Challenge and progress: palladium-catalyzed sp3 C–H activation
Hu
Li
a,
Bi-Jie
Li
a and
Zhang-Jie
Shi
*ab
aBeijing National Laboratory of Molecular Sciences (BNLMS) and Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry and Green Chemistry Center, Peking University, Beijing 100871, China. E-mail: zshi@pku.edu.cn; Web: http://www.chem.pku.edu.cn/zshi Fax: +86 10-6276-0890; Tel: +86 10-6276-0890
bState Key Laboratory of Organometallic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
First published on 10th February 2011
Abstract
Palladium-catalysis has been broadly applied to sp2 C–H activation. Recently, palladium-catalyzed sp3 C–H activation has also been considered as an important strategy to construct synthetically useful C–C/C–X bonds. Allylic sp3 C–H bonds can be successfully activated by Pd(II) species to produce η3-coordinated palladium species for further transformations, while activation of general sp3 C–H bonds mainly proceeds through directed pathways with the assistance of proper directing groups or initiated by oxidative addition. Various catalytic mechanisms were extensively investigated through either Pd(II)/Pd(0), Pd(II)/Pd(IV) or Pd(0)/Pd(II) catalytic cycles. The challenges faced in this area have also been addressed in this perspective article.
1. Introduction
The construction of C–C and C–X (X = O, N, etc.) bonds is the key task in organic synthesis. Traditionally, these goals were mainly approached through the transformation of pre-existing functional groups leaving C–H bonds untouched, which are ubiquitous in organic compounds. With recently much more attention to the atom-economical process and green chemistry goal, transition metal-catalyzed direct C–H activation has emerged as the most powerful and straightforward strategy for the production of organic molecules from easily available chemicals. In the last several decades, direct sp2 C–H activation of (hetero)arenes as well as some olefins has been extensively investigated and found impressive applications in organic synthesis, which has been the subject of many accounts and reviews.1,2 In comparison, much less research has been devoted to the activation of more “inert” sp3 C–H bonds of alkyl groups.The challenges faced in activation of inert C–H bonds of alkyl groups are concluded in three different aspects: high bond dissociation energy (BDE), lack of the “active” HOMO or LUMO to interact with transition metal catalytic centers, as well as difficulty to control the selectivity of C–H transformations. Compared with well-approached sp2 C–H activation, highly selective functionalization of sp3 C–H bonds is much more difficult due to the lack of the assistance of π-groups, which could efficiently interact with transition metal centers. To overcome the tremendous challenges faced in sp3 C–H activation, the broadly used directing group-oriented strategy in sp2 C–H activation is also preferred to promote both reactivity and selectivity of sp3 C–H activation. Among various pathways successfully involved to pursue these challenging goals, palladium-catalyzed sp3 C–H activation shows great advantages and potential applications due to its outstanding reactivity and controllable selectivity compared with other transition metal catalysis. Hence we mainly focus on the significant progress in palladium-catalyzed sp3 C–H activation up to late 2010, providing discussions of C–C/C–X (X = O, N, etc.) bond formation by cleavage of an sp3 C–H bond. We will separate these important studies into three relatively independent areas: allylic/benzylic sp3 C–H activation and common sp3 C–H activation.
2. Allylic C–H activation
The Tsuji–Trost reaction (Pd-catalyzed allylic alkylation) has been well developed and has become a powerful and applicable transformation to construct C–C bonds in organic synthesis with good efficiency, both regio- and stereoselectivity.3 Generally, pre-functionalized groups at the allylic position, for example, halogen atoms, acetates, carbonates, phosphates, alcohols, etc., are required to serve as reacting and leaving groups to process this important transformation. In contrast, direct construction of valuable compounds from easily available alkenes avoiding the pre-functionalization should be highly desirable for green and sustainable organic transformation due to both the atom and step economy.The reactivity of an allylic C–H bond benefits from the adjacent alkene group, which not only dramatically decreases the BDE of the allylic C–H bond, but also plays a critical role as a directing group to initially coordinate to the Pd center to stress the distance between the allylic C–H bond and Pd catalytic species (Scheme 1).1c Actually the idea to approach direct allylic C–H oxidation using stoichiometric palladium(II) species had been achieved in the 1960s,4a–c and the first successful example carried out to prepare π-allyl–Pd complexes and realize the allylic alkylation was reported by Trost and coworkers in the 1970s.4d–f However, the catalytic allylic C–H activation is not realized due to the challenges of finding proper ligands and efficient oxidants to facilitate the catalytic cycle until recently.
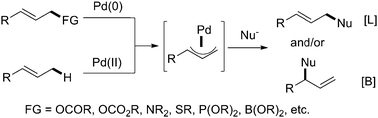 | ||
| Scheme 1 Traditional Tsuji–Trost alkylationversus direct allylic C–H activation. | ||
2.1 Direct carboxylation
In 2004, White and coworkers first reported that sulfoxide ligation to Pd(II) salts could selectively promote allylic C–H oxidation of terminal alkenes with good regioselectivity (Scheme 2).5 In their studies, DMSO showed some dramatic efficacy to promote Pd-catalyzed transformations. For example, the combination of Pd(OAc)2 as a catalyst and DMSO as a ligand in HOAc afforded linear (E)-allylic acetates in high regio- and stereoselectivities in good efficacy, and was compatible with a wide range of functionalities such as amides, carbamates, esters, and ethers (Scheme 2, [L]).5 This method could be further applied to the synthesis of linear (E)-allylic alcohol intermediates with fewer functional group manipulations, in a much shorter reaction sequence and in higher overall yields while the traditional alternative routes involve conventional Wittig-type C–C bond-forming and reduction sequences.6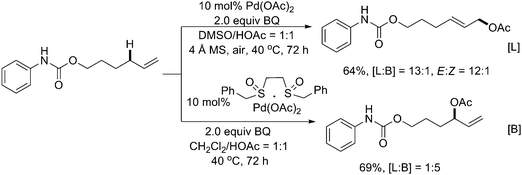 | ||
| Scheme 2 Pd(II)-catalyzed regioselective allylic C–H acetoxylation.5 | ||
In contrast, with the design of bis-sulfoxide ligands, the great reactivity and unusual selectivity were observed in direct allylic C–H acetoxylation. Compared to the Pd-DMSO system, the bis-sulfoxide Pd(OAc)2 complex was found to be an effective allylic C–H oxidation catalyst effecting a reverse of regioselectivity, which favored the formation of branched allylic acetates (Scheme 2, [B]).5 Interestingly, the application of phenyl vinyl sulfoxide with Pd(OAc)2 also furnished the branched allylic acetoxylation.7 Although the mechanism is still unclear, preliminary mechanistic studies suggested that the sulfoxide and benzoquinone (BQ) might act as serial ligands setting to promote the catalytic transformation (Scheme 3).7
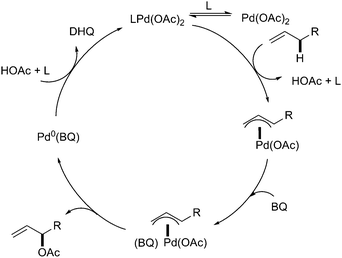 | ||
| Scheme 3 A plausible mechanism of Pd(II)-catalyzed regioselective allylic C–H oxidation promoted by serial ligands.7 | ||
Later on, the same group extended this chemistry to achieve intramolecular macrolactonization of linear ω-alkenoic acids (Scheme 4).8Aryl, alkyl, and (Z)-α,β-unsaturated acids were all competent nucleophiles for this macrolactonization without the observation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond isomerization. Most importantly, various functional groups were compatible, including biologically and medicinally relevant functionalities, such as ortho-substituted salicylate esters, bis(indoyl)maleimides, and peptides. The same operational simplicity could be scaled up to 1 g scale, which showed the great potential for industrial application. Currently provided evidences supported the hypothesis that the macrolactonization proceeded via inner-sphere functionalization from a templated π-allyl–Pd carboxylate intermediate.
C bond isomerization. Most importantly, various functional groups were compatible, including biologically and medicinally relevant functionalities, such as ortho-substituted salicylate esters, bis(indoyl)maleimides, and peptides. The same operational simplicity could be scaled up to 1 g scale, which showed the great potential for industrial application. Currently provided evidences supported the hypothesis that the macrolactonization proceeded via inner-sphere functionalization from a templated π-allyl–Pd carboxylate intermediate.
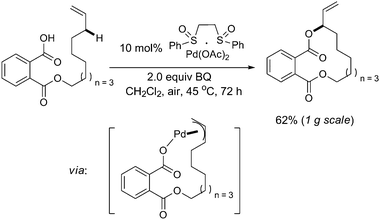
Besides, sequential allylic C–H oxidation/vinylic C–H arylation of α-olefins to furnish (E)-arylated allylic esters in high regio- and E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶Z selectivities (>20∶1) was further reported (Scheme 5).9 The broad scope of this method enabled the rapid assembly of densely functionalized fragments for complex molecule synthesis.
∶Z selectivities (>20∶1) was further reported (Scheme 5).9 The broad scope of this method enabled the rapid assembly of densely functionalized fragments for complex molecule synthesis.
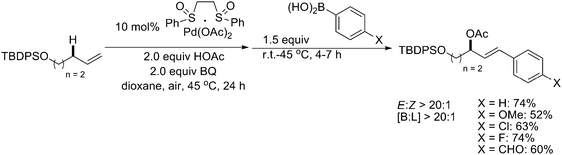
Based on these developments, the asymmetric allylic C–H oxidation of terminal alkenes with a heterobimetallic Pd(II)–bis(sulfoxide)/Cr(III)(salen) system that proceeded with high levels of enantioselectivity has been reported (Scheme 6).10 This represented the first report of the interaction of a chiral Lewis acid co-catalyst with an organometallic intermediate that influenced the stereochemical outcome of a catalytic process.
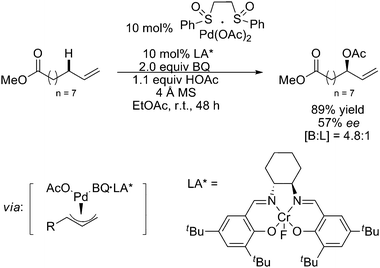
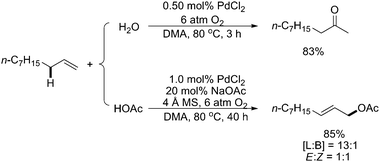
It is noteworthy that in 2006, Kaneda and coworkers reported a highly efficient and reusable catalytic system constituted by the combination of PdCl2 and N,N-dimethylacetamide (DMA), which used O2 as the sole reoxidant for liquid-phase Wacker oxidation and acetoxylation of terminal olefins to the corresponding methyl ketones and linear allylic acetates, respectively (Scheme 7).11
 | ||
| Scheme 7 Pd(II)-catalyzed Wacker oxidation and allylic C–H oxidation.11 | ||
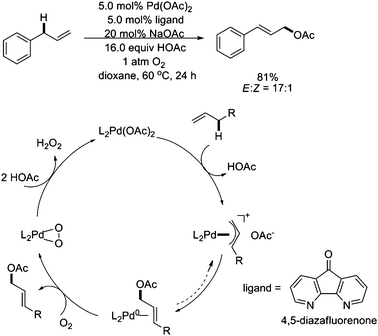
In 2010, Stahl and coworkers found that the use of 4,5-diazafluorenone as an ancillary ligand for Pd(OAc)2 enabled the allylic C–H acetoxylation of terminal alkenes to linear allylic acetates in good yields and selectivity under 1 atm O2 (Scheme 8).12 Mechanistic studies revealed that the ligand facilitated C–O reductive elimination from a π-allyl–Pd intermediate, thereby eliminating the requirement for benzoquinone in this key catalytic step.
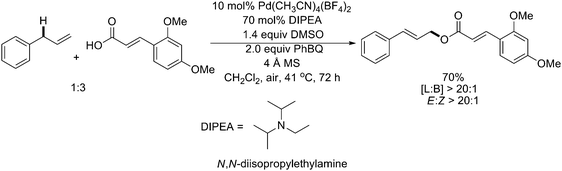
Very recently, White and coworkers extended the intermolecular allylic C–H oxidation method to construct allylic esters (Scheme 9).13 The reactions allowed a wide range of complex aryl and alkyl carboxylic acids to couple directly with terminal olefins to furnish (E)-allylic esters in outstanding yields and selectivities.
2.2 Direct amination
Besides acetoxylation, a diastereoselective allylic C–H amination reaction of chiral homoallylic N-tosyl carbamates was first reported by White's group in excellent yields and with good anti/syn diastereoselectivities that were controlled by the stereocenter (Scheme 10).14 Mechanistic studies provided evidences to imply that the transformation went through a Pd/sulfoxide-mediated allylic C–H cleavage to form a key π-allyl–Pd intermediate. Pd(II) counterion-assisted deprotonation of the nitrogen nucleophile was also highly preferred to fulfill this functionalization. This efficient direct amination was further successfully applied to prepare syn-1,3-amino alcohol motifs other than 1,2-amino alcohol scaffolds, by employing electron-deficient N-nosyl carbamate nucleophiles (Scheme 11).15 In the latter reaction, higher concentrations of anionic species generated in situ were considered to promote the transformation.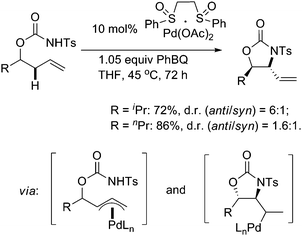
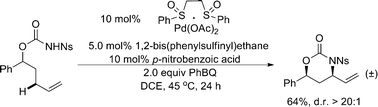
Shortly after, a similar successful intramolecular allylic C–H amination was developed by Liu and coworkers in the absence of sulfoxide ligands (Scheme 12).16 Notably, in this case, the Brønsted base and some interesting additives could modulate the regioselectivity favoring the formation of a 7-membered ring, which was not clearly understandable at this stage.
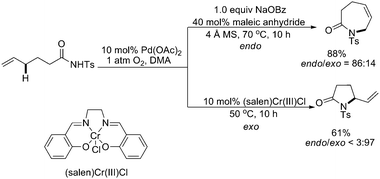
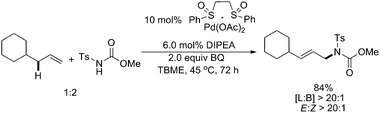
Significant progress had been achieved by both White and Liu, respectively, to extend the direct amination to intermolecular versions.17,18 White and coworkers first applied their heterobimetallic Pd(II)sulfoxide/(salen)Cr(III)Cl system to achieve the linear allylic C–H amination with good yields and outstanding regio- and stereoselectivities (>20∶1) (Scheme 13, top).17 Their subsequent investigations indicated that a Brønsted base N,N-diisopropylethylamine (DIPEA) also promoted the intermolecular amination (Scheme 14).19
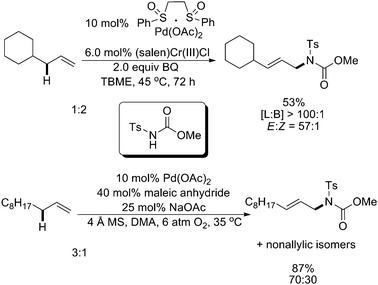
In Liu's studies, the employment of maleic anhydride and sodium acetate with the Pd(OAc)2 catalyst afforded highly regioselective linear allylic C–H amination products, although the reactions proceeded at a pressure of 6 atm of O2 and other non-allylic isomers were also observed (Scheme 13, bottom).18 This method was modified recently by enhancing the re-oxidation of the palladium catalyst with a strong oxidant PhI(OPiv)2 rather than high pressure of O2 (Scheme 15).20 Mechanistic studies indicated that 1,4-naphthoquinone (NQ) was essential for the allylic C–H activation to generate a π-allyl–Pd intermediate.
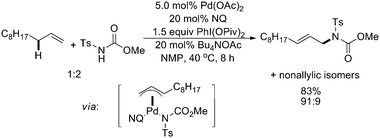 | ||
| Scheme 15 Pd(II)-catalyzed intermolecular linear allylic C–H amination with naphthoquinone and PhI(OPiv)2.20 | ||
Another outstanding example in C–N formation was reported by Baran and coworkers in 2009, in which indoles were successfully applied as nucleophiles other than carbamates or sulfonamides.21 In their report, direct prenylation of indoles at the N-1 position was processed by Pd-catalyzed allylic C–H activation (Scheme 16). These reactions tolerated a broad range of functional groups, and could be carried out in a gram scale, as demonstrated by the formal syntheses of a number of natural products. Mechanistic studies also indicated that a π-allyl–Pd intermediate was initiated by the allylic C–H activation, and then reacted with indole either by direct coordination of the indole nitrogen atom to the Pd center or palladation of the indole at C-3 to undergo a metallo-Claisen rearrangement.
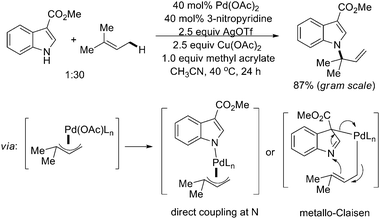 | ||
| Scheme 16 Pd(II)-catalyzed prenylation of indoles by allylic C–H amination.21 | ||
2.3 Direct C–C bond formation
In comparison, C–C bond formation viaPd-catalyzed allylic C–H activation was seldom reported although the stoichiometric version had been developed a long time ago. Until recently, Shi and White almost simultaneously reported the intermolecular allylic C–H alkylations to construct sp3–sp3 C–C bonds.22,23 Shi and co-workers found that, by using benzoquinone (BQ) as an oxidant under a balloon pressure of O2, the allylic C–H alkylation between allylic sp3 C–H bonds and methylenic sp3 C–H bonds yielded very good results with excellent regio- and stereoselectivities (Scheme 17a).22 Besides, polysubstituted cyclic compounds were also constructed by the intramolecular allylic C–H alkylation in excellent diastereoselectivities, which might offer an efficient way to construct carbocycles from linear olefins (Scheme 17b).22 Notably, preliminary mechanistic studies provided evidences that BQ might act both as an oxidant and a proton acceptor at the same time.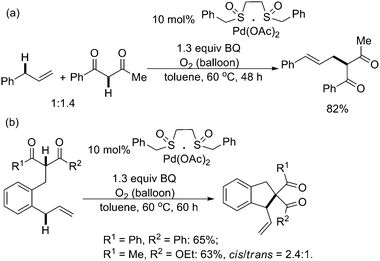 | ||
| Scheme 17 Pd(II)-catalyzed inter- and intramolecular allylic C–H alkylation.22 | ||
In White's studies, they found that using 2,6-dimethylbenzoquinone (DMBQ) as the oxidant, a wide range of aromatic and heteroaromatic linear (E)-α-nitro-arylpentenoates were obtained as single olefin isomers in excellent yields directly from terminal olefin substrates and methyl nitroacetate (Scheme 18).23 The use of DMSO as a π-acidic ligand was found to be crucial for promoting functionalization of the π-allyl–Pd intermediate.
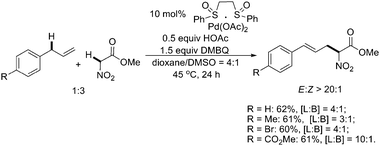 | ||
| Scheme 18 Pd(II)-catalyzed intermolecular allylic C–H alkylation.23 | ||
3. Benzylic C–H activation
Due to the structural similarity, a benzylic C–H bond is somehow considered like an equivalent allylic C–H bond. However, the reactivities of them in Pd-catalyzed C–H activation are completely different. In a benzylic system, the heteroatom-containing directed strategy should be applied to facilitate the C–H transformation viaPd catalysis, and direct C–H functionalizations to synthetic useful compounds have only been approached recently.3.1 Direct acetoxylation and fluorination
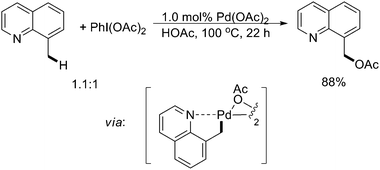
Although the redox chemistry involving Pd(IV) has been poorly understood in its early stage,24 a number of sp3 C–H activation reactions have been developed by considering Pd(II)/(IV) catalysis including arylation, acetoxylation, amination, and halogenation, which have gained much attention in recent years.In 2004, Sanford and coworkers reported the first example of Pd(II)-catalyzed acetoxylation of benzylic C–H bonds.25 The readily available quinoline directing groups were oxidized with extremely high levels of chemo- and regioselectivities (Scheme 19). The high selectivities were rationalized on the basis of the requirements of putative palladium alkyl intermediates, which supported a Pd(II)/Pd(IV) mechanism.
Later on, the same research group developed a successful Pd(II)-catalyzed fluorination of benzylic C–H bonds, which was achieved under oxidative conditions using electrophilic N-fluoropyridinium reagents (Scheme 20).26 Microwave irradiation served as the optimal condition for this fluorination of benzylic C–H bonds in substituted 8-methylquinoline derivatives.
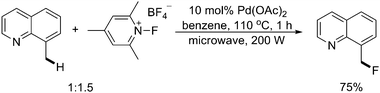 | ||
| Scheme 20 Pd(II)-catalyzed quinoline-directed fluorination of benzylic C–H bonds with N-fluoropyridinium salts.26 | ||
3.2 Direct C–C bond formation
With the similar strategy, in 2005, Sanford and coworkers described a Pd(II)-catalyzed direct arylation of benzylic C–H bonds with hypervalent iodine(III) arylating reagents (Scheme 21).27 Mechanistic experiments provided preliminary evidences in support of an unusual mechanism for this transformation involving a Pd(II)/Pd(IV) catalytic cycle. Further studies indicated that a bimetallic high oxidation state Pd species acted as a key catalytic intermediate in the reactions.28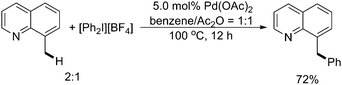 | ||
| Scheme 21 Pd(II)-catalyzed quinoline-directed arylation of benzylic C–H bonds with diaryliodonium(III) salts.27 | ||
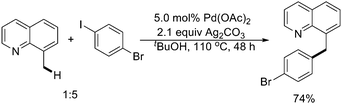
Simultaneously, Shabashov and Daugulis reported a different strategy for Pd(II)-catalyzed arylation of benzylic C–H bonds of quinoline derivatives with aryl iodides (Scheme 22).29 It is worth noting that a bromo substituent was tolerated on the aryl iodide coupling component.
In 2000, Catellani and coworkers showed another example of Pd(0)-catalyzed activation of sp3 C–H bonds of benzylic methyl groups with formation of cyclopentene rings (Scheme 23).30 The reaction was initiated by the oxidative addition of the C–I bond to the Pd(0) center, followed by insertion of norbornene. Then intramolecular palladation of the benzylic C–H bond and reductive elimination afforded the final product.
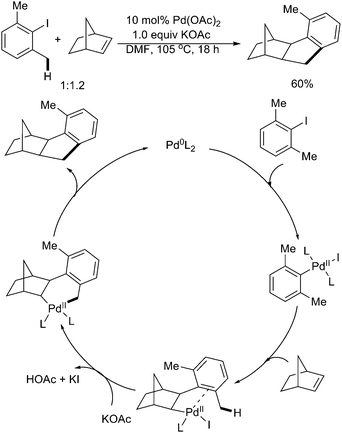 | ||
| Scheme 23 Pd(0)-catalyzed benzylic C–H activation with norbornene.30 | ||
In 2008, Fagnou and coworkers reported Pd(0)-catalyzed site selective arylation reactions of both sp2 and benzylic sp3 C–H bonds of azine and diazine N-oxide substrates (Scheme 24).31 The transformations occurred in good to excellent yields and with complete reversed selectivities at the desired positions. From these studies, the choice of the base emerged as a pivotal component for site selectivity, pointing to its intimate involvement in the mechanism of direct arylation.
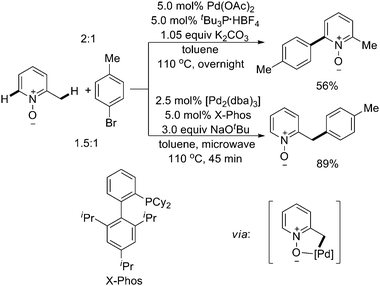 | ||
| Scheme 24 Pd(0)-catalyzed azineN-oxide-directed site selective arylation of C–H bonds with aryl halides.31 | ||
Recently, Huang and coworkers reported another interesting example of facilitating C–C bond formation by supposedly Pd(II)-catalyzed nucleophilic benzylic addition of 2-methyl-substituted azaarenes to N-sulfonyl aldimines under neutral conditions via benzylic C–H activation (Scheme 25).32 This reaction represented a very efficient protocol for the synthesis of heterocycle-containing amines with very good functional group tolerance. The N-sulfonyl imine was proposed to be activated via coordination to the palladium center.
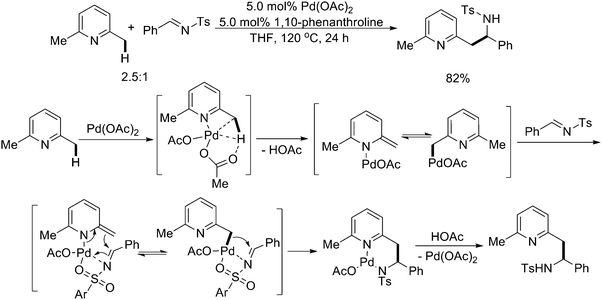
4. Common sp3 C–H activation
Compared with allylic and benzylic C–H activation, activation of common sp3 C–H bonds is much more challenging. As mentioned, allylic C–H activation can serve as an olefin-directed C–H activation process, where the η3-coordination of the palladium center can activate the inert sp3 C–H bond. Notably, for non-allylic C–H bonds, the majority of sp3 C–H activation, the strategy by utilizing heteroatom-containing chelating groups that can coordinate the metal center and direct C–H insertion is the most successful process up to date.33In general, Pd-catalyzed common sp3 C–H activation reactions are developed via two distinct catalytic pathways: initiated by Pd(II)-catalyzed sp3 C–H cleavage, which requires an external oxidant for reoxidation to proceed viaPd(II)/Pd(0) or Pd(II)/Pd(IV) catalytic cycles (Scheme 26a); and initiated by oxidative addition of aryl halide to Pd(0) to proceed viaPd(0)/Pd(II) catalytic cycles (Scheme 26b). For the latter pathway, heteroatom-directing groups may not be required.
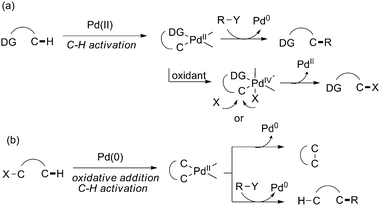 | ||
| Scheme 26 Two general pathways of Pd-catalyzed common sp3 C–H activation. | ||
4.1 Direct C–X bond formation (X = O, N, and I)
As mentioned above, Sanford and coworkers reported the first acetoxylation of benzylic C–H bonds (see Section 3.1),25 which could also be extended to other unactivated sp3 C–H bonds with readily available O-methyl oxime and/or pyridine as directing groups with extremely high levels of chemo-, regio-, and in some cases diastereoselectivities (Scheme 27a).34 Recently, O-acetyl oxime serving as an effective directing group for the similar sp3 C–H functionalization reactions had also been achieved (Scheme 27b).35 The C–H functionalization products could be subsequently transformed into β-functionalized ketones, alcohols, and amines.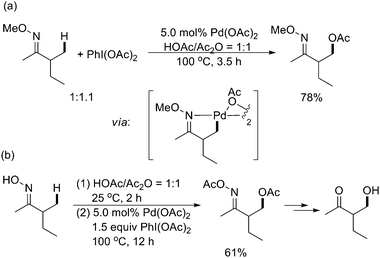
This chemistry was further extended to C–N bond formation in 2006 by Che and co-workers. They described a Pd(II)-catalyzed intermolecular amidation reaction of unactivated sp3 C–H bonds using nucleophilic primary sulfonamides or amides and potassium persulfate (Scheme 28).36 The substrates containing a pendent oxime or pyridine group were amidated with excellent chemo- and regioselectivities. It is noteworthy that primary sp3 C–H bonds were preferred to undergo β-amidation rather than secondary sp3 C–H bonds. Preliminary mechanistic studies suggested that the catalytic reaction was initiated by chelation-assisted cyclopalladation involving C–H activation, and then reacted with the reactive nitrene species, which was generated by the persulfate oxidation of primary sulfonamides or amides.
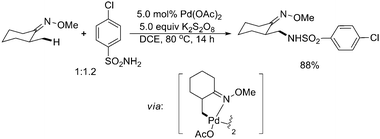 | ||
| Scheme 28 Pd(II)-catalyzed O-methyl oxime-directed amidation of sp3 C–H bonds with primary sulfonamides.36 | ||
In 2005, Yu and coworkers reported a successful iodination of sp3 C–H bonds at the β position of oxazoline, a removable chelating chiral auxiliary, which assisted in these asymmetric reactions (Scheme 29).37,38Pd(OAc)2 was an effective catalyst for the selective and asymmetric iodination of methyl and cyclopropyl groups at room temperature with IOAc as an efficient oxidant. The diiodination of similar substrates was further performed and applied as a novel route to the synthesis of cyclopropane derivatives.39
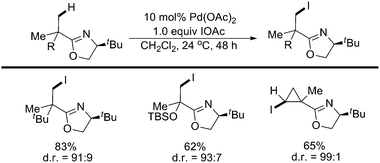 | ||
| Scheme 29 Pd(II)-catalyzed oxazoline-directed iodination of sp3 C–H bonds with IOAc.38 | ||
Moreover, the same or similar substrates could undergo acetoxylation using inexpensive oxidants, such as lauroyl peroxide and tert-butyl peroxyacetate, viaPd catalysis (Scheme 30).40Carboxylic anhydrides were essential for both the oxidation of the Pd–C bonds and regeneration of Pd(II). The use of Ac2O-d6 as the solvent showed that the acetyl group incorporated into the product was from acetic anhydride rather than the oxidant. Mechanistic studies indicated that Pd(IV) intermediates might be involved in the transformations. Later on, this methodology was extended to the acetoxylation of sp3 C–H bonds in Boc-protected N-methylamines with IOAc as the oxidant.41
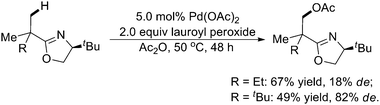 | ||
| Scheme 30 Pd(II)-catalyzed oxazoline-directed acetoxylation of sp3 C–H bonds with carboxylic anhydrides.40 | ||
More recently, Glorius and coworkers reported an efficient Pd(II)-catalyzed intramolecular direct amidation process via sp3 C–H activation (Scheme 31).42 Under the optimized conditions, the activation of inert sp3 C–H bonds was achieved by the assistance of an amide directing group that subsequently acted as a reaction partner in the same process. Numerous indoline derivatives were formed from anilines with an extraordinary tolerance of functional groups. The authors proposed that a Pd(IV) intermediate might exist in the catalytic transformation.
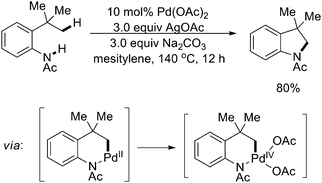 | ||
| Scheme 31 Pd(II)-catalyzed intramolecular amidation of sp3 C–H bonds.42 | ||
4.2 Direct C–C bond formation
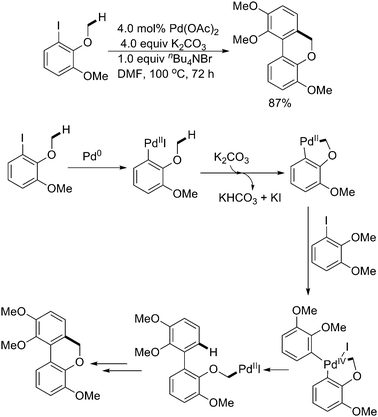 | ||
| Scheme 32 Pd(0)-catalyzed sequential sp3/sp2 C–H activation.43 | ||
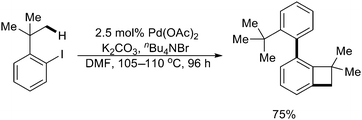 | ||
| Scheme 33 Pd(0)-catalyzed domino coupling of 2-iodo-tert-butylbenzene with sequential sp3/sp2 C–H activation.44 | ||
In 2005, Buchwald and coworkers observed an interesting arylation of sp3 C–H bonds in a bulky aryl bromide with aryl boronic acids. This unexpected arylation was considered to proceed through oxidative addition/1,4-Pd migration/Suzuki–Miyaura cross-coupling pathways (Scheme 34).45 The initial oxidative addition of aryl bromide to the Pd(0) center generated arylpalladium(II) species, followed by intramolecular sp3 C–H activation to form a five-membered palladacycle. Thereafter protonation of the arene, transmetalation of aryl boronic acid, and reductive elimination afforded the final product. In fact, such a kind of 1,4-Pd migration had been observed before and well studied (herein we will not discuss it in detail).46
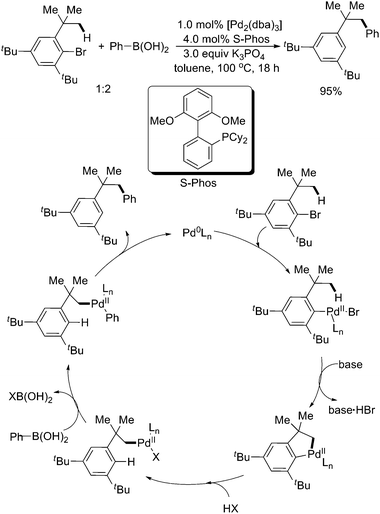 | ||
| Scheme 34 Pd(0)-catalyzed sequential sp3 C–H activation/Suzuki–Miyaura cross-coupling reaction.45 | ||
In 2009, Yu described a Pd(0)-catalyzed intermolecular arylation of sp3 C–H bonds using CONHC6F5 as the directing group (Scheme 35).47 The usage of the PR3 ligand with ArI suggested a Pd(0)/Pd(II) mechanism. This protocol could be used to arylate a variety of aliphatic carboxylic acid derivatives. Interestingly, this strategy could be further achieved viaPd(II)/Pd(IV) catalysis (Scheme 36).48
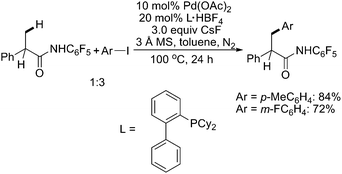 | ||
| Scheme 35 Pd(0)-catalyzed N-pentafluorophenyl amide-directed arylation of sp3 C–H bonds with aryl iodides.47 | ||
 | ||
| Scheme 36 Pd(II)-catalyzed N-pentafluorophenyl amide-directed arylation of sp3 C–H bonds with aryl iodides.48 | ||
Recently, Baudoin and coworkers reported a mild and efficient intermolecular β-arylation of unactivated sp3 C–H bonds of carboxylic esters in the presence of an appropriate Pd(0) catalyst (Scheme 37).49 In an asymmetric version with a chiral palladium ligand related to davephos, β-arylated products were obtained with good enantiomeric ratios (Scheme 38).49 Computational studies indicated that the mechanism involved a β-hydride elimination/Pd-η2(CC) bond rotation/hydride insertion/reductive elimination manifold, and that β arylation was kinetically favored over α-arylation.
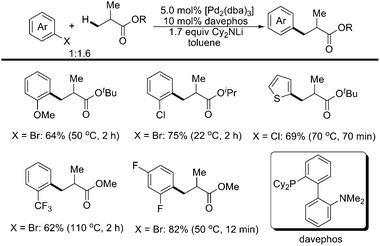 | ||
| Scheme 37 Pd(0)-catalyzed intermolecular β-arylation of sp3 C–H bonds with aryl halides.49 | ||
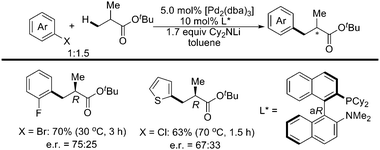 | ||
| Scheme 38 Pd(0)-catalyzed asymmetric β-arylation of sp3 C–H bonds with aryl halides.49 | ||
Besides intermolecular reactions, intramolecular sp3 C–H activation/C–C bond formation triggered by oxidative addition of C–X to Pd(0) was also well developed. In 2003, Baudoin and coworkers reported Pd(0)-catalyzed selective intramolecular sp3 C–H activation of gem-dialkyl groups on bromo- and iodobenzenes to give olefins or benzocyclobutenes with good regioselectivities (Scheme 39).50 For the formation of olefins, alkyl groups had at least two carbons; while for benzocyclobutenes, at least one alkyl group was methyl. The mechanism proposed was that after initial oxidative addition of the C–X bond to the Pd(0) center, intramolecular sp3 C–H activation took place to generate either a five-membered palladacycle or a six-membered palladacycle. The former underwent reductive elimination directly to generate benzocyclobutene, and the latter might undergo β-hydride elimination to form palladium hydride species, followed by reductive elimination to afford the olefin product.
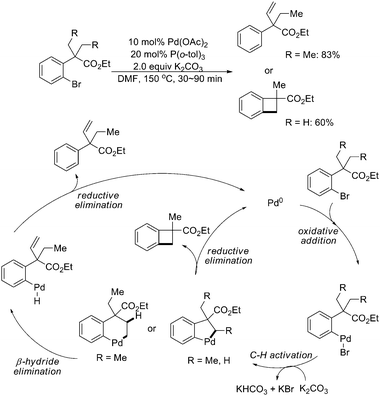 | ||
| Scheme 39 Pd(0)-catalyzed selective intramolecular sp3 C–H activation of gem-dialkyl groups.50 | ||
Further studies by the same group indicated that among other analogues of tris(2-methylphenyl)phosphane [P(o-tol)3], tris(5-fluoro-2-methylphenyl)phosphane (F-TOTP) was shown to have the optimal metal-bonding properties for this reaction (Scheme 40).51 This catalytic system operated under milder reaction conditions that allowed the regioselective production of various olefins adjacent to a quaternary benzylic carbon atom, as well as novel bi- and tricyclic molecules.
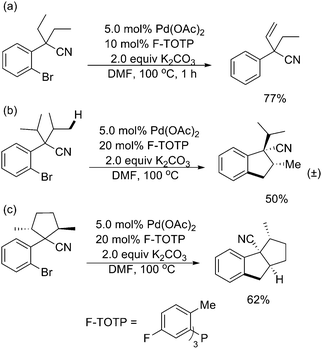 | ||
| Scheme 40 Pd(0)-catalyzed selective intramolecular sp3 C–H activation promoted by F-TOTP.51 | ||
Thereafter, they found that employing a combination of Pd(OAc)2 and tBu3P as the catalyst, K2CO3 as the base, and DMF as the solvent, a variety of substituted benzocyclobutenes were obtained efficiently by intramolecular sp3 C–H activation of methyl groups (Scheme 41).52 Several molecules that were hardly accessible by other methods could also be achieved, although the reaction was found to be limited to substrates bearing a quaternary benzylic carbon. The computationally mechanistic studies indicated that a carbonate-mediated concerted metalation–deprotonation (CMD)53 pathway might be involved.
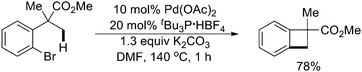 | ||
| Scheme 41 Pd(0)-catalyzed synthesis of benzocyclobutenes by sp3 C–H activation.52 | ||
Furthermore, the methodology could be extended to (hetero)aryl chloride substrates. The intramolecular sp3 C–H activation underwent efficiently, giving rise to a variety of valuable cyclobutarenes and indanes (Scheme 42).54
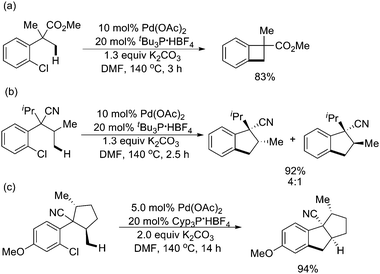 | ||
| Scheme 42 Pd(0)-catalyzed intramolecular sp3 C–H activation employing aryl chlorides.54 | ||
In 2007, Fagnou and coworkers found related Pd(0)-catalyzed intramolecular sp3 C–H activation reactions for the preparation of 2,2-dialkyl-dihydrobenzofuran derivatives by using an O-containing group as the tether (Scheme 43).55 These reactions occurred in excellent yield and very high selectivity for the formation of one sole product arising from a reaction at nearby methyl groups. Mechanistic and computational studies pointed to the involvement of a CMD pathway that was enabled by the presence of three-center agostic interactions at the transition state. Further studies indicated that using (hetero)aryl chlorides as substrates, the intramolecular sp3 C–H activation could also generate a variety of valuable dihydrobenzofurans and indanones with good efficiency (Schemes 43 and 44).54,55 It is worth noting that a similar example of preparation of indoline derivatives from N-alkyl-2-bromoanilines viaPd(0)-catalyzed intramolecular sp3 C–H activation was reported by Ohno's group (Scheme 45).56
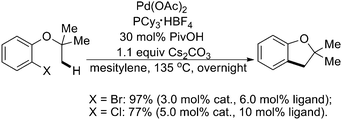 | ||
| Scheme 43 Pd(0)-catalyzed synthesis of dihydrobenzofurans by intramolecular sp3 C–H activation.55 | ||
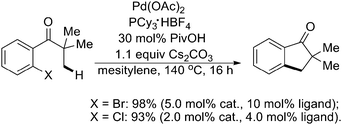 | ||
| Scheme 44 Pd(0)-catalyzed synthesis of indanones by intramolecular sp3 C–H activation.54 | ||
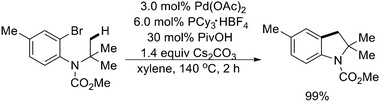 | ||
| Scheme 45 Pd(0)-catalyzed synthesis of indolines by intramolecular sp3 C–H activation.56 | ||
Recently, the intramolecular activation of sp3 C–H bonds adjacent to a nitrogen atom under Pd(0) catalysis had been reported by the same group (Scheme 46).57 Diminishing the Lewis basicity of the nitrogen lone pair was crucial for this catalytic activity. A range of N-methylamides and sulfonamides reacted exclusively at primary sp3 C–H bonds to afford the products of alkane arylation in good yields (Scheme 47).57 Mechanistic studies and DFT calculations provided support for a CMD transition state.
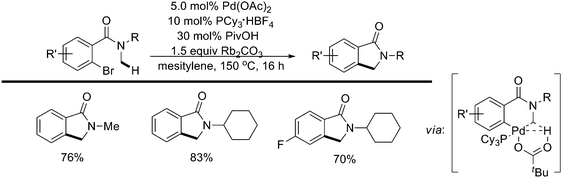 | ||
| Scheme 46 Pd(0)-catalyzed intramolecular sp3 C–H activation of N-methylamides.57 | ||
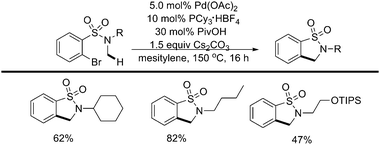 | ||
| Scheme 47 Pd(0)-catalyzed intramolecular sp3 C–H activation of N-methylsulfonamides.57 | ||
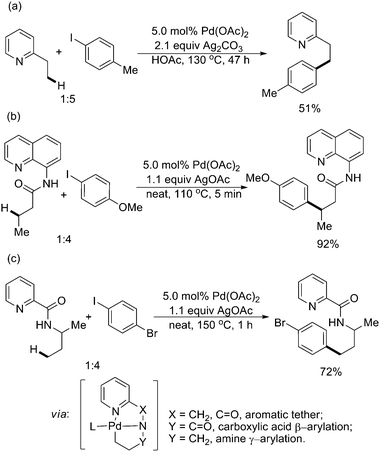 | ||
| Scheme 48 Pd(II)-catalyzed arylation of sp3 C–H bonds with chelating pyridine directing groups and aryl iodides.29,58 | ||
Thereafter, the same group developed a method for Pd(II)-catalyzed auxiliary-directed β-arylation and alkylation of sp3 C–H bonds in carboxylic acid derivatives (Scheme 49).60 By employing a 2-methylthioaniline auxiliary, selective monoarylation of primary sp3 C–H bonds could be achieved (Scheme 49a). If arylation of secondary sp3 C–H bonds was desired, an 8-aminoquinoline auxiliary might be used (Scheme 49b). For alkylation of sp3 C–H bonds, the 8-aminoquinoline auxiliary afforded the best results (Scheme 49c). With good functional group tolerance, amino- and hydroxy-acid derivatives could be functionalized using this methodology.
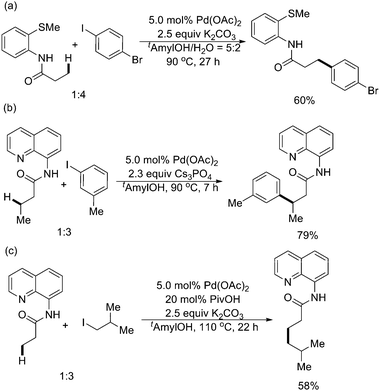 | ||
| Scheme 49 Pd(II)-catalyzed auxiliary-directed arylation and alkylation of sp3 C–H bonds with aryl iodides and alkyl iodides.60 | ||
In this field, Yu and coworkers made pioneering and significant contributions. In 2006, they reported the first example of Pd(II)-catalyzed pyridine-directed alkylations of sp3 C–H bonds with either methylboroxine or alkylboronic acids (Scheme 50).61 The mixture of benzoquinone (BQ) and Cu(OAc)2 or Ag2O was used as a crucial oxidant and promoter for the transmetalation step. Ether, ester, alcohol, and alkene functional groups were well tolerated.
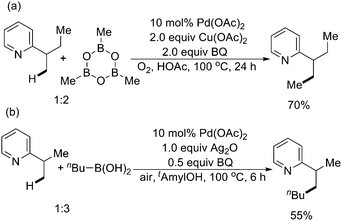 | ||
| Scheme 50 Pd(II)-catalyzed pyridine-directed alkylation of sp3 C–H bonds with methylboroxine and alkylboronic acids.61 | ||
Later on, the same group reported Pd(II)-catalyzed pyridine-directed enantioselective sp2 and sp3 C–H activation/C–C coupling reactions with boronic acids (Scheme 51).62 The relay of chiral information from the α-carbon atom of the amino acid ligand was believed to be crucial for chiral induction in the reactions. Although the C–C coupling of butylboronic acid with the sp3 C–H bond only afforded moderate yield and enantioselectivity, this constituted the first example of catalytic asymmetric sp3 C–H activation.
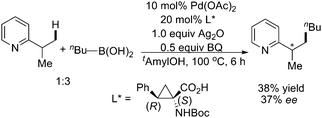 | ||
| Scheme 51 Pd(II)-catalyzed enantioselective alkylation of the sp3 C–H bond with boronic acid.62 | ||
Other than pyridine groups, Yu and coworkers investigated in the area of directed sp3 C–H activation focused on utilizing more synthetically practical functional groups, such as amino, amide, and carboxyl groups as directing groups. In 2007, they developed a protocol for Pd(II)-catalyzed carboxylic acid-directed arylation of β-C–H bonds in aliphatic acids using either a phenylboronate or ArI (Scheme 52).63 Here, the use of in situ generated potassium carboxylates as substrates led to the first observation of facile Pd-insertions into sp3 β-C–H bonds in simple aliphatic acids.
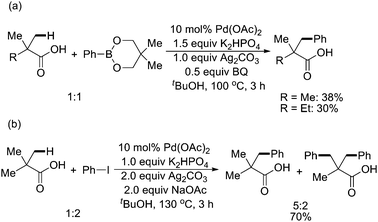 | ||
| Scheme 52 Pd(II)-catalyzed carboxylic acid-directed arylation of sp3 C–H bonds with phenylboronate and aryl iodides.63 | ||
The O-methyl hydroxamic acids, readily available from carboxylic acids, were also extremely reactive for β-C–H activationviaPd catalysis (Scheme 53).64 This reactivity was exploited to develop the first example of cross-coupling sp3 C–H bonds with alkyl boronic acids when 2,2,5,5-tetramethyltetrahydrofuran was used as the solvent, which served as a sterically bulky ligand preventing homocoupling and β-hydride elimination (Scheme 53b). Air was also shown to be a suitable stoichiometric oxidant for the catalytic oxidative coupling reaction with a high pressure and longer reaction time (Scheme 53c). Since CONHOMe groups can be readily converted to esters and amides or reduced to alkane fragments, C–H activation using this functionality will be synthetically useful.
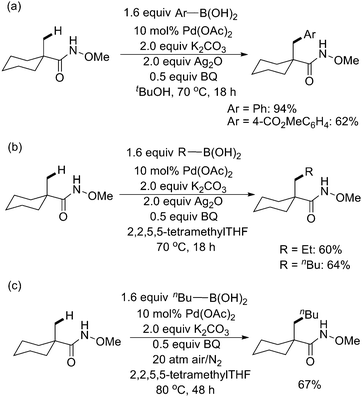 | ||
| Scheme 53 Pd(II)-catalyzed O-methyl hydroxamic acid-directed arylation and alkylation of sp3 C–H bonds with boronic acids.64 | ||
Very recently, they further successfully developed the first Pd(II)-catalyzed sp3 C–H olefination reaction using N-arylamide directing groups (Scheme 54).65 Following olefination, the resulting intermediates were found to undergo rapid 1,4-addition to give the corresponding γ-lactams. Notably, this method was effective with substrates containing α-hydrogen atoms and could be applied to effect methylene C–H olefination of cyclopropane substrates.
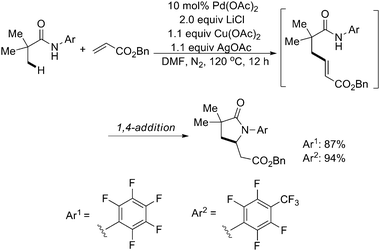 | ||
| Scheme 54 Pd(II)-catalyzed N-arylamide-directed olefination of sp3 C–H bonds with benzyl acrylate.65 | ||
With the same directing group, Pd(II)-catalyzed β-C(sp3)–H carbonylation of N-arylamides under 1 atm of CO had been achieved (Scheme 55).66 Following sp3 C–H cleavage and insertion of CO into the resulting [Pd(II)–C(sp3)] bond, intramolecular C–N reductive elimination gave the corresponding succinimides, which could be readily converted to 1,4-dicarbonyl compounds. Similarly, this method was also found to be effective with substrates containing α-hydrogen atoms and could be applied to effect methylene sp3 C–H carbonylation of cyclopropanes.
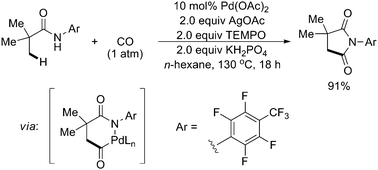 | ||
| Scheme 55 Pd(II)-catalyzed N-arylamide-directed carbonylation of sp3 C–H bonds with CO.66 | ||
In this field, another important aspect for sp3 C–H activation is to construct the C–C bond by cross-dehydrogenative coupling (CDC) reactions.67 In 2008, Liégault and Fagnou described outstanding Pd(II)-catalyzed arene–alkane coupling reactions between an azole ring and an unactivated methyl substituent (Scheme 56).68 The reactions exhibited high regioselectivity with respect to both the azole and the alkane moieties, and could be performed in an open flask using air as the terminal oxidant, which constituted an atom-economical process. Mechanistic studies indicated that a six-membered palladacycle was involved as the key intermediate generated by sequential sp2/sp3 C–H activation.
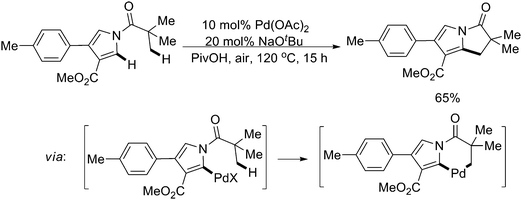 | ||
| Scheme 56 Pd(II)-catalyzed intramolecular sequential sp2/sp3 C–H activation.68 | ||
5. Direct transformation of methane
Natural gas, which is composed primarily of methane, is one of the most readily available, abundant and inexpensive C1 feedstocks. However, efficient technology has not yet been developed to economically, selectively, and directly convert methane to useful functionalized molecules. Consequently, the development of high efficient catalysis for selectively direct conversion of methane to valuable chemicals is the desirable goal of chemists today.To date, several transition metal catalysts, such as Rh,69Pt,70Hg,71etc. have been applied to realize direct homogeneous transformation of methane. In this important field, Pd also played a key role in the oxidation of methane to a methanol derivative.72 In 2003, Periana and coworkers reported a direct, selective, oxidative condensation of two methane molecules to acetic acid at 180 °C in liquid sulfuric acidviaPdSO4 catalysis (Scheme 57).73 Along with methanol, acetic acid was produced in comparable amounts with a combined selectivity of >90% and >10% yield based on the amount of methane converted. Sulfuric acid acted as the oxidant in the reaction on the basis of the observed formation of SO2. 13C isotopic labeling studies showed that both carbons of acetic acid originated from methane. A Pd(II)-catalyzed tandem process was proposed, initiated by sp3 C–H activation of methane to generate Pd–CH3 species, followed by efficient oxidative carbonylation with methanol, generated in situ from methane, to produce acetic acid.
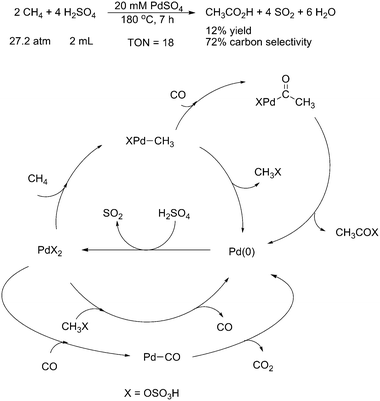 | ||
| Scheme 57 Pd(II)-catalyzed tandem oxidative condensation of methane to acetic acidvia sp3 C–H activation.73 | ||
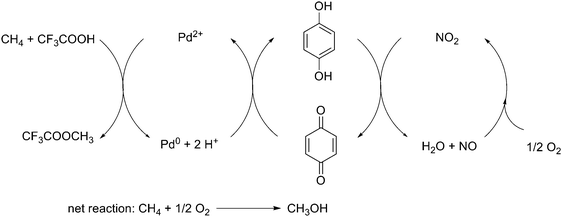
Later studies by Bao and coworkers indicated that the selectively catalytic oxidation of methane to methanol could be performed with the combination of three redox couples Pd(II)/Pd(0)–quinone/hydroquinone–NO2/NO as an electron-transfer loop, which facilitated this one-pot oxidation of methane with dioxygen as the terminal oxidant at 80 °C (Scheme 58).74 This was the first example where an organic cocatalyst significantly increased the catalytic efficiency of a transition metal in the selective oxidation of methane, which made such a transformation cleaner and more applicable. Although great achievements had been made in this field, there is still a long way to reach industrial application.
6. Summary and perspective
As a broadly used metal, palladium has been proved to play a “king” role in many organic transformations. The summary here also concludes that Pd-catalyzed sp3 C–H activation has been achieved via several pathways to construct synthetically useful C–C/C–X bonds. For allylic sp3 C–H bonds, the η3-coordination of the palladium center can activate the inert sp3 C–H bond; for non-allylic sp3 C–H bonds, the activation occurs through various pathways, such as Pd(II)/Pd(0) and Pd(II)/Pd(IV), in which heteroatom-containing chelating groups play crucial roles, as well as Pd(0)/Pd(II) catalysis, initiated by the oxidative addition of aryl halides to Pd(0).To meet the requirement of green and sustainable development, direct C–H transformation becomes more and more important. The state-of-the-art investigations in this field exhibit the promising future for clean and efficient chemical production. Obviously, although significant progresses have been achieved in the past decades with chemists' efforts, Pd-catalyzed sp3 C–H activation still seems to represent a tremendous challenge compared with sp2 C–H activation and other chemical transformations. In particular, efforts to decrease the catalyst loadings and to conduct the reactions under milder conditions are expected. Besides, more inexpensive and environment-friendly oxidants such as air are expected to be employed instead of silver salts. In addition, site-selective activation of sp3 C–H bonds for a broader scope of substrates, especially for completely unactivated alkanes, would be of great interest. Finally, the development of enantioselective sp3 C–H activation and its application in organic synthesis of natural products still face tremendous challenges. We believe that the fast development in this field will promote the chemistry in both academia and industry.
References
- For reviews on C–H bonds functionalization, see: (a) R. H. Crabtree, Chem. Rev., 1985, 85, 245 CrossRef CAS; (b) M. Beller and C. Bolm, Transition Metals for Organic Synthesis, Wiley-VCH, Weinheim, 2004 Search PubMed; (c) G. Dyker, Handbook of C–H Transformations. Applications in Organic Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed; (d) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS; (e) L.-C. Campeau and K. Fagnou, Chem. Commun., 2006, 1253 RSC; (f) R. G. Bergman, Nature, 2007, 446, 391 CrossRef CAS; (g) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS; (h) B.-J. Li, S.-D. Yang and Z.-J. Shi, Synlett, 2008, 949 CAS; (i) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS; (j) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS; (k) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (l) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS; (m) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS; (n) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16, 2654 CrossRef CAS.
- For selected examples for functionalization of aromatic C–H bonds, see: (a) E. J. Hennessy and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 12084 CrossRef CAS; (b) F. Faccini, E. Motti and M. Catellani, J. Am. Chem. Soc., 2004, 126, 78 CrossRef CAS; (c) N. P. Grimster, C. Gauntlett, C. R. A. Godfrey and M. J. Gaunt, Angew. Chem., Int. Ed., 2005, 44, 3125 CrossRef CAS; (d) L.-C. Campeau, S. Rousseaux and K. Fagnou, J. Am. Chem. Soc., 2005, 127, 18020 CrossRef CAS; (e) H. A. Chiong, Q.-N. Pham and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 9879 CrossRef CAS; (f) K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 11904 CrossRef CAS; (g) D.-H. Wang, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 17676 CrossRef CAS; (h) B.-J. Li, S.-L. Tian, Z. Fang and Z.-J. Shi, Angew. Chem., Int. Ed., 2008, 47, 1115 CrossRef CAS; (i) X. Zhao and Z. Yu, J. Am. Chem. Soc., 2008, 130, 8136 CrossRef CAS; (j) S. H. Cho, S. J. Hwang and S. Chang, J. Am. Chem. Soc., 2008, 130, 9254 CrossRef CAS.
- For reviews of Tsuji–Trost reactions, see: (a) J. Tsuji, Palladium Reagents and Catalysts New Perspectives for the 21st Century, John Wiley & Sons, Chichester, 2004 Search PubMed; (b) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS; (c) Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E.-i. Negishi, Wiley-Interscience, New York, 2002 Search PubMed; (d) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258 CrossRef CAS.
- (a) W. Kitching, Z. Rappoport, S. Winstein and W. G. Young, J. Am. Chem. Soc., 1966, 88, 2054 CrossRef CAS; (b) S. Wolfe and P. G. C. Campbell, J. Am. Chem. Soc., 1971, 93, 1497 CrossRef CAS; (c) S. Wolfe and P. G. C. Campbell, J. Am. Chem. Soc., 1971, 93, 1499 CrossRef CAS; (d) B. M. Trost and T. J. Fullerton, J. Am. Chem. Soc., 1973, 95, 292 CrossRef CAS; (e) B. M. Trost, P. E. Strege, L. Weber, T. J. Fullerton and T. J. Dietsche, J. Am. Chem. Soc., 1978, 100, 3407 CrossRef CAS; (f) B. M. Trost and P. J. Metzner, J. Am. Chem. Soc., 1980, 102, 3572 CrossRef CAS.
- M. S. Chen and M. C. White, J. Am. Chem. Soc., 2004, 126, 1346 CrossRef CAS.
- K. J. Fraunhoffer, D. A. Bachovchin and M. C. White, Org. Lett., 2005, 7, 223 CrossRef CAS.
- M. S. Chen, N. Prabagaran, N. A. Labenz and M. C. White, J. Am. Chem. Soc., 2005, 127, 6970 CrossRef CAS.
- K. J. Fraunhoffer, N. Prabagaran, L. E. Sirois and M. C. White, J. Am. Chem. Soc., 2006, 128, 9032 CrossRef CAS.
- J. H. Delcamp and M. C. White, J. Am. Chem. Soc., 2006, 128, 15076 CrossRef CAS.
- D. J. Covell and M. C. White, Angew. Chem., Int. Ed., 2008, 47, 6448 CrossRef CAS.
- T. Mitsudome, T. Umetani, N. Nosaka, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, Angew. Chem., Int. Ed., 2006, 45, 481 CrossRef CAS.
- A. N. Campbell, P. B. White, I. A. Guzei and S. S. Stahl, J. Am. Chem. Soc., 2010, 132, 15116 CrossRef CAS.
- N. A. Vermeulen, J. H. Delcamp and M. C. White, J. Am. Chem. Soc., 2010, 132, 11323 CrossRef CAS.
- K. J. Fraunhoffer and M. C. White, J. Am. Chem. Soc., 2007, 129, 7274 CrossRef CAS.
- G. T. Rice and M. C. White, J. Am. Chem. Soc., 2009, 131, 11707 CrossRef CAS.
- L. Wu, S. Qiu and G. Liu, Org. Lett., 2009, 11, 2707 CrossRef CAS.
- S. A. Reed and M. C. White, J. Am. Chem. Soc., 2008, 130, 3316 CrossRef CAS.
- G. Liu, G. Yin and L. Wu, Angew. Chem., Int. Ed., 2008, 47, 4733 CrossRef CAS.
- S. A. Reed, A. R. Mazzotti and M. C. White, J. Am. Chem. Soc., 2009, 131, 11701 CrossRef CAS.
- G. Yin, Y. Wu and G. Liu, J. Am. Chem. Soc., 2010, 132, 11978 CrossRef CAS.
- M. R. Luzung, C. A. Lewis and P. S. Baran, Angew. Chem., Int. Ed., 2009, 48, 7025 CrossRef CAS.
- S. Lin, C.-X. Song, G.-X. Cai, W.-H. Wang and Z.-J. Shi, J. Am. Chem. Soc., 2008, 130, 12901 CrossRef CAS.
- A. J. Young and M. C. White, J. Am. Chem. Soc., 2008, 130, 14090 CrossRef CAS.
- (a) R. Uson, J. Fornies and R. Navarro, J. Organomet. Chem., 1975, 96, 307 CrossRef CAS; (b) B. M. Trost and G. J. Tanoury, J. Am. Chem. Soc., 1987, 109, 4753 CrossRef CAS; (c) A. J. Canty, Acc. Chem. Res., 1992, 25, 83 CrossRef CAS; (d) P. K. Byers, A. J. Canty, B. W. Skelton and A. H. White, J. Chem. Soc., Chem. Commun., 1986, 1722 RSC; (e) A. J. Canty, M. C. Denney, G. van Koten, B. W. Skelton and A. H. White, Organometallics, 2004, 23, 5432 CrossRef CAS; (f) L.-M. Xu, B.-J. Li, Z. Yang and Z.-J. Shi, Chem. Soc. Rev., 2010, 39, 712 RSC.
- A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300 CrossRef CAS.
- K. L. Hull, W. Q. Anani and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 7134 CrossRef CAS.
- D. Kalyani, N. R. Deprez, L. V. Desai and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 7330 CrossRef CAS.
- N. R. Deprez and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 11234 CrossRef CAS.
- D. Shabashov and O. Daugulis, Org. Lett., 2005, 7, 3657 CrossRef CAS.
- M. Catellani, E. Motti and S. Ghelli, Chem. Commun., 2000, 2003 RSC.
- L.-C. Campeau, D. J. Schipper and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 3266 CrossRef CAS.
- B. Qian, S. Guo, J. Shao, Q. Zhu, L. Yang, C. Xia and H. Huang, J. Am. Chem. Soc., 2010, 132, 3650 CrossRef CAS.
- A. D. Ryabov, Chem. Rev., 1990, 90, 403 CrossRef CAS.
- L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542 CrossRef CAS.
- S. R. Neufeldt and M. S. Sanford, Org. Lett., 2010, 12, 532 CrossRef CAS.
- H.-Y. Thu, W.-Y. Yu and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 9048 CrossRef CAS.
- For a lead review on the use of chiral auxiliaries, see: Y. Gnas and F. Glorius, Synthesis, 2006, 1899 Search PubMed.
- R. Giri, X. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 2112 CrossRef.
- R. Giri, M. Wasa, S. P. Breazzano and J.-Q. Yu, Org. Lett., 2006, 8, 5685 CrossRef CAS.
- R. Giri, J. Liang, J.-G. Lei, J.-J. Li, D.-H. Wang, X. Chen, I. C. Naggar, C. Guo, B. M. Foxman and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 7420 CrossRef CAS.
- D.-H. Wang, X.-S. Hao, D.-F. Wu and J.-Q. Yu, Org. Lett., 2006, 8, 3387 CrossRef CAS.
- J. J. Neumann, S. Rakshit, T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2009, 48, 6892 CrossRef CAS.
- G. Dyker, Angew. Chem., Int. Ed. Engl., 1992, 31, 1023 CrossRef.
- G. Dyker, Angew. Chem., Int. Ed. Engl., 1994, 33, 103 CrossRef.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685 CrossRef CAS.
- J. Zhao, M. Campo and R. C. Larock, Angew. Chem., Int. Ed., 2005, 44, 1873 CrossRef CAS , and references therein.
- M. Wasa, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 9886 CrossRef CAS.
- M. Wasa and J.-Q. Yu, Tetrahedron, 2010, 66, 4811 CrossRef CAS.
- A. Renaudat, L. Jean-Gérard, R. Jazzar, C. E. Kefalidis, E. Clot and O. Baudoin, Angew. Chem., Int. Ed., 2010, 49, 7261 CrossRef CAS.
- O. Baudoin, A. Herrbach and F. Guéritte, Angew. Chem., Int. Ed., 2003, 42, 5736 CrossRef CAS.
- J. Hitce, P. Retailleau and O. Baudoin, Chem.–Eur. J., 2007, 13, 792 CrossRef CAS.
- M. Chaumontet, R. Piccardi, N. Audic, J. Hitce, J.-L. Peglion, E. Clot and O. Baudoin, J. Am. Chem. Soc., 2008, 130, 15157 CrossRef CAS.
- (a) D. García-Cuadrado, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 1066 CrossRef CAS; (b) M. Lafrance, C. N. Rowley, T. K. Woo and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 8754 CrossRef.
- S. Rousseaux, M. Davi, J. Sofack-Kreutzer, C. Pierre, C. E. Kefalidis, E. Clot, K. Fagnou and O. Baudoin, J. Am. Chem. Soc., 2010, 132, 10706 CrossRef CAS.
- M. Lafrance, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2007, 129, 14570 CrossRef CAS.
- T. Watanabe, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2008, 10, 1759 CrossRef CAS.
- S. Rousseaux, S. I. Gorelsky, B. K. W. Chung and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 10692 CrossRef CAS.
- V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS.
- B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391 CrossRef CAS.
- D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef CAS.
- X. Chen, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 12634 CrossRef CAS.
- B.-F. Shi, N. Maugel, Y.-H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882 CrossRef CAS.
- R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510 CrossRef CAS.
- D.-H. Wang, M. Wasa, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 7190 CrossRef CAS.
- M. Wasa, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 3680 CrossRef CAS.
- E. J. Yoo, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 17378 CrossRef CAS.
- For reviews on CDC reactions, see: (a) C.-J. Li and Z. Li, Pure Appl. Chem., 2006, 78, 935 CrossRef CAS; (b) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS; (c) C. Scheuermann, Chem.–Asian J., 2010, 5, 436 CrossRef CAS.
- B. Liégault and K. Fagnou, Organometallics, 2008, 27, 4841 CrossRef CAS.
- D. A. Hickman and L. D. Schmidt, Science, 1993, 259, 343 CrossRef CAS.
- (a) R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh and H. Fujii, Science, 1998, 280, 560 CrossRef CAS; (b) M. Lin, C. Shen, E. A. Garcia-Zayas and A. Sen, J. Am. Chem. Soc., 2001, 123, 1000 CrossRef CAS; (c) I. Bar-Nahum, A. M. Khenkin and R. Neumann, J. Am. Chem. Soc., 2004, 126, 10236 CrossRef CAS.
- R. A. Periana, D. J. Taube, E. R. Evitt, D. G. Löffler, P. R. Wentrcek, G. Voss and T. Masuda, Science, 1993, 259, 340 CAS.
- L.-C. Kao, A. C. Hutson and A. Sen, J. Am. Chem. Soc., 1991, 113, 700 CrossRef CAS.
- R. A. Periana, O. Mironov, D. Taube, G. Bhalla and C. J. Jones, Science, 2003, 301, 814 CrossRef CAS.
- Z. An, X. Pan, X. Liu, X. Han and X. Bao, J. Am. Chem. Soc., 2006, 128, 16028 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |