalyst particles with different copper doping concentrations (Fig. 8). The TiO2 molar percentage was fixed at 2%, while the copper molar percentages were varied from 0 to 1%. For pure TiO2–SiO2, the absorption peak wavelength was located at around 367 nm, which is slightly shorter than the standard absorption wavelength of bulk anatase, i.e. 387 nm, due to the quantum size confinement (blue shift) of nanosized TiO2. With Cu doping up to 0.1%, there is no significant bandgap shift due to the very low doping concentration. In addition, no absorption peaks in the visible range could be observed for all of the above three cases. However, the absorption in the visible range was observed when increasing Cu molar percentages from 0.1 to 1%. This was due to the formation of CuO (Eb = 1.2 eV) inside the particles, which was the reason for the decreased photoreduction efficiency as explained before. Additional information for CuO formation inside the aerosol reactor from the copper nitrate precursor can be found in the ESI† (Fig. S2).
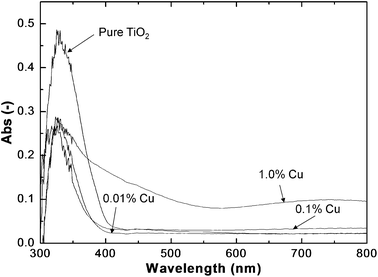 | ||
| Fig. 8 UV-VIS spectra of the as-prepared composite particles with different Cu molar percentages. The particles were obtained with a TiO2 molar percentage of 2% and an air flow rate of 8 l min−1 at 800 °C. | ||
4. Conclusions
Cu–TiO2–SiO2 mesoporous composite particles were successfully synthesized by the FuAR method in a rapid manner. The size, composition, and porosity of the composite particles were tailored by manipulating the precursor concentration, stoichiometric ratio, and temperature profile, respectively. The results revealed that the as-prepared particles were submicrometre-sized mesoporous spheres having optimal molar percentages of TiO2 and Cu to the whole particle of 2% and 0.01%, respectively. The TiO2 nanocrystals were efficiently utilized by selectively distributing them on the SiO2 mesoporous matrix surface by evaporation driven self-assembly, resulting in a relatively high CO2 conversion efficiency with a maximal CO yield of approximately 20 μmol/g TiO2/h. It should be noted that these catalyst particles were obtained within merely several seconds, much faster than those obtained by time/energy consuming wet-chemical methods. The mesoporous structure was directly formed in the FuAR process without any templates and post-treatment, thus avoiding contamination and reducing cost.Acknowledgements
The work was supported by the Consortium for Clean Coal Utilization at Washington University in St. Louis. Partial support from the Jens Endowment and AAQRL is gratefully acknowledged.References
- M. Halmann and M. Steinberg, Greenhouse Gas Carbon Dioxide Mitigation: Science and Technology, Lewis Publishers, Boca Raton, FL, 2000 Search PubMed.
- P. Usubharatana, D. McMartin, A. Veawab and P. Tontiwachwuthikul, Ind. Eng. Chem. Res., 2006, 45, 2558–2568 CrossRef CAS.
- B. Metz, O. Davidson, H. de Coninck, M. Loos and C. Meyer, IPCC Special Report on Carbon Dioxide Capture and Storage, New York, 2005 Search PubMed.
- C. S. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- M. Kitano, M. Matsuoka, M. Ueshima and M. Anpo, Appl. Catal., A, 2007, 325, 1–14 CrossRef CAS.
- V. P. Indrakanti, J. D. Kubicki and H. H. Schobert, Energy Environ. Sci., 2009, 2, 745–758 RSC.
- M. Anpo, H. Yamashita, Y. Ichihashi, Y. Fujii and M. Honda, J. Phys. Chem. B, 1997, 101, 2632–2636 CrossRef CAS.
- M. Anpo, H. Yamashita, K. Ikeue, Y. Fujii, S. G. Zhang, Y. Ichihashi, D. R. Park, Y. Suzuki, K. Koyano and T. Tatsumi, Catal. Today, 1998, 44, 327–332 CrossRef CAS.
- H. Yamashita, M. Honda, M. Harada, Y. Ichihashi, M. Anpo, T. Hirao, N. Itoh and N. Iwamoto, J. Phys. Chem. B, 1998, 102, 10707–10711 CrossRef CAS.
- K. Ikeue, H. Yamashita, M. Anpo and T. Takewaki, J. Phys. Chem. B, 2001, 105, 8350–8355 CrossRef CAS.
- M. Ogawa, K. Ikeue and M. Anpo, Chem. Mater., 2001, 13, 2900–2904 CrossRef CAS.
