Received
5th November 2010
, Accepted 19th January 2011
First published on 16th February 2011
1. Introduction
Chlorinated aromatic compounds are organic wastes and are harmful to the human body. These chemical substances are found in nature as persistent organic pollutants. Chlorinated aromatic compounds are generally treated by incineration or biodegradation. Since these methods aim to mineralize the chlorinated aromatic compounds, a large amount of carbon dioxide (CO2) is produced. In addition, we cannot recover and re-use aromatic rings of the original chlorinated aromatic compounds by using these methods. Therefore, recovery of aromatics by dechlorination of chlorinated aromatic compounds is one of the promising methods for treatment of chlorinated compounds from the viewpoints of the environment and chemical resources. Although catalytic dechlorination of chlorinated aromatic compounds has been examined under various conditions,1–5 most of the methods used required specific reaction conditions such as high temperatures, high pressure and the presence of molecular hydrogen (H2). Recently, it has been reported that aromatic compounds were recovered from various chlorinated aromatic compounds under ordinary pressure by a catalytic process in a 2-propanol–sodium hydroxide (NaOH) system.3–5 However, elevated temperatures were still required under these conditions. On the other hand, photocatalytic dechlorination of chlorinated compounds has attracted attention because photocatalytic reaction occurs under mild conditions (at room temperature and under atmospheric pressure) and can utilize solar energy. Since strong oxidation power of holes can be used, photocatalytic oxidation of chlorinated compounds has been studied widely. However, selective recovery of a dechlorinated product(s) has been difficult.6,7 Although reductive treatment of chlorinated aromatic compounds by heterogeneous photocatalytic reaction has been reported, the recovery of dechlorinated aromatic compounds was insufficient.8–11 Previous studies on photocatalytic reaction have suggested that strong and selective electron injection into chlorinated aromatic compounds and effective removal of chloride ions from the reaction system are important for quantitative recovery of aromatics from chlorinated aromatic compounds. Based on the above considerations, we previously examined photocatalytic reductive dechlorination of chlorobenzene in a 2-propanol suspension of metal-loaded titanium(IV) oxide (TiO2) nanocrystals in the presence of dissolved NaOH and briefly reported that benzene and chloride ions were recovered quantitatively when palladium (Pd)-loaded titanium(IV) oxide (TiO2) nanocrystals were used as the photocatalyst.12 If the oxidation process in dechlorination of chlorobenzene is also effectively utilized for production of valuable compounds, dechlorination of chlorobenzene becomes more valuable. Dechlorination of chlorobenzene without the use of corrosive compounds is preferable for practical use. Here we show stoichiometric formation of benzene and ketones by photocatalytic dechlorination of chlorobenzene in secondary alcohol suspensions of palladium-loaded titanium(IV) oxide powder in the presence of sodium ion (Na+) sources.
2. Experimental
2.1 Preparation of photocatalysts
All of the reagents were commercial materials of reagent grade and used without further purification. Nanocrystalline TiO2 powder was prepared at 573 K using the HyCOM method.13 Titanium(IV) butoxide was used as the starting material and the product was calcined at 823 K. The HyCOM-TiO2 powder (148.5 mg) was suspended in 80 vol% aqueous solution of acetic acid containing palladium(II) acetate in a test tube. The test tube was sealed with a rubber septum under Ar and then photoirradiated at a wavelength λ > 300 nm by a 400 W high-pressure mercury arc (Eiko-sha, Osaka, Japan) with continuous magnetic stirring in a water bath kept at 298 K. The Pd source was reduced by photogenerated electrons and the Pd metal was deposited on HyCOM-TiO2 particles, resulting in the formation of Pd-loaded HyCOM-TiO2 (Pd–TiO2). The resultant powder was washed with acetone and distilled water and then dried at 310 K overnight under air. For comparison, HyCOM-TiO2 samples were calcined at various temperatures and commercial TiO2 samples were also used as TiO2 photocatalysts and loading of Pd was conducted using the photodeposition method. Various metals (Cu, Ag, Ru, Rh, Pt and Au) other than Pd were also deposited on HyCOM-TiO2 particles using the same method.
