Non-covalent immobilization of asymmetric organocatalysts
Long
Zhang
,
Sanzhong
Luo
* and
Jin-Pei
Cheng
Beijing National Laboratory for Molecular Sciences (BNLMS), CAS Key Laboratory of Molecular Recognition and Function, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: luosz@iccas.ac.cn; Fax: +86-10-82613774; Tel: +86-10-62554446
First published on 8th March 2011
Abstract
The immobilization of organocatalysts is one of the most explored approaches to overcome the limitations associated with organocatalytic systems such as high catalyst loading, difficult product separation and catalyst recycling. When compared with the commonly used covalent immobilization methods, which usually involve tedious synthetic manipulations, the non-covalent attachment of organocatalysts to supports offers facile and modular construction of immobilized chiral catalysts with maintained or even improved activity and stereoselectivity. This review summarizes the successful application of non-covalent interactions, such as acid–base interaction, ion–pair interaction, hydrophobic interaction and so on, in assembling recoverable and reusable organocatalysts.
 Long Zhang | Long Zhang graduated from Nankai University in 2005 with a major of chemistry. He spent five years studying for a Ph.D in a joint project between Nankai University and the Institute of Chemistry, Chinese Academy of Sciences (ICCAS) under the supervision of Prof. Jin-Pei Cheng and Prof. Sanzhong Luo. Afterwards, he joined Prof. Luo's group as a research assistant in July, 2010. His work focuses on de novo design and synthesis of functionalized chiral ionic liquids and their applications as asymmetric organocatalysts. |
 Sanzhong Luo | Sanzhong Luo graduated from Zhengzhou University in 1999, and then spent his graduate studies at Nankai University, the Chinese Academy of Sciences and the Ohio State University and received his PhD under the supervision of Prof. Jin-Pei Cheng in 2005. He has been at the Institute of Chemistry, Chinese Academy of Sciences (ICCAS) since 2005. He was a visiting scholar at Stanford University in 2009. He won the National Science Fund for Distinguished Young Scholars in 2010. His research is to develop novel, viable and reliable asymmetric catalysts and asymmetric synthesis of natural products. |
 Jin-Pei Cheng | Jin-Pei Cheng received his PhD from Northwestern University under the supervision of Prof. F. G. Bordwell in 1987. He then spent one year as a postdoctoral fellow at Duke University. Since 1989, he has been a full professor at Nankai University. In 2001, he was elected as a fellow of the Chinese Academy of Sciences. He is an adjunct professor at ICCAS since 2002. His research interests include physical organic chemistry, chemistry of organic hydrides and NO donors, and asymmetric catalysis. |
Introduction
The past decade has witnessed a renaissance of organocatalysis in the field of asymmetric catalysis.1 Compared with the well-established asymmetric transition metal catalysts and natural enzymes, organocatalysts are readily available, much easier to handle and free of metal contaminations which are problematic in pharmaceutical processes. Consequently, organocatalytic reactions are frequently deemed to be environmentally-benign and sustainable processes. Notwithstanding these attractive features, organocatalysts have been seldom applied in industry,2 a fact that can be largely ascribed to their insufficient efficiency and the difficulties in catalyst separation and recycling.3 One viable approach to address these challenges would be the heterogenization of organocatalysts. Indeed, catalyst immobilization has been widely explored with asymmetric transition metal catalysts and enzymes aiming to improve their applicability and practicability. Similarly, the immobilization strategy has also been frequently attempted for organocatalysis even before its renaissance in 2000s.4 Typically, the immobilization of organocatalysts has been achieved via covalent attachment onto solid supports such as polystyrene, poly(ethylene glycol) (PEG), dendrimers and inorganic solids.5 However, most of these supported organocatalysts demonstrated reduced activity and selectivity comparing with their small molecular parent catalysts, consequently higher catalyst loading (both w/w% and mol%) is required to attain reasonable yields. In addition, multiple synthetic manipulations are necessary to achieve the covalent immobilization and significant structural perturbations to the parent catalyst skeletons are generally not avoidable in such circumstances.With their multiple catalytic moieties residing in rather small skeletons, small structural modifications of organocatalysts may lead to serious deterioration of their catalytic behaviours. It becomes even more challenging when connecting those privileged organocatalysts onto large solid supports. Therefore, an ideal catalyst immobilization strategy should provide (1) minimal synthetic modification and structural perturbations to the parent catalysts, and (2) a facile catalyst linkage that allows for combinatorial screening of (3) a synergistic support to achieve optimal activity and selectivity. To this end, non-covalent immobilization strategies recently appeared as attractive solutions with their powers being well-demonstrated in asymmetric transition metal catalysis.6 There has also been increasing examples of supported organocatalysts via non-covalent strategies starting to show their potentials in this field. This perspective summarizes the major progresses regarding the development of non-covalently supported asymmetric organocatalysts since 2005, with a focus on the immobilization methods, their advantages, applications and limitations. The classification of this review is based on different modes of immobilization, such as acid–base, ion–pair, hydrophobic as well as self-assembled gel-type organocatalysts. The classic biphasic immobilization of organocatalysts is also incorporated in this review owing to their non-covalent features.
1 Acid–base immobilization
Acid–base assembly of chiral amines has been proven to be one of the most efficient bifunctional amine catalysts.7 The acids used in these examples were essential units that dramatically impacted the catalytic activity and stereoselectivity. Taking advantage of the acid–base principle, Luo and Cheng developed a non-covalent immobilization strategy for chiral amines by utilizing solid acids.8 Similar solid acid–base strategy has previously been successfully applied in attaching transition-metal catalysts onto solid supports with good activity and reusability.9 The acid–base immobilization of chiral amine organocatalysts is distinct from these previous reports as a result of the dual functions of the solid-acids: first, they act as an anchor for the chiral diamines and second they act as a critical modulator for the catalytic activity and stereoselectivity.In Luo's study, the main solid acids they used were polyoxometalates (POMs) and polystyrene sulfonic acids. POMs belong to a large family of metal-oxide clusters, best-known for their strong acidity and redox properties. The POM-supported organocatalyst could be realized by simply mixing the two components in certain ratio and a plethora of readily available chiral diamines could be immobilized on POMs without any additional modifications (Scheme 1). The non-covalent feature of acid–base interactions allows for screening different POMs, chiral amines and their combinations for optimal catalysts. Several thus identified optimal catalysts are listed in Scheme 1. These hybrid solids have biphasic properties and are soluble in polar organic solvents such as acetone, DMP, DMF and DMSO, but insoluble in less polar solvents like hexane, toluene and diethyl ether. Another interesting feature is that they are well dispersed in water. Optical microscopy shows uniform micelle-like aggregates with 0.4 μm mean diameter (Scheme 1, bottom), rendering these hybrid solids potential asymmetric surfactant-type catalysts in aqueous media.