- I. H. Tseng, W. C. Chang and J. C. S. Wu, Appl. Catal., B, 2002, 37, 37–48 CrossRef CAS.
- T. V. Nguyen and J. C. S. Wu, Sol. Energy Mater. Sol. Cells, 2008, 92, 864–872 CrossRef CAS.
- M. Anpo, S. G. Zhang, H. Mishima, M. Matsuoka and H. Yamashita, Catal. Today, 1997, 39, 159–168 CrossRef CAS.
- H. Yamashita, Y. Fujii, Y. Ichihashi, S. G. Zhang, K. Ikeue, D. R. Park, K. Koyano, T. Tatsumi and M. Anpo, Catal. Today, 1998, 45, 221–227 CrossRef CAS.
- Y. J. Xu, T. F. Xie, D. J. Wang, Y. Wang and T. J. Li, Chem. J. Chin. Univ., 1999, 20, 461–463 CAS.
- W. Y. Lin, H. X. Han and H. Frei, J. Phys. Chem. B, 2004, 108, 18269–18273 CrossRef CAS.
- X. H. Xia, Z. H. Jia, Y. Yu, Y. Liang, Z. Wang and L. L. Ma, Carbon, 2007, 45, 717–721 CrossRef CAS.
- Y. Li, W. N. Wang, Z. L. Zhan, M. H. Woo, C. Y. Wu and P. Biswas, Appl. Catal., B, 2011, 100, 386–392.
- S. Basak, K. S. Rane and P. Biswas, Chem. Mater., 2008, 20, 4906–4914 CrossRef CAS.
- W. N. Wang, W. Widiyastuti, T. Ogi, I. W. Lenggoro and K. Okuyama, Chem. Mater., 2007, 19, 1723–1730 CrossRef CAS.
- W. N. Wang, F. Iskandar, K. Okuyama and Y. Shinomiya, Adv. Mater., 2008, 20, 3422–3426 CrossRef CAS.
- W. N. Wang, Y. Kaihatsu, F. Iskandar and K. Okuyama, Chem. Mater., 2009, 21, 4685–4691 CrossRef CAS.
- F. Iskandar, L. Gradon and K. Okuyama, J. Colloid Interface Sci., 2003, 265, 296–303 CrossRef CAS.
- D. Sen, S. Mazumder, J. S. Melo, A. Khan, S. Bhattyacharya and S. F. D'Souza, Langmuir, 2009, 25, 6690–6695 CrossRef CAS.
- S. T. Chang and O. D. Velev, Langmuir, 2006, 22, 1459–1468 CrossRef CAS.
- R. B. Bird, W. E. Stewart and E. N. Lightfoot, Transport Phenomena, John Wiley & Sons, Inc., New York, 2002 Search PubMed.
- T. T. Kodas and M. Hampden-Smith, Aerosol Processing of Materials, Wiley-VCH, New York, 1999 Search PubMed.
- S. Lowell, J. E. Shields, M. A. Thomas and M. Thommes, Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density, Springer, Dordrecht, The Netherlands, 2006 Search PubMed.
- O. D. Velev, A. M. Lenhoff and E. W. Kaler, Science, 2000, 287, 2240–2243 CrossRef CAS.
- W. N. Wang, W. Widiyastuti, I. W. Lenggoro, T. O. Kim and K. Okuyama, J. Electrochem. Soc., 2007, 154, J121–J128 CrossRef CAS.
- W. Widiyastuti, S. Y. Lee, F. Iskandar and K. Okuyama, Adv. Powder Technol., 2009, 20, 318–326 Search PubMed.
- F. Iskandar, H. W. Chang and K. Okuyama, Adv. Powder Technol., 2003, 14, 349–367 Search PubMed.
- S. P. Fisenko, W. N. Wang, I. W. Lenggoro and K. Okyuama, Chem. Eng. Sci., 2006, 61, 6029–6034 CrossRef CAS.
- J. J. Hegseth, N. Rashidnia and A. Chai, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1996, 54, 1640–1644 CrossRef CAS.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS.
- A. H. Yahaya, M. A. Gondal and A. Hameed, Chem. Phys. Lett., 2004, 400, 206–212 CrossRef CAS.
- T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2132–2133 CrossRef CAS.
- S. K. Friedlander, Smoke, Dust, and Haze: Fundamental of Aerosol Dynamics, Oxford University Press, New York, 2000 Search PubMed.
- W. N. Wang, A. Purwanto, I. W. Lenggoro, K. Okuyama, H. Chang and H. D. Jang, Ind. Eng. Chem. Res., 2008, 47, 1650–1659 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c0cy00091d |
| This journal is © The Royal Society of Chemistry 2011 |