2.2 Characterization
XRD patterns of the TiO2 samples were recorded using a Rigaku Multi Flex (Carbon monochromator, CuKα, 40 V–30 mA). Diffraction patterns were collected from 20° to 70°. Specific surface areas of the TiO2 samples were determined using the BET single-point method on the basis of nitrogen uptake measured at 77 K.
Photocatalytic dechlorination of chlorobenzene was carried out in a 2-propanol (or 2-butanol) suspension of Pd-loaded TiO2 powder in the presence of dissolved NaOH. A small amount of NaOH dissolved in the solvent previously dried with molecular sieves was used to remove chloride ions eliminated from chlorobenzene as sodium chloride (NaCl) because of the low solubility of NaCl in 2-propanol or 2-butanol. Pd-loaded TiO2 powder (50 mg) was suspended in 2-propanol (or 2-butanol) (5 cm3) containing NaOH (220 μmol) in a test tube. After the mixture had been bubbled with argon and sealed with a rubber septum, chlorobenzene (98 μmol) was injected into the mixture. The test tube was photoirradiated in the same way as that for photodeposition. During photoirradiation, the test tube was set in a water bath kept at 298 K to avoid thermal reaction. For comparison, a commercial LTA-type zeolite containing Na+ (molecular sieves 4 Å) (200 mg) was also used as an Na+ source other than NaOH. The amount of H2 in the gas phase was measured using a Shimadzu GC-8A gas chromatograph equipped with an MS-5A column. The amounts of benzene and chlorobenzene in the liquid phase were determined with a Shimadzu GC-14B gas chromatograph equipped with a DB-1 capillary column (30 m × 0.25 mm). The amount of acetone in the liquid phase was determined with a Shimadzu GC-14A gas chromatograph equipped with a fused silica capillary column (HiCap-CBP20, 25 m × 0.22 mm). Toluene was used as an internal standard sample, and the amounts of benzene, chlorobenzene and acetone were determined using the ratios of the peak areas of benzene, chlorobenzene and acetone to the peak area of toluene. The reaction solution (1 cm3) was added to a diethyl ether/water mixture (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 v/v, 3 cm3). After the mixture had been stirred for 10 min, benzene, chlorobenzene and acetone in the ether phase were analyzed with Shimadzu GC-14B and GC-14A.
∶1 v/v, 3 cm3). After the mixture had been stirred for 10 min, benzene, chlorobenzene and acetone in the ether phase were analyzed with Shimadzu GC-14B and GC-14A.
3. Results and discussion
3.1 Effect of metal loading onto TiO2 on photocatalytic dechlorination of chlorobenzene
Since chloride ions were not observed in the liquid phase (in 2-propanol solution) after photoirradiation, water (2 cm3) was added to the reaction mixture and the resultant liquid phase was analyzed using an ion chromatograph. Fig. 1 shows the effect of loaded metal (1.0 wt%) on the yield of chloride ions eliminated from chlorobenzene in a 2-propanol suspension in the presence of dissolved NaOH. For comparison, results for a bare TiO2 sample are also shown in Fig. 1. When a bare TiO2 sample was used, no chloride ions and no other products were observed, indicating that neither hole scavenging by 2-propanol nor electron scavenging by chlorobenzene occurred under the present conditions. When Cu-, Ag-, Ru-, Rh-, Pt- and Pd-loaded TiO2 samples were used under the reaction conditions, chloride ions were observed in the liquid phase after addition of water. Since NaOH was soluble in 2-propanol and NaCl was not, these results indicate that chloride ions eliminated from chlorobenzene were solidified with NaOH as NaCl, i.e., chloride ions were removed from the present photocatalytic reaction system. These results also indicate that these metals loaded on TiO2 particles worked as co-catalysts (reduction sites) in photocatalytic dechlorination of chlorobenzene. The extent of dechlorination increased in the order of loaded metals: Ag ≈ Cu < Pt < Ru ≪ Rh ≈ Pd. It should be noted that Rh- and Pd-loaded TiO2 samples showed extraordinarily high levels of activity in photocatalytic dechlorination of chlorobenzene. The hydrogen over-voltage (HOV) of Pd and Rh electrodes is slightly higher than that of Pt and much lower than that of Ag and Cu, suggesting that a slightly higher HOV, moderately suppressing hydrogen formation,14 is important for selective reduction of chlorobenzene. However, Au did not show any effects on photocatalytic dechlorination of chlorobenzene. From these results, it can be concluded that Rh and Pd were efficient co-catalysts for reductive dechlorination of chlorobenzene. Here we focus on only Pd–TiO2 and details of Rh–TiO2 will be reported separately.
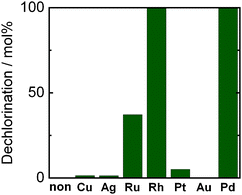 |
| | Fig. 1 Dechlorination in photocatalytic reaction of chlorobenzene in a 2-propanol suspension of 1.0 wt% metal-loaded HyCOM-TiO2 in the presence of dissolved NaOH (220 μmol) under UV light irradiation for 30 min. | |
| | | (CH3)2CHOH + 2h+ → (CH3)2CO + 2H+ | (1) |
Therefore, in the present system, chlorobenzene was effectively reduced by photogenerated electrons as shown in eqn (2).| | | PhCl + H+ + 2e− → PhH + Cl− | (2) |
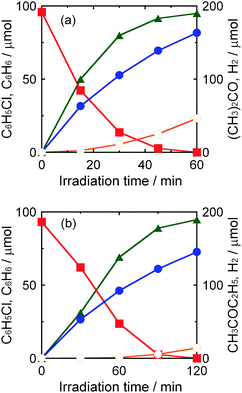
As mentioned in the previous section, chloride ions were not observed in the liquid phase (in 2-propanol solution). Water (2 cm3) was added to the reaction mixture after the reaction and the amount of chloride ions in the resultant liquid phase was determined using an ion chromatograph. The amount of chloride ions (98 μmol) was in good agreement with that of benzene determined by GC, indicating that chlorobenzene was completely dechlorinated, resulting in the formation of benzene and chloride ions. The photocatalytic reaction shown in Fig. 2(a) is expressed as eqn (3).
| | | PhCl + (CH3)2CHOH + NaOH → PhH + (CH3)2CO + NaCl ↓ + H2O | (3) |
Chloride ions eliminated from
chlorobenzene were immediately solidified as
NaCl by
NaOH dissolved in
2-propanol and separated from other products such as
benzene and
acetone dissolved in
2-propanol. Isolation of
chloride from the reaction system suppressed consumption of
chloride by other side reactions that caused insufficient stoichiometry in
dechlorination.
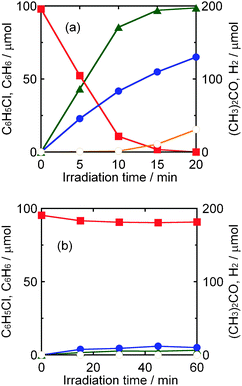 |
| | Fig. 2 Time courses of the amounts of chlorobenzene (squares), benzene (triangles), acetone (closed circles) and H2 (open circles) in a 2-propanol suspension of 1.0 wt% Pd–TiO2 in the presence of dissolved NaOH (220 μmol) under (a) UV light irradiation and (b) dark conditions. | |
Although chlorobenzene was almost completely consumed at 15 min, the yield of acetone still increased and a small amount of H2 formed after 15 min (Fig. 2(a)). These results indicate that hole scavenging by 2-propanol (eqn (1)) still continued and that photogenerated electrons were used to reduce H+ as shown in eqn (4).
Formation of H
2 is totally expressed as
eqn (5):
| | | (CH3)2CHOH → (CH3)2CO + H2. | (5) |
From these results, it can be concluded that photogenerated electrons were almost selectively used for
dechlorination of
chlorobenzene just before its consumption in the present system.
Acetone can be further oxidized by holes, resulting in the formation of CO
2; however, CO
2 was not observed at any reaction time. Since a large excess of
2-propanol was present in this reaction system, the rate of
acetone oxidation was probably much smaller than that of
2-propanol oxidation.
Since an almost stoichiometric material balance among the amounts of consumed chlorobenzene and formed benzene and chloride ions was confirmed, the redox balance (ROB), i.e., ratio of the amount of consumed photogenerated electrons to that of consumed holes, in this reaction system was defined in eqn (6):
| | | ROB = [n(benzene) + n(H2)]/n(acetone), | (6) |
where
n(benzene),
n(H
2) and
n(acetone) are yields of
benzene, H
2 and
acetone, respectively. The value of ROB at 10 min, at which time only
dechlorination of
chlorobenzene occurred (no H
2 formation), was determined to be 1.04, indicating that photocatalytic
reduction (
dechlorination of
chlorobenzene to
benzene) and photocatalytic
oxidation of
2-propanol to
acetone took place with an ROB close to unity. The values of ROB at 15 and 20 min were determined to be 0.98 and 1.00, respectively. The values of ROB close to unity indicate that both of the photocatalytic
redox reactions shown in
eqn (3) and (5),
i.e.,
dechlorination of
chlorobenzene and formation of H
2, occurred with a high stoichiometry without side reaction(s) and formation of byproduct(s).
3.3 Effect of the amount of Pd loading on photocatalytic dechlorination of chlorobenzene
Fig. 3 shows the effect of the amounts of Pd on the yield of benzene in photocatalytic dechlorination of chlorobenzene in 2-propanol containing NaOH under irradiation of UV light, in which Pd–TiO2 (50 mg) samples having different Pd loadings (0.1, 0.5 and 1.0 wt%) were used. The rate of benzene formation increased with increase in the amount of Pd loaded. When 0.5 wt% and 1.0 wt% of Pd were loaded on TiO2, chlorobenzene was completely dechlorinated to benzene within 60 min. The reductive dechlorination of chlorobenzene to benzene was promoted even when a very small amount of Pd such as 0.1 wt% was supported.
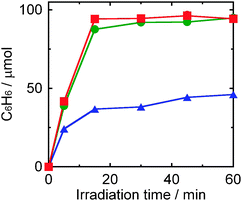 |
| | Fig. 3 Effect of loading amounts of Pd (0.1 wt%: triangles, 0.5 wt%: circles, 1.0 wt%: squares) on the yield of benzene in photocatalytic dechlorination of chlorobenzene (98 μmol) in a 2-propanol suspension of Pd–TiO2 (50 mg) in the presence of dissolved NaOH (220 μmol) under UV light irradiation. | |
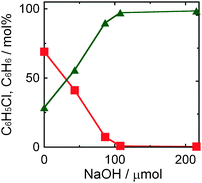
3.4 Effect of the amount of NaOH on yield of benzene in photocatalytic dechlorination of chlorobenzene in a 2-propanol suspension of 1.0 wt% Pd–TiO2
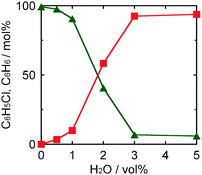
Fig. 4 shows the effect of the amount of NaOH on yield of benzene in photocatalytic dechlorination of chlorobenzene in a 2-propanol suspension of 1.0 wt% Pd–TiO2 under irradiation of UV light for 30 min. Although dechlorination of chlorobenzene occurred in the absence of NaOH, the yield of benzene was low. Time courses of photocatalytic dechlorination of chlorobenzene in the absence of NaOH were shown in Fig. S1 (ESI†). The reaction rate was small and chlorobenzene remained even after UV irradiation for 3 h. In addition, the yield of chloride ions was lower than that of benzene. These results suggest that chloride ions eliminated from chlorobenzene were used for side-reaction(s). The reaction rate and benzene yield were improved with maintenance of a high material balance by increasing the amount of NaOH up to ca. 100 μmol. The benzene yield was constant at higher concentrations of NaOH because almost 100% conversion of chlorobenzene had been achieved at ca. 100 μmol. These results indicate that reductive dechlorination of chlorobenzene in 2-propanol was promoted by addition of a small amount of NaOH and that chlorobenzene can be almost completely dechlorinated only in the presence of NaOH, satisfying the stoichiometry shown in eqn (3). Therefore, it should be noted that removal of chloride ions from the reaction system, i.e., solidification of chloride, avoiding consumption of chloride by other reactions, is important for effective recovery of benzene from chlorobenzene in the photocatalytic process.
3.5 Effects of the physical properties of TiO2 on photocatalytic dechlorination of chlorobenzene to benzene
Effects of the physical properties of various TiO2 samples on the yield of benzene in photocatalytic dechlorination of chlorobenzene in a 2-propanol suspension of 1.0 wt% Pd–TiO2 containing NaOH under irradiation of UV light were investigated. The XRD patterns of various TiO2 samples are shown in Fig. 5. Table 1 shows the specific surface areas and crystal structures of various TiO2 samples and the yield of benzene using 1.0 wt% Pd–TiO2. Although dechlorination of chlorobenzene to benzene occurred in all TiO2 samples, the benzene yield was low when rutile TiO2 samples were used. On the other hand, a relatively high benzene yield was obtained when TiO2 samples containing the anatase phase were used. When HyCOM TiO2 samples calcined at 723 K, 823 K and 1073 K were used, almost complete recovery of benzene from chlorobenzene was achieved within 15 min. Since the conduction band of rutile TiO2 is smaller than that of anatase TiO2, reduction activity of rutile TiO2 is thought to be inferior to that of anatase TiO2.
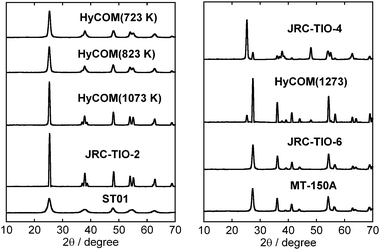 |
| | Fig. 5 XRD patterns of HyCOM-TiO2 samples calcined at 723 K, 823 K, 1073 K and 1273 K and various commercial TiO2 samples. | |
Table 1 Physical properties of various TiO2 samples and benzene yield in photocatalytic dechlorination of chlorobenzene (98 μmol) in a 2-propanol suspension of 1.0 wt% Pd–TiO2 in the presence of dissolved NaOH (220 μmol) under irradiation of UV light
| TiO2 |
T
cal
/K |
S
BET
/m2 g−1 |
Crystal structurec |
Reaction time/min |
Benzene yield/mol% |
|
Calcination temperature.
BET surface area.
A: anatase-type TiO2, R: rutile-type TiO2.
|
| HyCOM |
723 |
115 |
A |
15 |
>99 |
| HyCOM |
823 |
101 |
A |
15 |
>99 |
| HyCOM |
1073 |
39 |
A |
15 |
>99 |
| HyCOM |
1273 |
8 |
A, R |
20 |
19 |
| JRC-TIO-2 |
— |
10 |
A |
20 |
28 |
| JRC-TIO-4 |
— |
37 |
A, R |
20 |
70 |
| JRC-TIO-6 |
— |
70 |
R |
20 |
43 |
| JRC-TIO-8 |
— |
219 |
A |
20 |
98 |
| MT-150A |
— |
77 |
R |
20 |
36 |
Effects of specific surface areas of various anatase TiO2 samples on the initial rate of benzene formation are shown in Fig. 6. The rate increased with increase in specific surface area and the rate reached a maximum (9.9 mol% min−1) at 115 m2g−1. These results indicate that control of physical properties of anatase-type TiO2, i.e., balance of specific surface area and crystallinity of anatase TiO2, is important for effective recovery of benzene from chlorobenzene in photocatalytic reductive dechlorination.
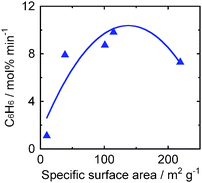 |
| | Fig. 6 Effects of specific surface areas of various anatase-type TiO2 samples on the initial rate of benzene formation in photocatalytic dechlorination of chlorobenzene (98 μmol) in a 2-propanol suspension of 1.0 wt% Pd–TiO2 in the presence of dissolved NaOH (220 μmol) under irradiation of UV light. | |
| | | CH3CH(OH)C2H5 + 2h+ → CH3COC2H5 + 2H+ | (7) |
Butanone is used widely in various chemical industries. Therefore, photocatalytic dechlorination of chlorobenzene using 2-butanol solvent is significant if butanone is obtained in addition to benzene. Photocatalytic dechlorination of chlorobenzene using 2-butanol solvent in the presence of dissolved NaOH is expected to proceed as shown in eqn (8).| | | PhCl + CH3CH(OH)C2H5 + NaOH → PhH + CH3COC2H5 + NaCl↓ + H2O | (8) |
Fig. 8 shows time courses of photocatalytic dechlorination of chlorobenzene in a 2-butanol suspension of 1.0 wt% Pd–TiO2 containing NaOH. Photocatalytic reductive dechlorination of chlorobenzene using 2-butanol solvent also exhibited high activity of chlorobenzene dechlorination in the early stage of the reaction. However, the rate of benzene production gradually decreased with time and complete dechlorination was not achieved. Slightly lower mass balance was also observed. Although dechlorination of chlorobenzene was almost saturated at 30 min, butanone and H2 were still produced after 30 min. These results indicate that photogenerated electrons were preferentially consumed for reduction of H+, i.e., Pd–TiO2 was not deactivated. Since water produced in this reaction (eqn (8)) probably increased the solubility of NaCl in the liquid phase, dechlorination of chlorobenzene was inhibited and chloride ions eliminated from chlorobenzene were used for side-reaction(s). Therefore, removal of product water is important for complete dechlorination of chlorobenzene and efficient recovery of both products, benzene and butanone, in 2-butanol.
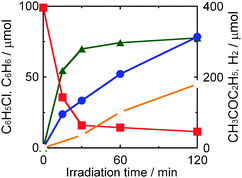 |
| | Fig. 8 Time courses of the amounts of chlorobenzene (squares), benzene (triangles), butanone (closed circles) and H2 (open circles) in a 2-butanol suspension of 1.0 wt% Pd–TiO2 in the presence of dissolved NaOH (220 μmol) under UV light irradiation. | |
Fig. 9(b) shows the time courses of photocatalytic dechlorination of chlorobenzene in a 2-butanol suspension of 1.0 wt% Pd–TiO2 containing the Na+-containing LTA-type zeolite (200 mg) under irradiation of UV light. It should be noted that complete dechlorination of chlorobenzene was achieved within 120 min in 2-butanol solvent in contrast to the reaction using NaOH as the Na+ source (Fig. 8). High stoichiometry of the reduced and oxidized products was also attained by using Na+-containing LTA-type zeolite. These results indicate that the Na+-containing LTA-type zeolite effectively worked as a Na+ source to solidify chloride ions. Photocatalytic reactions using the Na+-containing LTA-type zeolite (Na-zeolite) shown in Fig. 9 proceed as shown in eqn (9).
| | | PhCl + (CH3)2CHOH (or CH3CH(OH)C2H5) + Na-zeolite → PhH + (CH3)2CO (or CH3COC2H5) + (Na, Cl, H)-zeolite | (9) |
According to
eqn (9),
water does not form in contrast to the reaction using
NaOH (
eqn (8)). Therefore, the decrease in reaction rate due to
water formation and release of
chloride ions to the liquid phase were avoided by the use of the
Na+-containing LTA-type
zeolite. Replacement of corrosive
NaOH with the
Na+-containing LTA-type
zeolite in this reaction system is useful for practical application. Since we have no information on where
NaCl is fixed, we are now characterizing the solid phase.
4. Conclusions
Photocatalytic dechlorination of chlorobenzene (98 μmol) in alcohol suspensions of metal-loaded TiO2 in the presence of Na+ sources under irradiation of UV light was examined. When 1.0 wt% palladium (Pd) (or rhodium), 2-propanol and NaOH were used as the metal co-catalyst, alcohol and Na+ source, respectively, dechlorination of chlorobenzene was promoted by addition of a small amount of NaOH. The reaction rate and benzene yield were improved with maintenance of a high material balance by increasing the amount of NaOH, and benzene was obtained almost quantitatively in a very short time at ca. 100 μmol. Chloride ions eliminated from chlorobenzene were immediately solidified as NaCl by NaOH dissolved in 2-propanol and separated from other products such as benzene and acetone dissolved in 2-propanol. Isolation of chloride from the reaction system suppressed consumption of chloride by other side reactions that caused insufficient stoichiometry in dechlorination. Effects of the physical properties of TiO2 and water addition on photocatalytic dechlorination of chlorobenzene were examined, and it was found that the reaction rate reached a maximum at a specific surface area of TiO2 of 115 m2 g−1 and that the reaction rate monotonously decreased with increase in the amount of water. When 2-butanol was used as an alcohol solvent, a more useful oxidized product, i.e., butanone, was successfully obtained in addition to benzene as the reduction product in photocatalytic dechlorination of chlorobenzene. To avoid formation of water in dechlorination of chlorobenzene in 2-propanol and 2-butanol, a Na+-containing LTA-type zeolite (molecular sieves 4 Å) was used as the Na+ source, and almost quantitative recovery of benzene was achieved along with stoichiometric formation of the corresponding ketones.
Acknowledgements
One of the authors (H. K.) is grateful for financial support from the Faculty of Science and Engineering, Kinki University.
References
- R. B. Lapierre, L. Guczi, W. L. Kranich and A. H. Weiss, J. Catal., 1978, 52, 230 CrossRef CAS.
- T. Hara, T. Kaneta, K. Mori, T. Mitsudome, T. Mizugaki, K. Ebitani and K. Kaneda, Green Chem., 2007, 9, 1246 RSC.
- Y. Ukisu and T. Miyadera, Appl. Catal., A, 2004, 271, 165 CrossRef CAS.
- Y. Ukisu and T. Miyadera, J. Hazard. Mater., 2005, 122, 1 CrossRef CAS.
- Y. Ukisu, J. Hazard. Mater., 2008, 152, 287 CrossRef CAS.
- S. Moroi, K. Hirano, Y. Asami and R. Takagi, Electrochemistry, 1989, 57, 1207 CAS.
- P. Kluson, M. Drobek, S. Krejcikova, J. Krysa, A. Kalaji, T. Cajthaml and J. Rakusan, Appl. Catal., B, 2008, 80, 321 CrossRef CAS.
- H. Yin, Y. Wada, T. Kitamura and S. Yanagida, Environ. Sci. Technol., 2001, 35, 227 CrossRef CAS.
- H. Yin, Y. Wada, T. Kitamura, T. Sakata, H. Mori and S. Yanagida, Chem. Lett., 2001, 334 CrossRef CAS.
- N. Serpone, I. Texier, A. V. Emeline, P. Pichat, H. Hidaka and J. Zhao, J. Photochem. Photobiol., A, 2000, 136, 145 CrossRef CAS.
- H. Park and W. Choi, J. Phys. Chem. B, 2004, 108, 4086 CrossRef CAS.
- K. Fuku, K. Hashimoto and H. Kominami, Chem. Commun., 2010, 46, 5118 RSC.
- H. Kominami, M. Kohno, Y. Takada, M. Inoue, T. Inui and Y. Kera, Ind. Eng. Chem. Res., 1999, 38, 3925 CrossRef CAS.
- H. Kominami, A. Furusho, S.-Y. Murakami, H. Inoue, Y. Kera and B. Ohtani, Catal. Lett., 2001, 76, 31 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c0cy00040j |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.