 | ||
| Scheme 1 POM immobilized chiral amines and their suspension in water. (Left: optical microscopy of 1a; right: SEM picture of 3.) | ||
The POM supported secondary–tertiary diamine catalyst 1a was found to catalyze the direct aldol reaction of ketones effectively7a,b (Scheme 2, eqn (1)). The catalyst loading could be reduced to less than 0.33 mol% (1 mol% chiral amine loading), such a low loading is extremely rare in enamine-based organocatalysts, particularly for reusable ones. The reaction could be conducted in both neat and wet conditions. Under neat condition, the catalyst could be recycled by precipitation with the addition of diethyl ether. When aqueous condition was used, the aqueous phase containing the catalyst after product extraction could be used directly in the next run. In both cases, the catalysts could be reused for six runs with similar enantioselectivity but slightly reduced activity. POM hybrid catalyst 1b with H4SiW12O40 as the solid support could also catalyze the asymmetric Michael addition of ketones to nitrostyrenes under aqueous condtions7b (Scheme 2, eqn (2)).
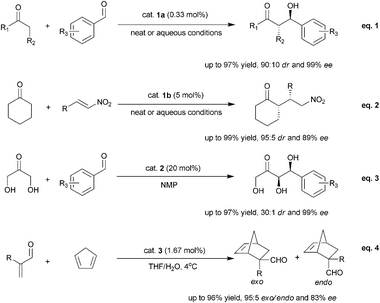 | ||
| Scheme 2 POM supported organocatalyst catalyzed reactions. | ||
The POM supported primary–tertiary diamine catalyst 2 showed very good catalytic activities in the direct aldol reactions of α-hydroxyketones with 20 mol% loading7c (Scheme 2, eqn (3)). Up to 97% yield, 99% ee and 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 syn diastereoselectivity could be obtained. The catalyst could be reused up to four times with slightly decreased activity and selectivity in the third and fourth run.
:1 syn diastereoselectivity could be obtained. The catalyst could be reused up to four times with slightly decreased activity and selectivity in the third and fourth run.
Unlike catalyst 1 and 2, a multidentate chiral amine was employed in catalyst 3.7d It was found that self-assembled catalyst 3 could efficiently catalyze the Diels–Alder reactions of α-substituted acroleins under aqueous condition (Scheme 2, eqn (4)). The Diels–Alder products were obtained with up to 96% yield, 95:5 exo/endo ratio and 83% ee. In addition, the catalyst could be easily recycled and reused six times with slightly reduced activity and selectivity.
In 2008, Luo and Cheng reported the PS-sulfonic acid supported diamine catalysts via acid–base strategy7e (Scheme 3). A series of PS-sulfonic acid with different acid loading was synthesized and used in the immobilization. Surprisingly, the one with medium loading gave the best catalytic results in the direct aldol reaction. In their study, the PS-sulfonic acid supported primary–tertiary diamine catalyst 4a and 4b showed great capability in direct aldol reactions, while the PS-sulfonic acid supported secondary–tertiary diamine catalyst 5 was preferred in Michael addition reactions of nitrostyrenes. The catalyst can be recovered via filtration and reused up to 6 rounds. Notably, the deactivated catalyst can be reactivated via an acid-washing/amine-recharge protocol.
 | ||
| Scheme 3 Polystyrene supported organocatalysts via acid–base interaction. | ||
Recently, Li and co-workers reported a SO3H-hollow nanosphere immobilized primary–tertiary diamine catalyst and evaluated its catalytic capability in direct aldol reactions10 (Scheme 4, left). Although good yields and selectivities could be obtained, the recyclability was rather poor for obvious loss of activity was observed in the third and fourth runs. Itsuno and co-workers also reported a series of sulfonic acid functionalized polymer immobilized MacMillan catalysts for the Diels–Alder reactions of cinnamaldeyhde11 (Scheme 4, right). However, both the catalytic activity and selectivity was inferior to the non-immobilized catalyst.
 | ||
| Scheme 4 Other solid sulfonic acids supported organocatalysts. | ||
2 Ion–pair immobilization
Ion–pair strategy probably represents one of the most early explored non-covalent immobilization methods due to the ubiquitous nature of ion pairs in many catalytic processes.12 In a broad sense, acid–base immobilization would belong to the ion–pair category as described here. Therefore, we will limit this section to those immobilized catalysts prepared via typical ion–exchange processes. In this regard, the section is further classified according to the different ionic support such as ionic polymer, clays and ionic liquids.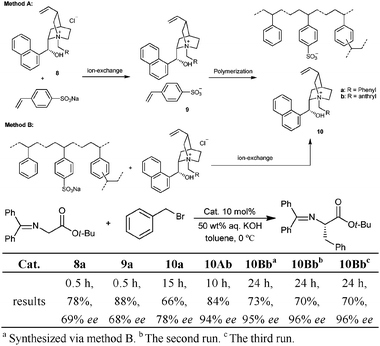 | ||
| Scheme 5 Methods for non-covalent immobilization of quaternary ammonium salts. | ||
Besides the side–chain immobilized organocatalysts mentioned above, Itsuno and co-workers also developed a main–chain functionalized polymer via ion–pair interactions 16 (Scheme 6), i.e., self-assembly of a bis-quaternary ammonium salts 11 and disulfonate salts 12 to form a polymeric ions, reminiscent of self-supported catalysts developed by Ding et al.17 Mixing aqueous solution of 12 with powdered 11 led to the formation of polymeric ionic 13, for which NMR spectra and inherent viscosity ([η] 0.1–0.2) serve as direct proofs of a polymeric structure. The catalytic ability of this polymer was also tested in the benzylation of N-diphenylmethylene gylcine tert-butyl ester, showing again increased enantioselectivity but loss of activity. Catalyst 13 can be easily recycled and reused with no loss of activity and enantioselectivity in the second and third runs. It remains unclear how the polymeric ion–pair participates in the phase transfer process.
 | ||
| Scheme 6 Main–chain functionalized polymers. | ||
 | ||
| Scheme 7 Montmorillonite immobilized organocatalysts. | ||
Chiral anionic organocatalysts have also been shown to promote the transformations such as Michael addition reaction and cyanosilylation reaction.21 One conceivable way to immobilize this type of catalysts would be ion–exchange with cationic supports. Layered double hydroxide (LDH) appears as such a type of inorganic solid support. LDH consists of stacks of positively charged metal hydroxide layers and interlayer anions. Both inorganic and organic anions could be entrapped into the interlayer. In 2002, Choudary and co-workers reported the synthesis of LDH entrapped proline via anion–exchange method22 (Scheme 8, left). LDH-supported proline 17 was found to promote several transformations including aldol reaction and Michael addition reaction with good activity but low enantioselectivity. Nakayama reported a calcination–reconstruction method for the immobilization of amino acids including L-proline onto LDH,23 but no catalytic performance was presented. Using this method, He and co-workers synthesized the LDH supported proline 1824 (Scheme 8, right) and examined its catalytic ability in asymmetric direct aldol reactions. Surprisingly, the direct aldol reaction of acetone and benzaldehyde proceeded extremely well in the presence of catalyst 18 with up to 94% enantioselectivity, standing in sharp contrast with that of catalyst 17. The reason for the different performances of 17 and 18 is not very clear, but may be ascribed to the intrinsic structure discrepancy caused by different preparation methods. Later, Pitchumani and co-workers prepared a similar catalyst like He's and examined its catalytic ability in the asymmetric Michael addition, showing low to moderate enantioselectivity.25
 | ||
| Scheme 8 LDH immobilized proline catalyzed aldol reactions. | ||
 | ||
| Scheme 9 Proline anion type IL catalyzed asymmetric reactions. | ||
The supported ionic liquids (SIL) could also be used as an cationic supports for organocatalysts. In 2007, You and co-workers reported the synthesis of a polymer supported ionic liquid 21 bearing proline anions and studied their applications in metal scavenging and heterogeneous metal catalysis30 (Scheme 10, left). However, their potential application in organocatalytic process was initially ignored. Recently, Salunkhe and co-workers reported a similar supported ionic liquid 22 containing proline anion31 (Scheme 10, right). SIL 22 was shown to promote the Mannich type reaction of 2-naphthol with good activity but no enantioselectivity. The supported ionic liquid catalyst could be easily recycled and reused without any significant loss in chemical yield of the products.
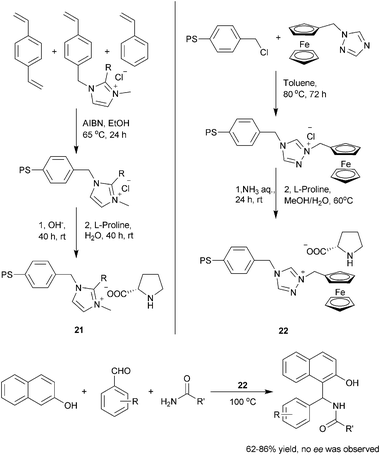 | ||
| Scheme 10 SIL supported L-proline via ion–pair interactions. | ||
3 Immobilization via hydrophobic interaction
The marriage of supramolecular principles and systems with organocatalytic motifs has recently been shown to viable and powerful strategy to evolve new type of asymmetric supramolecular catalysts on one hand32,33 and to enable facile catalyst recycling and reuse on the other hand. For the latter purpose, Zhang and co-workers utilized the well known cyclodextrin host and its inner hydrophobic cavity to immobilize organocatalysts via hydrophobic effect34 (Scheme 11, 23). In their study, the apolar phenyl ring of 4-phenoxyproline served as the inclusion handle toward the β-cyclodextrin cavity. In the presence of 10 mol% of the immobilized catalysts, the direct aldol reaction of acetone and o-nitrobenzaldehyde was completed in 16 h with 90% yield and 83% ee. The enantioselectivity is slightly improved compared with that of the parent unsupported catalyst, suggesting the host–guest assembly is synergistic for catalysis. The assembled catalyst 23 could be recovered by filtration and employed for three subsequent runs with unchanged enantioselectivity and a slightly decreased yields, a result comparable with PEG-supported proline.35 Using the same principle, Armstrong and co-workers later reported a similar catalytic system 2436 (Scheme 11, 24), wherein a sulfated β-CD was employed as the host and tert-butylphenyl serves as the inclusion handle. Meanwhile, Woggon and co-workers utilized the adamantyl group as the including handle and also realized the immobilization of proline via β-CD binding37 (Scheme 11, 25). The catalytic activity and selectivity are similar with catalyst 24.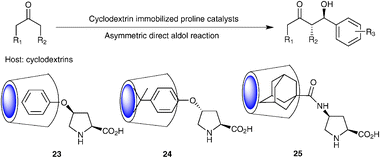 | ||
| Scheme 11 Cyclodextrins immobilized proline catalysts. | ||
4 Self-supported gel-type organocatalysts
Supramolecular gels are nanoscalic aggregates of small organic molecules formed via intermolecular non-covalent interactions such as solvophobic, ionic, H-bonding, van der Waals, π–π interactions.38 The functional applications of gels have been a recent research focus. In this regard, the use of a supramolecular gel as asymmetric organocatalyst can be traced back to 1990, when Inoue and co-workers reported asymmetric cyanide addition catalyzed by a cyclodipeptide in the gel state.39 However, subsequent studies suggested that gel-nature of the catalyst was a “significant obstacle” to elucidate the catalytic processes. The aggregation of small molecular catalyst was later considered as a problem to be avoided in many examples.40. It is not until very recently that the work by Escuder and Miravet shed new light on supramolecular gel-type asymmetric organocatalysts.41 In fact, the heterogeneous nature of the gel was found to be an additional advantageous feature that allows easy recovery and recycling of the catalyst. Importantly, the highly ordered structures of a gel could result in enhanced catalytic efficiency associated with the aligned cooperative catalytic units or with conformational restrictions.Miravet, Escuder and co-workers successfully developed a series of organic gel systems 26 and 27 (Scheme 12) and examined their catalytic properties in enamine-based transformations. The gels 26a–c were found to effectively promote Henry reactions as a result of the enhanced basicity caused by cooperation of neighbouring proline moieties in the gel. In sharp comparison, the non-aggregated catalysts in solution showed no catalytic activity (Scheme 13).41d The aggregated status of organocatalyst may also impact the stereocontrol due to the aggregation-driven conformational change around the catalytic moieties. For example, in the asymmetric Michael addition catalyzed by 26, a reversal of enantioselectivity was observed with 27b when going from solution phase to the aggregated gel status (Scheme 14).

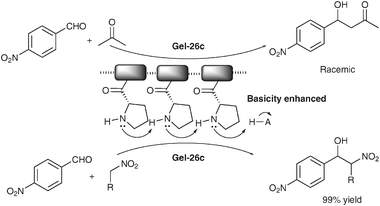 | ||
| Scheme 13 Supramolecular gel catalyzed aldol and Henry reactions. | ||
 | ||
| Scheme 14 Supramolecular gels catalyzed Michael addition reaction. | ||
In another study, the gelator 27b was found to form an almost transparent hydrogel in water.41g The direct aldol reaction between cyclohexanone and 4-nitrobenzaldehyde was tested in the hydrogel phase (Scheme 15), the reagents dissolved in toluene were easily loaded onto the gel and the reaction was quantitatively completed after 24 h at 5 °C with high stereoselectivity (anti:syn = 92:8, 88% ee). Meanwhile, the catalytic hydrogel could be reused after decantation of the toluene phase for at least three times with the same efficiency and stereoselectivity. Interestingly, gel 26a–c in acetonitrile showed good activity but only poor enantioselectivity in the same direct aldol reaction,41c indicating dramatic solvent effect in gel catalysis. Provided with its tunable, reversible and modular features, the potential of organocatalytic gelators as self-supported catalysts remains to be further explored.
 | ||
| Scheme 15 Hydrogel catalyzed aldol reaction. | ||
5 Biphasic immobilization
The environmentally benign solvents such as ionic liquids, PEGs, etc. have been widely used in organic reactions to replace volatile organic solvents.42 They are also widely explored in the biphasic immobilization of organocatalysts due to their polar natures. One of the most investigated organocatalyst is the ionic proline catalyst. As early as 2002, Loh and Toma independently reported the use of an ionic liquid media for proline catalyzed aldol reactions.43,44 In both cases, they have found that the reactions in imidazolium ionic liquids such as [BMIM]PF6 gave comparable or even higher enantioselectivities than traditional DMSO solvent.45 Ionic liquids such as [BMIM]BF446 and guanidine-derived ionic liquids47 are also suitable solvent for the direct aldol reactions with generally better yield and enantioselectivity in guanidine type ionic liquid (Scheme 16). Other reactions such as cross-aldol reaction between aldehydes48 and asymmetric α-aminoxylations49,50 could also be performed in similar reaction systems (Scheme 17). The proline derivatives such as L-prolinamide (28),51 bis(prolinamide)cyclohexane (29),52 secondary-tertiary diamine (30)53 and chiral zwitterions (31)54 could also be immobilized in IL with good catalysts' reusability (Scheme 18). In most of the catalytic system mentioned above, the catalyst could be recycled and reused for at least 3 times. In a typical recycle procedure, the reaction mixture is extracted with organic solvents such as diethyl ether, the highly polarized ionic liquids and organocatalysts, which have low solubility in the extracting solvents, will be separated from the products. After removal of the residual organic solvents, the catalysts are directly used in the next cycle.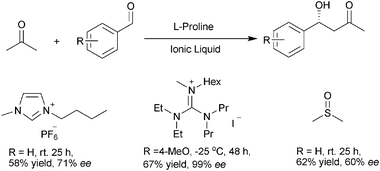 | ||
| Scheme 16 Ionic liquid as phase supports for L-proline catalyzed aldol reactions. | ||
 | ||
| Scheme 17 Ionic liquid as phase supports for L-proline catalyzed cross-aldol reactions and α-aminoxylations. | ||
 | ||
| Scheme 18 Other immobilized organocatalysts in ILs. | ||
In biphasic catalysis in ionic liquids, the leaching of catalyst, contamination of products in ionic liquids, as well as the high cost of ionic liquids, are issues to be further addressed. One of the explored solutions is to use solid surface supported ionic liquid monolayer as the catalyst immobilization phase with or without additional ionic liquid.55 In 2004, Gruttadauria and co-workers covalently attached an ionic liquid moiety to the surface of silica gel56,57 (Scheme 19). This monolayer was then treated with more ionic liquid before being used to immobilize proline for aldol reactions. The ionic layer acted as the reaction media and enabled the easy recovery of catalysts upon filtration. Up to 8 recycles could be reached and the catalyst could be regenerated by recharging with fresh proline. In 2007, this methodology was extended to other linkers and to peptide organocatalysts.58,59 Recently, a non-covalently supported ionic liquid on SiO2 bed was used for MacMillan catalysts immobilization60 with good reusability for asymmetric Diels–Alder cycloaddition (Scheme 20). Another interesting immobilization system is the (S)-proline/polyelectrolyte system,61 where the proline catalyst was immobilized on the surface of solid poly(diallyldimethylammonium) salts, however, the catalyst could only be reused for twice.
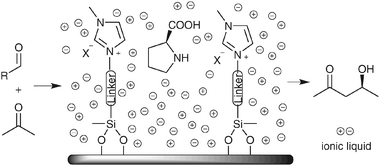 | ||
| Scheme 19 Supported ionic liquids for proline immobilization and their application in aldol reactions. | ||
 | ||
| Scheme 20 Supported ionic liquids for imidazolidinone immobilization. | ||
Liquid poly(ethylene glycol)s (PEGs) could also be used as phase immobilization supports. Chandrasekhar and co-workers reported the utilization of low-weight poly(ethylene glycol) (PEG MW = 400 Da) as immobilization phase for the proline catalyzed aldol reactions.62,63 The catalytic system proved to be as efficient as DMSO system and could be reused for more than ten times with similar enantioselectivity. Luo and Cheng also reported the PEG-200 as reaction media for the zwitterions catalyzed aldol reactions.54 Taking advantage of the cation coordination ability of PEGs,64 Xu and co-workers developed a host–guest complex of PEG with organocatalysts containing a positive charge unit65 (Scheme 21), Asymmetric Michael addition reactions of unmodified ketones to nitroalkenes were tested, showing up to 97% yield and 99% enantioselectivity. A series of spectroscopy studies verified the existence of host–guest complex.66
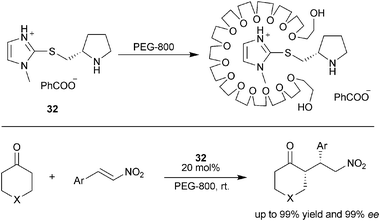 | ||
| Scheme 21 PEGs as phase immobilization supports. | ||
Flouro phase has also been well explored in biphasic catalysis,67 however, further modification of the organocatalysts were usually needed, thus these will not be discussed here.
Summary and outlook
The examples covered in this perspective illustrate the powers of non-covalent strategies for evolving more practical supported organocatalysts. Intriguing features about non-covalent immobilization of organocatalyst include minimal modifications of the parent catalysts, facile catalyst linkage and combinatorial flexibility for the identifications of a optimal supported catalyst. It's even possible for de-novo design and discovery of a new organocatalyst itself by employing the non-covalent strategies.7 Although still early in their developments, the non-covalent immobilization is anticipated to provide a viable solution to enhance the applications of organocatalysts with “practical” credentials. Future catalysts development should be more oriented toward real problems in synthetic processes. It would be also interesting to see the explorations of other non-covalent interactions such as charge-transfer,68 π–π stacking immobilization69 and adsorption and entrapment in nanosized materials70 in developing supported organocatalysts.Acknowledgements
This work was supported by the Natural Science Foundation of China (NSFC 20632060, 20972163 and 21025208), the Ministry of Science and Technology (MoST) of China (2011CB808600) and the Chinese Acedemy of Sciences.Notes and references
- (a) Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis, ed. A. Berkessel and H. Gröger, Wiley-VCH, Weinheim, 2005 Search PubMed.
- For a recent example, see: F. Xu, Michael Zacuto, Naoki Yoshikawa, Richard Desmond, Scott Hoerrner, T. Itoh, M. Journet, G. R. Humphrey, C. Cowden, N. Strotman and P. Devine, J. Org. Chem., 2010, 75, 7829–7841 Search PubMed.
- (a) H.-U. Blaser and E. Schmidt, in Asymmetric Catalysis on Industrial Scale, ed. H.-U. Blaser and E. Schmidt, Wiley-VCH, Weinheim, Germany, 2004, pp. 1–19 Search PubMed; (b) H.-U. Blaser, B. Pugin and F. Spindler, J. Mol. Catal. A: Chem., 2005, 231, 1 CrossRef CAS; (c) H.-U. Blaser, Chem. Commun., 2003, 293 RSC.
- (a) Handbook of Asymmetric Heterogeneous Catalysis, ed. K. Ding and Y. Uozumi, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) Chiral Catalysts Immobilization and Recycling, ed. D. E. De Vos, I. F. J. Vankelecom, P. A. Jacobs, Wiley-VCH, Weinheim, Germany, 2000 Search PubMed; (c) K. Kondo, T. Yamano and K. Takemoto, Makromol. Chem., 1985, 186, 1781 CrossRef CAS.
- (a) T. E. Kristensen and T. Hansen, Eur. J. Org. Chem., 2010, 3179 Search PubMed; (b) M. Gruttadauria, F. Giacalone and R. Noto, Chem. Soc. Rev., 2008, 37, 1666 RSC; (c) F. Cozzi, Adv. Synth. Catal., 2006, 348, 1367 CrossRef CAS; (d) M. Benaglia, New J. Chem., 2006, 30, 1525 RSC; (e) M. Benaglia, A. Puglisi and F. Cozzi, Chem. Rev., 2003, 103, 3401 CrossRef CAS.
- (a) C. Saluzzo and M. Lemaire, Adv. Synth. Catal., 2002, 344, 915 CrossRef CAS; (b) J. Horn, F. Michalek, C. C. Tzschucke and W. Bannwarth, Top. Curr. Chem., 2004, 242, 43 CAS; (c) R. Van de Coevering, R. J. M. K. Gebbink and G. Van Koten, Prog. Polym. Sci., 2005, 30, 474 CrossRef CAS; (d) M. Heitbaum, F. Glorius and I. Escher, Angew. Chem., Int. Ed., 2006, 45, 4732 CrossRef CAS; (e) A. F. Trindade, P. M. P. Gois and C. A. M. Afonso, Chem. Rev., 2009, 109, 418 CrossRef CAS; (f) J. M. Fraile, J. I. García and J. A. Mayoral, Chem. Rev., 2009, 109, 360 CrossRef CAS.
- For a review, see: (a) S. Saito and H. Yamamoto, Acc. Chem. Res., 2004, 37, 570 CrossRef CASfor selected examples, see: (b) M. Nakadai, S. Saito and H. Yamamoto, Tetrahedron, 2002, 58, 8167 CrossRef CAS; (c) T. Ishii, S. Fujioka, Y. Sekiguchi and H. Kotsuki, J. Am. Chem. Soc., 2004, 126, 9558 CrossRef CAS; (d) N. Mase, K. Watanabe, H. Yoda, K. Takabe, F. Tanaka and C. F. Barbas III, J. Am. Chem. Soc., 2006, 128, 4966 CrossRef CAS; (e) N. Mase, Y. Nakai, N. Ohara, H. Yoda, K. Takabe, F. Tanaka and C. F. Barbas III, J. Am. Chem. Soc., 2006, 128, 734 CrossRef CAS; (f) S. V. Pansare and K. Pandya, J. Am. Chem. Soc., 2006, 128, 9624 CrossRef CAS; (g) D. Gryko, M. Zimnicka and R. Lipinski, J. Org. Chem., 2007, 72, 964 CrossRef CAS; (h) S. Luo, H. Xu, J. I. L. Zhang and J.-P. Cheng, J. Am. Chem. Soc., 2007, 129, 3073; (i) S. Luo, H. Xu, J. Li, L. Zhang, X. Mi, X. Zheng and J.-P. Cheng, Tetrahedron, 2007, 63, 11307 CrossRef CAS; (j) S. Luo, H. Xu, L. Zhang, J. Li and J.-P. Cheng, Org. Lett., 2008, 10, 653 CrossRef CAS; (k) S. Luo, H. Xu, L. Chen and J.-P. Cheng, Org. Lett., 2008, 10, 1775 CrossRef CAS; (l) S. Luo, Y. Qiao, L. Zhang, J. Li, X. Li and J.-P. Cheng, J. Org. Chem., 2009, 74, 9521 CrossRef CAS; (m) J. Li, N. Fu, X. Li, S. Luo and J.-P. Cheng, J. Org. Chem., 2010, 75, 4501 CrossRef CAS; (n) S. Luo, P. Zhou, J. Li and J.-P. Cheng, Chem.–Eur. J., 2010, 16, 4457 CrossRef CAS.
- (a) S. Luo, J. Li, H. Xu, L. Zhang and J.-P. Cheng, Org. Lett., 2007, 9, 3675 CrossRef CAS; (b) J. Li, S. Hu, S. Luo and J.-P. Cheng, Eur. J. Org. Chem., 2009, 132 CrossRef CAS; (c) J. Li, S. Luo and J.-P. Cheng, J. Org. Chem., 2009, 74, 1747 CrossRef CAS; (d) J. Li, X. Li, P. Zhou, L. Zhang, S. Luo and J.-P. Cheng, Eur. J. Org. Chem., 2009, 4486 CrossRef CAS; (e) S. Luo, J. Li, L. Zhang, H. Xu and J.-P. Cheng, Chem.–Eur. J., 2008, 14, 1273 CrossRef CAS.
- For a concept article, see: (a) P. Barbaro, Chem.–Eur. J., 2006, 12, 5666 CrossRef CASfor a recent example, see: (b) A. Michrowsak, K. Mennecke, U. Kunz, A. Kirschning and K. Grela, J. Am. Chem. Soc., 2006, 128, 13261 CrossRef CAS.
- J. Gao, J. Liu, S. Bai, P. Wang, H. Zhong, Q. Yang and C. Li, J. Mater. Chem., 2009, 19, 8580 RSC.
- N. Haraguchi, Y. Takemura and S. Itsuno, Tetrahedron Lett., 2010, 51, 1205 CrossRef CAS.
- M. Mazzei, W. Marconi and M. Riocci, J. Mol. Catal., 1980, 9, 381 CrossRef CAS.
- For early studies on immobilized quanternary ammonium salts, see: (a) E. Chiellini and R. Solaro, J. Chem. Soc., Chem. Commun., 1977, 231 RSC; (b) S. Colonna, R. Fornasier and U. Pfeiffer, J. Chem. Soc., Perkin Trans. 1, 1978, 8 RSC; (c) N. Kobayashi and K. Iwai, Makromol. Chem. Rapid Commun., 1981, 2, 105 CrossRef CAS; (d) P. Hodge, E. Khoshdel and J. Waterhouse, J. Chem. Soc., Perkin Trans. 1, 1983, 2205 RSC; (e) R. Chinchilla, P. Mazón and C. Nájera, Tetrahedron: Asymmetry, 2000, 11, 3277 CrossRef; (f) R. Chinchilla, P. Mazón and C. Nájera, Adv. Synth. Catal., 2004, 346, 1186 CrossRef CAS; (g) B. Thierry, J. C. Plaquevent and D. Cahard, Tetrahedron: Asymmetry, 2001, 12, 983 CrossRef CAS; (h) B. Thierry, T. Perrard, C. Audouard, J. C. Plaquevent and D. Cahard, Synthesis, 2001, 1742 CrossRef CAS; (i) Q. Shi, Y. J. Lee, H. Song, M. Cheng, S. S. Jew, H. G. Park and B. S. Jeong, Chem. Lett., 2008, 37, 436 CrossRef CAS; (j) B. Thierry, J. C. Plaquevent and D. Cahard, Tetrahedron: Asymmetry, 2003, 14, 1671 CrossRef CAS; (k) T. Danelli, R. Ammunziata, M. Benaglia, M. Cinquini, F. Cozzi and G. Tocco, Tetrahedron: Asymmetry, 2003, 14, 461 CrossRef CAS; (l) X. Wang, L. Yin, T. Yang and Y. Wang, Tetrahedron: Asymmetry, 2007, 18, 108 CrossRef CAS.
- (a) Y. Arakawa, N. Haraguchi and S. Itsuno, Tetrahedron Lett., 2006, 47, 3239 CrossRef CAS; (b) Y. Arakawa, A. Chiba, N. Haraguchi and S. Itsuno, Adv. Synth. Catal., 2008, 350, 2295 CrossRef.
- Y. Arakawa, N. Haraguchi and S. Itsuno, Angew. Chem., Int. Ed., 2008, 47, 8232 CrossRef CAS.
- S. Itsuno, D. K. Paul, M. A. Salam and N. Haraguchi, J. Am. Chem. Soc., 2010, 132, 2864 CrossRef CAS.
- (a) Z. Wang, G. Chen and K. Ding, Chem. Rev., 2009, 109, 322 CrossRef CAS; (b) K. Ding, Z. Wang, X. Wang, Y. Liang and X. Wang, Chem.–Eur. J., 2006, 12, 5188 CrossRef CAS.
- T. Mitsudome, K. Nose, T. Mizugaki, K. Jitsukawa and K. Kaneda, Tetrahedron Lett., 2008, 49, 5464 CrossRef CAS.
- N. Hara, S. Nakamura, N. Shibata and T. Toru, Adv. Synth. Catal., 2010, 352, 1621 CrossRef CAS.
- V. Srivastava, K. Gaubert, M. Pucheault and M. Vaultier, ChemCatChem, 2009, 1, 94 Search PubMed.
- (a) M. Yamaguchi, T. Shiraishi and M. Hirama, Angew. Chem., Int. Ed. Engl., 1993, 32, 1176 CrossRef; (b) M. Yamaguchi, T. Shiraishi and M. Hirama, J. Org. Chem., 1996, 61, 3520 CrossRef CAS; (c) M. Yamaguchi, T. Shiraishi, Y. Igarashi and M. Hirama, Tetrahedron Lett., 1994, 35, 8233 CrossRef CAS; (d) M. Yamaguchi, Y. Igarashi, R. S. Reddy, T. Shiraishi and M. Hirama, Tetrahedron, 1997, 53, 11223 CrossRef CAS; (e) Y. Xiong, Y. Wen, F. Wang, B. Gao, X. Liu, X. Huang and X. Feng, Adv. Synth. Catal., 2007, 349, 2156 CrossRef CAS; (f) X. Liu, B. Qin, X. Zhou, B. He and X. Feng, J. Am. Chem. Soc., 2005, 127, 12224 CrossRef CAS; (g) M. Hatano, T. Ikeno, T. Matsumura, S. Torii and K. Ishihara, Adv. Synth. Catal., 2008, 350, 1776 CrossRef CAS; (h) T. Darbre and M. Machuqueiro, Chem. Commun., 2003, 1090 RSC.
- B. M. Choudary, B. Kavita, N. S. Chowdari, B. Sreedhar and M. L. Kantam, Catal. Lett., 2002, 78, 373 CrossRef CAS.
- H. Nakayama, N. Wada and M. Tsuhako, Int. J. Pharm., 2004, 269, 469 CrossRef CAS.
- Z. An, W. Zhang, H. Shi and J. He, J. Catal., 2006, 241, 319 CrossRef CAS.
- S. Vijaikumar, A. Dhakshinamoorthy and K. Pitchumani, Appl. Catal., A, 2008, 340, 25 CrossRef CAS.
- For selected reviews, see: (a) B. Ni and A. D. Headley, Chem.–Eur. J., 2010, 16, 4426 CrossRef CAS; (b) S. Luo, L. Zhang and J.-P. Cheng, Chem.–Asian J., 2009, 4, 1184 CrossRef CAS; (c) P. D. de Maria, Angew. Chem., Int. Ed., 2008, 47, 6960 CrossRef; (d) R. Šebesta, I. Kmentová and S. Toma, Green Chem., 2008, 10, 484 RSC; (e) K. Bica and P. Gaertner, Eur. J. Org. Chem., 2008, 3235 CrossRef CAS.
- S. Hu, T. Jiang, Z. Zhang, A. Zhu, B. Han, J. Song, Y. Xie and W. Li, Tetrahedron Lett., 2007, 48, 5613 CrossRef CAS.
- K. Fukumoto, M. Yoshizawa and H. Ohno, J. Am. Chem. Soc., 2005, 127, 2398 CrossRef CAS.
- (a) Y. Qian, S. Xiao, L. Liu and Y. Wang, Tetrahedron: Asymmetry, 2008, 19, 1515 CrossRef CAS; (b) X. Zheng, Y. Qian and Y. Wang, Eur. J. Org. Chem., 2010, 515 CrossRef CAS; (c) X. Zheng, Y. Qian and Y. Wang, Catal. Commun., 2010, 11, 567 CrossRef CAS; (d) Y. Qian, X. Zheng and Y. Wang, Eur. J. Org. Chem., 2010, 3672 CrossRef CAS.
- W. Chen, Y. Zhang, L. Zhu, J. Lan, R. Xie and J. You, J. Am. Chem. Soc., 2007, 129, 13879 CrossRef CAS.
- G. Rashinkar and R. Salunkhe, J. Mol. Catal. A: Chem., 2010, 316, 146 CrossRef CAS.
- Supramolecular Catalysis, ed. P. W. N. M. Van Leeuwen, Wiley-VCH, Weinheim, 2008 Search PubMed.
- For a recent examples, see: S. Hu, J. Li, J. Xiang, J. Pan, S. Luo and J.-P. Cheng, J. Am. Chem. Soc., 2010, 132, 7216 Search PubMed.
- Z. Shen, J. Ma, Y. Liu, C. Jiao, M. Li and Y. Zhang, Chirality, 2005, 17, 556 CrossRef CAS.
- (a) M. Benaglia, M. Cinquini, F. Cozzi, A. Puglisi and G. Celentano, Adv. Synth. Catal., 2002, 344, 533 CrossRef CAS; (b) M. Benaglia, M. Cinquini, F. Cozzi, A. Puglisi and G. Celentano, J. Mol. Catal. A: Chem., 2003, 204–205, 157 CrossRef CAS.
- J. Huang, X. Zhang and D. W. Armstrong, Angew. Chem., Int. Ed., 2007, 46, 9073 CrossRef CAS.
- K. Liu, D. Haeussinger and W. D. Woggon, Synlett., 2007, 2298 CAS.
- B. Escuder, F. Rodríguez-Llansola and J. F. Miravet, New J. Chem., 2010, 34, 1044 RSC.
- (a) K. Tanaka, A. Mori and S. Inoue, J. Org. Chem., 1990, 55, 181 CrossRef CAS; (b) H. Danda, Synlett, 1991, 263 CrossRef CAS.
- (a) H. S. Rho, S. H. Oh, J. W. Lee, J. Y. Lee, J. Chin and C. E. Song, Chem. Commun., 2008, 1208 RSC; (b) S. H. Oh, H. S. Rho, J. W. Lee, J. E. Lee, S. H. Youk, J. Chin and C. E. Song, Angew. Chem., 2008, 120, 7990 CrossRef ; Angew. Chem. Int. Ed., 2008, 47, 7872.
- (a) B. Escuder, S. Martí and J. F. Miravet, Langmuir, 2005, 21, 6776 CrossRef CAS; (b) F. Rodríguez-Llansola, J. F. Miravet and B. Escuder, Chem. Commun., 2009, 209 RSC; (c) F. Rodríguez-Llansola, J. F. Miravet and B. Escuder, Org. Biomol. Chem., 2009, 7, 3091 RSC; (d) F. Rodríguez-Llansola, B. Escuder and J. F. Miravet, J. Am. Chem. Soc., 2009, 131, 11478 CrossRef CAS; (e) D. S. Tsekova, J. A. Sáez, B. Escuder and J. F. Miravet, Soft Matter., 2009, 5, 3727 RSC; (f) F. Rodríguez-Llansola, J. F. Miravet and B. Escuder, Chem. Eur. J., 2010, 16, 8480 CrossRef CAS; (g) F. Rodríguez-Llansola, J. F. Miravet and B. Escuder, Chem. Commun., 2009, 7303 RSC.
- For reviews, see: (a) T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS; (b) J. Dupont, R. F. De Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667 CrossRef CAS; (c) D. Zhao, M. Wu, Y. Kou and E. Min, Catal. Today, 2002, 74, 157 CrossRef CAS; (d) J. S. Wilkes, J. Mol. Catal. A: Chem., 2004, 214, 11 CrossRef CAS; (e) T. Welton, Coord. Chem. Rev., 2004, 248, 2459 CrossRef CAS; (f) Z. Zhang, Adv. Catal., 2006, 49, 153; (g) V. I. Pârvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615 CrossRef CAS; (h) R. Giernoth, Top. Curr. Chem., 2007, 276, 1 CAS; (i) J. W. Lee, J. Y. Shin, Y. S. Chun, H. B. Jang, C. E. Song and S. Lee, Acc. Chem. Res., 2010, 43, 985 CrossRef CAS.
- T.-P. Loh, L.-C. Feng, H.-Y. Yang and J.-Y. Yang, Tetrahedron Lett., 2002, 43, 8741 CrossRef CAS.
- P. Kotrusz, I. Kmentová, B. Gotov, Š. Toma and E. Solčniová, Chem. Commun., 2002, 2510 RSC.
- B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef CAS.
- K. R. Reddy, L. Chakrapani, T. Ramani and C. V. Rajasekhar, Synth. Commun., 2007, 34, 4301 CrossRef.
- J. Shah, H. Blumenthal, Z. Yacob and J. Liebscher, Adv. Synth. Catal., 2008, 350, 1267 CrossRef CAS.
- A. Córdova, Tetrahedron Lett., 2004, 45, 3949 CrossRef CAS.
- H.-M. Guo, H.-Y. Niu, M.-X. Xue, Q.-X. Guo, L.-F. Cun, A.-Q. Mi, Y.-Z. Jiang and J.-J. Wang, Green Chem., 2006, 8, 682 RSC.
- K. Huang, Z.-Z. Huang and X.-J. Li, J. Org. Chem., 2006, 71, 8320 CrossRef CAS.
- H.-M. Guo, L.-F. Cun, L.-Z. Gong, A.-Q. Mi and Y.-Z. Jiang, Chem. Commun., 2005, 1450 RSC.
- A. S. Kucherenko, D. E. Siyutkin, V. O. Muraviev, M. I. Struchkova and S. G. Zlotin, Mendeleev Commun., 2007, 17, 277 Search PubMed.
- H. Hagiwara, T. Okabe, T. Hoshi and T. Suzuki, J. Mol. Catal. A: Chem., 2004, 214, 167 CrossRef CAS.
- Y. Qiao, L. Zhang, S. Luo and J.-P. Cheng, Synlett., 2010 Search PubMed , accepted.
- C. V. Doorslaer, J. Wahlen, P. Mertens, K. Binnemans and D. D. Vos, Dalton Trans., 2010, 39, 8377 RSC.
- M. Gruttadauria, S. Riela, P. L. Meo, F. D'Anna and R. Noto, Tetrahedron Lett., 2004, 45, 6113 CrossRef CAS.
- M. Gruttadauria, S. Riela, C. Aprile, P. L. Meo, F. D'Anna and R. Noto, Adv. Synth. Catal., 2006, 348, 82 CrossRef CAS.
- C. Aprile, F. Giacalone, M. Gruttadauria, A. M. Marculescu, R. Noto, J. D. Revell and H. Wennemers, Green Chem., 2007, 9, 1328 RSC.
- Z. Wang, J. Yan, X. Zhang and L. Wang, Synthesis, 2009, 3744 CAS.
- H. Hagiwara, T. Kuroda, T. Hoshi and T. Suzuki, Adv. Synth. Catal., 2010, 352, 909 CAS.
- A. S. Kucherenko, M. I. Struchkova and S. G. Zlotin, Eur. J. Org. Chem., 2006, 2000 CrossRef CAS.
- S. Chandrasekhar, C. Narsihmulu, N. R. Reddy and S. S. Sultana, Tetrahedron Lett., 2004, 45, 4581 CrossRef CAS.
- S. Chandrasekhar, N. R. Reddy, S. S. Sultana, C. Narsihmulu and K. V. Reddy, Tetrahedron, 2006, 62, 338 CrossRef CAS.
- For recent reviews, see: (a) T. J. Dickerson, N. N. Reed and K. D. Janda, Chem. Rev., 2002, 102, 3325 CrossRef CAS; (b) J. Chen, S. K. Spear, J. G. Huddleston and R. D. Rogers, Green Chem., 2005, 7, 64 RSC.
- D.-Q. Xu, S.-P. Luo, Y.-F. Wang, A.-B. Xia, H.-D. Yue, L.-P. Wang and Z.-Y. Xu, Chem. Commun., 2007, 4393 RSC.
- S.-P. Luo, S. Zhang, Y.-F. Wang, A.-B. Xia, G.-C. Zhang, X.-H. Du and D.-Q. Xu, J. Org. Chem., 2010, 75, 1888 CrossRef CAS.
- For a review, see: (a) W. Zhang, Green Chem., 2009, 11, 911 RSC ; for some selected examples, see: J. K. Park, H. G. Lee, C. Bolm and B. M. Kim, Chem.–Eur. J., 2005, 11, 945 Search PubMed; (b) Z. Dalicsek, F. Pollreisz, A. Gömöry and T. Soós, Org. Lett., 2005, 7, 3243 CrossRef CAS; (c) L. Zu, J. Wang, H. Li and W. Wang, Org. Lett., 2006, 8, 3077 CrossRef CAS; (d) L. Zu, H. Li, J. Wang, X. Yu and W. Wang, Tetrahedron Lett., 2006, 47, 5131 CrossRef CAS; (e) Q. L. Chu, W. Zhang and D. P. Curran, Tetrahedron Lett., 2006, 47, 9287 CrossRef CAS; (f) S. Goushi, K. Funabiki, M. Ohta, K. Hatano and M. Matsui, Tetrahedron, 2007, 63, 4061 CrossRef CAS; (g) H. Cui, Y. Li, C. Zheng, G. Zhao and S. Zhu, J. Fluorine Chem., 2008, 129, 45 CrossRef CAS; (h) Q. Chu, M. S. Yu and D. P. Curran, Org. Lett., 2008, 10, 749 CrossRef CAS; (i) L. Zu, H. Xie, H. Li, J. Wang and W. Wang, Org. Lett., 2008, 10, 1211 CrossRef CAS; (j) T. Miura, K. Imai, M. Ina, N. Tada, N. Imai and A. Itoh, Org. Lett., 2010, 12, 1620 CrossRef CAS.
- G. Chollet, F. Rodriguez and E. Schulz, Org. Lett., 2006, 8, 539 CrossRef CAS.
- L. Xing, J.-H. Xie, Y.-S. Chen, L.-X. Wang and Q.-L. Zhou, Adv. Synth. Catal., 2008, 350, 1013 CrossRef CAS.
- R. Akiyama and S. Kobayashi, Chem. Rev., 2009, 109, 594–642 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |