Heterogeneous catalysis of the glycerol hydrogenolysis
Yoshinao
Nakagawa
and
Keiichi
Tomishige
*
Department of Applied Chemistry, School of Engineering, Tohoku University, 6-6-07 Aoba, Aramaki, Aoba-ku, Sendai, 980-8579, Japan. E-mail: tomi@erec.che.tohoku.ac.jp; Fax: +81 22-795-7214
First published on 10th February 2011
Abstract
Glycerol has become an important feedstock for chemicals. Selective hydrogenolysis of glycerol has been attempted using several types of catalysts: non-noble-metal catalysts, noble-metal catalysts with an acid as an additive, noble-metal catalysts combined with a base, and metal-oxide-modified noble-metal catalysts. The former three approaches give 1,2-propanediol as a main product, while the mechanisms may be different. Only the last one can selectively give the more valuable 1,3-propanediol.
 Yoshinao Nakagawa | Yoshinao Nakagawa obtained his PhD in 2005 from Graduate School of Engineering, The University of Tokyo, under the guidance of Prof. N. Mizuno. After 4 years of postdoctoral research at the University of Tokyo, he joined the research group of Keiichi Tomishige at University of Tsukuba. In 2010, he moved to Tohoku University and became an assistant professor. His current research interests are catalytic oxidations and reductions of bio-related chemicals. |
 Keiichi Tomishige | Keiichi Tomishige received his BS, MS and PhD from Graduate School of Science, The University of Tokyo, with Prof. Y. Iwasawa. During his PhD course in 1994, he moved to Graduate School of Engineering, The University of Tokyo, as a research associate and worked with Prof. K. Fujimoto. In 1998, he became a lecturer, and then he moved to Institute of Materials Science, University of Tsukuba, as a lecturer in 2001. In 2004, he became an associate professor, Graduate School of Pure and Applied Sciences, University of Tsukuba. Since 2010, he has been a professor, School of Engineering, Tohoku University. His research interests are catalytic conversions of biomass, CO2, and natural gas. |
1. Introduction
The use of renewable resources as replacements for fossil fuels is essential for the sustainable development of our society. Because biomass is the only renewable source of organic carbon, replacements for petroleum-derived liquid fuels and plastics require the use of biomass.1–4Alkyl-fatty esters, or biodiesel, are produced from vegetable oils (or animal fats) with transesterification and can be used directly in diesel engines with no or minor engine modifications. The production of biodiesel is rapidly growing, from <1 million tons in 2000 (annual production capacity)5 to ca. 10 million tons in 2010.6 The transesterification process co-generates glycerol at a rate of 1 mol for every 3 mol of alkyl-fatty esters synthesized; approximately 10 wt% of the total product. The amount of glycerol produced from the biodiesel process is comparable to or even larger than that produced from the traditional processes such as soap manufacture (0.5 million tons per year).7 Because of the oversupply of glycerol, the market price of glycerol has been dropped sharply (1.5–2 $ kg−1 to 0.4 $ kg−1-purified glycerol).8 Some biodiesel companies have severe problems in the quite expensive disposal processes of excess glycerol. The value-addition of glycerol will contribute to the cost-competitiveness of biodiesel processes. As a consequence, glycerol has been recognized as one of the most important building blocks in the conversion of biomass-derived feedstock to value-added chemicals.Glycerol is a multifunctionalized compound and a number of conversion processes of glycerol have been reported: fermentation, oxidation, reduction, dehydration, gasification, carboxylation, esterification and so on (Scheme 1).6,9 Hydrogenolysis is a class of reduction that involves chemical bond dissociation in an organic substrate and simultaneous addition of hydrogen to the resulting molecular fragments. Because glycerol is a saturated compound and has higher O/C content than most commodity chemicals, C–O hydrogenolysis is clearly one of the most attractive routes in the chemical conversion of glycerol.10 The pathways and products of hydrogenolysis of glycerol are shown in Scheme 2. Among the products in the glycerol hydrogenolysis, 1,3-propanediol (1,3-PD), 1,2-propanediol (1,2-PD; propylene glycol), 1-propanol (1-PrOH), 2-propanol (2-PrOH) and ethylene glycol (EG) are industrially important. 1,3-PD can be used as a monomer for polycondensations to produce plastics with special properties, i.e. polyesters, polyethers and polyurethanes. In plastics that use 1,3-PD as a monomer, polypropylene terephthalate (PPT) has gained increasing attention. PPT is a biodegradable polyester and has great potential for use in carpet and textile manufacturing. Shell and DuPont have recently started commercial production processes of PPT. In the Shell process, 1,3-PD is chemically obtained from hydroformylation of ethylene oxide and subsequent hydrogenation. In the DuPont process, fermentation of glucose is operated to produce 1,3-PD. Another chemical synthesis using hydration of acrolein is operated. The annual world production of 1,3-PD is in the order of 105 tons.11 While more 1,3-PD is produced via chemical syntheses, the harsh conditions and costly catalysts make chemical syntheses expensive. Because of the high price and large demands, 1,3-PD is the most attractive target in the conversion of biodiesel glycerol. 1,2-PD is a non-toxic chemical, which is extensively used as a monomer for polyester resins, as an antifreeze agent, and in paints, liquid detergent, cosmetics, and food, etc. The global production of 1,2-PD is 1.4 million tons per year.12 Currently, 1,2-PD is commercially produced from propylene oxide with hydration and sold at 1.0–2.2 $ kg−1.8 Although cost-competitiveness is yet a key barrier, the conversion of glycerol into 1,2-PD is a promising process. Archer Daniels Midland Company has very recently opened the first commercial plant with a capacity of 0.1 million tons 1,2-PD from biodiesel glycerol. Other projects for the production of 1,2-PD have been announced. Overhydrogenolysis products, 1- and 2-PrOHs, are also useful chemicals. 1-PrOH is currently produced viahydroformylation of ethylene and subsequent hydrogenation and is used mainly as a solvent, a printing ink, and a chemical intermediate for the production of n-propyl acetate. The annual global production of 1-PrOH is 0.2 million tons13 and the market price is around 1.2 $ kg−1.82-PrOH is produced by the hydration of propylene and is used mainly as a solvent. The global production of 2-PrOH is 2 million tons14 and the market price is 0.6–1.1 $ kg−1.8 EG is massively produced via hydration of ethylene oxide and is mainly used as an antifreeze and a raw material for polyethylene terephthalate. The global production capacity of EG is 7 million tons15 and the market price is around 1 $ kg−1. PrOHs and EG have not been regarded as main targets in the conversion of glycerol because of the lower market prices and poorer atom efficiency in the production from glycerol than from 1,3- and 1,2-PD, i.e. hydrogenolysis to PrOHs consumes two hydrogen molecules; production of EG utilizes only 2 of 3 carbon atoms in a glycerol molecule. However, the shortage of fossil fuels or the need to decrease CO2 emission may highlight glycerol-based production processes for PrOHs or EG. In this paper, the development of the catalytic systems for the selective hydrogenolysis of glycerol is examined. The proposed mechanisms of these systems and the applicability to other biomass-derived substrates are also discussed.
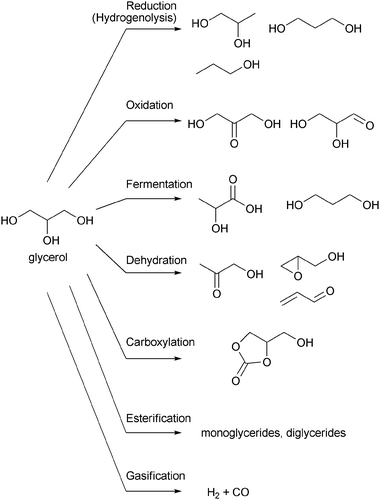 | ||
| Scheme 1 Examples of the conversions of glycerol. | ||
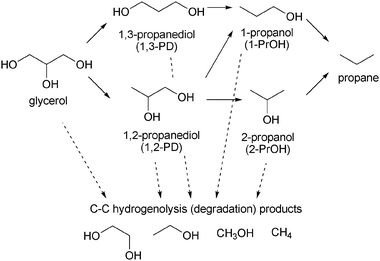 | ||
| Scheme 2 Pathways of hydrogenolysis of glycerol. | ||
2. Noble-metal catalysts
2.1 Monometallic catalysts
Since hydrogenolysis uses hydrogen as a reactant, the hydrogenolysis catalyst must have an ability to activate hydrogen molecules. Noble metals are well known to be able to activate hydrogen molecules and are widely used in hydrogenation catalysts. In the hydrogenolysis of glycerol (Table 1), ruthenium is frequently used as an active component. An early study by Montassier et al. showed that the hydrogenolysis of aqueous glycerol over Ru/C at 483 K gives EG (ca. 50% selectivity), ethane (ca. 25%) and 1,2-PD (ca. 12%).16 The lost carbon in the C–C hydrogenolysis (to EG or ethane) is released as methane. The poisoning of sulfur to the Ru surface increases the selectivity to 1,2-PD (up to 79%). Later studies showed that in milder conditions the main product is also 1,2-PD over the carbon-supported Ru catalyst.17–19 However, the formation of EG is always accompanied and the selectivity to 1,2-PD is not higher than 70% without additives or co-catalyst. Montassier et al. proposed a mechanism for the formation of 1,2-PD and EG over the Ru catalyst (Scheme 3):16 First, glycerol is reversibly dehydrogenated to glyceraldehyde. Dehydration of glyceraldehyde to 2-hydroxyacrolein and subsequent hydrogenation gives 1,2-PD. Retro-aldol reaction of glyceraldehyde to glycolaldehyde and subsequent hydrogenation gives EG. Other hydrogenolysis mechanisms have been also proposed, especially for 1,2-PD formation, as discussed later. Dasari et al. compared the activities of various commercial hydrogenation catalysts in the hydrogenolysis of glycerol at 473 K.17Pd/C showed low activity. Pt/C showed higher selectivity to 1,2-PD (82.7%) and higher yield (28.6%) than Ru/C (40.0% selectivity and 17.5% yield). However, the non-noble-metal copper-chromite catalyst showed even higher activity and selectivity (85.0%; 46.6% yield), and the catalysis of Pt/C was not investigated further by the authors. The higher selectivity to 1,2-PD over Pt/C than over Ru/C was also reported by Maris and Davis.20 Furikado et al. compared the activity of various supported noble-metal catalysts (Rh, Ru, Pt and Pd over C, SiO2 and Al2O3) in the hydrogenolysis of aqueous glycerol at a much lower temperature of 393 K.21 All the supported Pd and Pt catalysts exhibited very low activity (<1% conversion) in these mild conditions. All the Al2O3-supported metal catalysts also showed low activity (<1% conversion). In the case of Ru catalysts, active carbon (3.5% conversion) is a much more suitable support than SiO2 (0.2% conversion) and Al2O3. Of all the catalysts tested, Rh/SiO2 gave the highest glycerol conversion (7.2% conversion). In addition, the selectivity to C–C hydrogenolysis (degradation) products on Rh/SiO2 (total 7.1%) was much lower than that on Ru/C (total 42%). At higher reaction temperatures, the selectivity to degradation products on Rh/SiO2 increased significantly while the increase of glycerol conversion was smaller than that on Ru/C. These results show that the reaction over Rh/SiO2 should be carried out at lower reaction temperature, where Rh/SiO2 showed much higher activity and higher hydrogenolysis selectivity than Ru/C. The product distribution of Rh/SiO2-catalyzed hydrogenolysis was 1,3-PD![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1,2-PD∶1-PrOH∶2-PrOH∶degradation products = 8∶35∶38∶13∶7 at the 7.2% conversion level. In order to explain the product distribution, hydrogenolyses of 1,2- and 1,3-PDs over Rh/SiO2 were also tested. The hydrogenolysis of 1,2-PD can give 1-PrOH and 2-PrOH, and the hydrogenolysis of 1,3-PD can give only 1-PrOH. The hydrogenolysis of 1,2-PD proceeded with higher selectivity to 1-PrOH (66.5%) than to 2-PrOH (20.9%). The 1-PrOH/2-PrOH selectivity ratio in the 1,2-PD hydrogenolysis was almost the same as that in the glycerol hydrogenolysis, suggesting that 1-PrOH and 2-PrOH are mainly formed via1,2-PD. The three-step mechanism as shown in Scheme 3 cannot be applied to the formation of 1,3-PD or the overhydrogenolysis of 1,2-PD to 2-PrOH. Other mechanisms should be considered at least for Rh catalysts.
∶1,2-PD∶1-PrOH∶2-PrOH∶degradation products = 8∶35∶38∶13∶7 at the 7.2% conversion level. In order to explain the product distribution, hydrogenolyses of 1,2- and 1,3-PDs over Rh/SiO2 were also tested. The hydrogenolysis of 1,2-PD can give 1-PrOH and 2-PrOH, and the hydrogenolysis of 1,3-PD can give only 1-PrOH. The hydrogenolysis of 1,2-PD proceeded with higher selectivity to 1-PrOH (66.5%) than to 2-PrOH (20.9%). The 1-PrOH/2-PrOH selectivity ratio in the 1,2-PD hydrogenolysis was almost the same as that in the glycerol hydrogenolysis, suggesting that 1-PrOH and 2-PrOH are mainly formed via1,2-PD. The three-step mechanism as shown in Scheme 3 cannot be applied to the formation of 1,3-PD or the overhydrogenolysis of 1,2-PD to 2-PrOH. Other mechanisms should be considered at least for Rh catalysts.
| Catalyst | H2/MPa | Glycerol/water/catalyst/g | Temp./K | Time/h | Conv. (%) | Products (selectivity (%)) | Ref. |
|---|---|---|---|---|---|---|---|
| a PD = propanediol, EG = ethylene glycol, PrOH = propanol, n.r. = not reported. b Selectivities were reported at low conversion. | |||||||
| Ru/C, 10 wt% | 6 | 6.5/154/n.r. | 483 | b | b | 1,2-PD (12), EG (47), propane (8), ethane (26) | 16 |
| Ru–S/C, 10 wt% | 6 | 6.5/154/n.r. | 483 | b | b | 1,2-PD (79), EG + methane (21) | 16 |
| Ru/CNT, 5 wt% | 4 | 4/16/0.25 | 473 | 12 | 42.3 | 1,2-PD (60.2), 1-PrOH (2), EG (20.4) | 19 |
| Pt/C, 5 wt% | 1.38 | 50/10/2.5 | 473 | 24 | 34.6 | 1,2-PD (82.7) | 17 |
| Pd/C, 5 wt% | 1.38 | 50/10/2.5 | 473 | 24 | 5 | 1,2-PD (72.0) | 17 |
| Ru/C, 5 wt% | 1.38 | 50/10/2.5 | 473 | 24 | 43.7 | 1,2-PD (40.0) | 17 |
| Rh/SiO2, 4 wt% | 8 | 4/16/0.15 | 393 | 10 | 7.2 | 1,2-PD (38.1), 1,3-PD (7.9), 1-PrOH (35.2), 2-PrOH (12.6) | 21 |
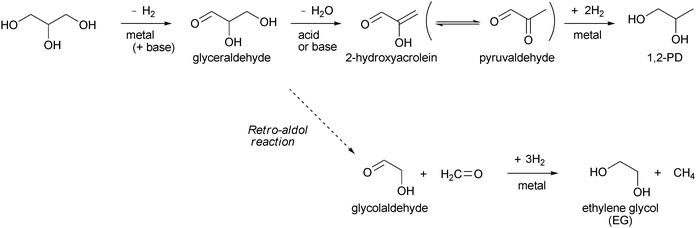 | ||
| Scheme 3 Glyceraldehyde-based mechanism. | ||
Supported Pt catalysts are also active in the reforming of aqueous glycerol to hydrogen and carbon dioxide at around 500 K (eqn (1)). D'Hondt et al. combined the hydrogenolysis of glycerol with in situ production of H2 by the reforming of glycerol.22 Over Pt/NaY as a catalyst for both reactions, 57% yield of 1,2-PD was obtained at 503 K under inert conditions, while 9.1% of glycerol was consumed by the reforming reaction. Roy et al. expanded the concept by using the mixture of Ru/Al2O3 and Pt/Al2O3 as catalyst for hydrogenation/hydrogenolysis and reforming, respectively.23 The main products were 1,2-PD (47.2% selectivity), EG (6.3%), CH4 (18.3%) and CO2 (14.3%), at a 50.2% conversion of glycerol at 493 K.
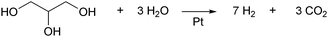 | (1) |
2.2 Noble-metal catalysts combined with an acid
As stated above, noble-metal catalysts without additives generally give more than one product. There are several approaches to improve the selectivity to a sole product, and the most studied one is the addition of an acid (Table 2). The addition of acid catalysts to Ru/C was very effective in the increase of conversion and selectivity to 1,2-PD.24,25 Acid-type cation-exchange resin Amberlyst 15 was the most effective co-catalyst in various Brønsted acids such as sulfonated zirconia, zeolites, and homogeneous H2SO4. Addition of HCl significantly decreased the activity because of the poisoning of Ru surface with Cl− ions. The addition of Amberlyst to Rh/C or Rh/SiO221 also enhanced the catalytic activity; however, the effects were smaller than for Ru/C. In the case of Pd/C and Pt/C, the addition of Amberlyst was less effective. A Ru/C catalyst on carbon support with a low surface area (ca. 250 m2 g−1) showed better performance than those on the active carbon with a high surface area.18 It is suggested that Ru species supported on the graphite phase with less surface functional groups can be active. In addition, treatment of Ru/C catalysts prepared from Ru(NO)(NO3)3 with Ar flowing at the appropriate temperature (573 K) enhanced the performance compared to that of the commercially available Ru/C catalysts. Using the combination of the home-made Ru/C and Amberlyst 15 co-catalyst, 79.3% conversion of dilute aqueous glycerol and 74.7% selectivity to 1,2-PD were achieved at 393 K. The other products were 1-PrOH (7.7% selectivity), 2-PrOH (1.6%), and degradation products (16.9%). The formation of 1,3-PD was negligible. The reaction tests of 1,2-PD and 1,3-PD showed that the conversion in the 1,3-PD reaction was comparable to that of the glycerol reaction; in contrast, the conversion of 1,2-PD was much lower than that of glycerol. The low reactivity of 1,2-PD is related to the high yield of 1,2-PD in the glycerol reaction, and the high reactivity of 1,3-PD causes the very low yield of 1,3-PD in the glycerol reaction. The hydrogenolysis of 1,2-PD proceeded with similar selectivities to 1-PrOH (35.7%) and 2-PrOH (30.9%) while that of glycerol gave higher selectivities to 1-PrOH, suggesting that 1-PrOH was formed mainly via1,3-PD in the glycerol hydrogenolysis over Ru/C + Amberlyst. From the comparison of Ru/C with and without Amberlyst, it was found that the TOF of 1,3-PD + 1-PrOH was not influenced by the presence of Amberlyst. These results suggest that the formation of 1,3-PD can be catalyzed by only Ru/C. In contrast, the TOF of the 1,2-PD formation was enhanced remarkably by the presence of Amberlyst. This result suggests that Amberlyst and Ru/C cooperatively catalyze 1,2-PD formation. The formation of 1,2-PD by the metal–acid bifunctional catalyst system is explained by the mechanism where the acid function plays a role of dehydration reaction and the metal catalyzes the hydrogenation reaction (Scheme 4).17 The dehydration of glycerol produces acetol (hydroxyacetone), and the successive hydrogenation produces 1,2-PD. In fact, Ru/C can catalyze the hydrogenation of acetol to 1,2-PD very efficiently. In a similar way, 1,3-PD might be produced by the dehydration of glycerol to 3-hydroxypropanal and subsequent hydrogenation. However, the formation of 1,3-PD was mainly catalyzed by Ru/C in this system as explained above, and the indirect dehydration–hydrogenation to 1,3-PD hardly proceeded. The preferable formation of acetol to 3-hydroxypropanal is probably due to the thermodynamic stability of acetol. The fact that acetol was not detected in the reaction solutions is probably due to the large amount of water and low reaction temperature. The dehydrogenation–dehydration–hydrogenation mechanism as shown in Scheme 3 may participate. However, the C–O hydrogenolysis rate over Ru/C + Amberlyst increases with increasing hydrogen pressure, which contradicts the existence of the initial reversible dehydrogenation step; high hydrogen pressure should shift the equilibrium to the reactant side and thus lower the rate of later steps.| Catalysts | H2/MPa | Glycerol/water/catalyst/g | Temp./K | Time/h | Conv. (%) | Products (selectivity (%)) | Ref. |
|---|---|---|---|---|---|---|---|
| a PD = propanediol, EG = ethylene glycol, PrOH = propanol. b Homogeneous catalysts. | |||||||
| Ru/C + Amberlyst 15 | 8 | 4/16/0.15 + 0.3 | 393 | 10 | 79.3 | 1,2-PD (74.7), 1-PrOH (7.7), 2-PrOH (1.6), EG (6.8) | 18 |
| Ru/C + Amberlyst 70 | 8 | 4/16/0.015 + 140 μmol H+ | 453 | 10 | 48.8 | 1,2-PD (70.2), 1,3-PD (1.3), 1-PrOH (7.1), 2-PrOH (1.0), EG (8.3) | 26 |
| Ru/C, 5 wt% + Nb2O5 | 6 | 10/40/0.15 + 0.30 | 453 | 8 | 44.6 | 1,2-PD (60.9), EG (29.1) | 27 |
| Ru/C, 5 wt% + H3PW12O40/ZrO2, 15 wt% | 6 | 10/40/0.15 + 0.30 | 453 | 8 | 44.0 | 1,2-PD (64.3), EG (27.0) | 27 |
| Ru/Cs2.5H0.5PW12O40, 5 wt% | 0.5 | 1/4/0.2 | 423 | 10 | 21 | 1,2-PD (95.8), 1-PrOH (4.2) | 28 |
| Ru/Al2O3, 5 wt% | 8 | 120/0/0.9 | 513 | 5 | 69 | 1,2-PD (37.9), 1,3-PD (0.7), propanols (45) | 29 |
| Pt/SiO2–Al2O3, 1 wt% | 4.5 | 8/32/1.33 | 493 | 24 | 19 | 1,2-PD (31.9), 1,3-PD (4.5), PrOHs (53.8), acetol (1,2) | 30 |
| [Ru(OH2)3(4’-phenyl-2,2’:6’,2’’-terpy)](OTf)2 + H2WO4b | 5.5 | 0.5 M glycerol, 0.5 mol% Ru, 4 mol% W | 523 | 24 | 100 | 1-PrOH (24), propane (76) | 31 |
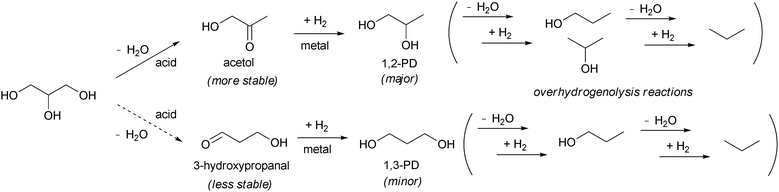 | ||
| Scheme 4 Dehydration–hydrogenation mechanism. | ||
A weak point in the system of Ru/C with Amberlyst 15 is that the reaction temperature is limited by the thermal stability of the ion-exchange resin. A heat-resistant ion-exchange resin Amberlyst 70 was applied to the co-catalyst with Ru/C.26 The Ru/C + Amberlyst 70 system could be operated at 453 K and showed much higher reaction rate than the Ru/C + Amberlyst 15 system at 393 K. Almost the same selectivity to 1,2-PD (70.2%) was achieved. Inorganic acids are thermally more stable than ion-exchange resins and can be used at higher temperature. Balaraju et al. used the combination of the commercial Ru/C catalyst with different inorganic solid acids such as niobia, zirconia-supported 12-tungstophosphoric acid, and acid caesium 12-tungstophosphate for the hydrogenolysis of aqueous glycerol at 453 K.27 The co-catalysts that possess moderate acid sites such as niobia and zirconia-supported 12-tungstophosphoric acid exhibited a better effect. A conversion of 62.8% and 1,2-PD selectivity of 66.5% was achieved using niobia as a co-catalyst. Instead of the addition of an external acid, noble-metal catalysts supported on the solid acid have been also tested. Alhanash et al. prepared ruthenium-doped acidic heteropoly salt Cs2.5H0.5[PW12O40] and used it as a catalyst for the hydrogenolysis of aqueous glycerol.28 At 423 K, an excellent selectivity to 1,2-PD (95.8%) was achieved at a 21% conversion level. At higher temperature, formation of EG started and the selectivity to 1,2-PD became lower. It should be noted that significant amount of acetol was detected after the reaction at higher temperature (5.7 and 8.6% selectivity at 453 and 473 K, respectively). Vasiliadou et al. prepared alumina-supported ruthenium catalysts with moderate acid sites and tested them in the hydrogenolysis of neat glycerol at 513 K.29 In these harsh conditions, overhydrogenolysis to 1- and 2-PrOHs proceeded significantly (45% selectivity to 1- or 2-PrOHs at a 69% conversion level). Both the conversion of glycerol and the selectivity to overhydrogenolysis products increased with the increase of the total acidity of the catalytic materials. Gandarias et al. reported the hydrogenolysis of aqueous glycerol over the platinum catalyst supported on the acidic amorphous silica-alumina.30 At 493 K, the selectivity to 1,2-PD was 31.9% at the 19% conversion level. The overhydrogenolysis to 1- and 2-PrOHs proceeded significantly (53.8% selectivity), while the degradation side-reactions were minor (total 8.3% selectivity). At higher 513 K, the conversion of glycerol reached 90%. However, selectivity to 1,2-PD dropped to 11.2% and both overhydrogenolysis and degradation reactions proceeded more significantly (59.7% selectivity to PrOHs and 23.0% to degradation products). Taher et al. examined the catalysis of homogeneous Ru complexes in the presence of triflic acid or tungstic acid.31 At 473 K, glycerol conversions of around 20% were obtained and the main products were 1-PrOH and propane. At 523 K and with sufficient amount of acid, glycerol was totally converted and propane was dominantly formed. These results showed that the combination of an acid and metal catalyst (especially Ru) gives 1,2-PD as a main product from aqueous glycerol at lower temperature (≤453 K). In harsh conditions (≥513 K), the overhydrogenolysis reactions proceed significantly and the main products are shifted to PrOHs and even propane.
2.3 Noble-metal catalysts combined with a base
Bases are also used as co-catalysts in the hydrogenolysis of glycerol (Table 3), although the cases are fewer than using acidic co-catalysts. Feng et al. reported the hydrogenolysis of glycerol catalyzed by Ru/TiO2 in the presence of various bases at 443 K.32 The addition of lithium or sodium base dramatically increased the conversion of glycerol and the selectivity to 1,2-PD and decreased the selectivity to EG. The highest conversion of glycerol (89.6%) and the highest selectivity to 1,2-PD (86.8%) were observed using LiOH. The effect of the amount of base on the reaction was investigated using LiOH as an additive. As the amount of LiOH increased, the conversion of glycerol and the selectivity to 1,2-PD initially exhibited an increase and ultimately reached a maximum. After that, they began to decrease. In contrast, the selectivity to EG decreased with an increase of LiOH amount. The effect of LiOH addition is explained based on the glyceraldehyde-based mechanism as shown in Scheme 3: the base can aid the initial dehydrogenation of glycerol to glyceraldehyde. The decrease in the selectivity to EG can be due to the fact that the retro-aldol reaction of glyceraldehyde is very difficult in basic conditions. Maris and Davis et al. investigated the addition effect of a base to Ru/C- or Pt/C-catalyzed hydrogenolysis of very dilute glycerol (1 wt% aq.) at 473 K.33,34 The presence of 0.8 M NaOH or CaO enhanced the rate of glycerol hydrogenolysis over both catalysts, and the extent of enhancement was greater over Pt/C than Ru/C. The main products were 1,2-PD, EG and lactates, while lactates or lactic acid were not formed under neutral conditions. Formation of lactate was explained by a mechanism that is based loosely on the glyceraldehyde-based mechanism. The base-catalyzed dehydration of glyceraldehyde yields pyruvaldehyde, the keto-form of 2-hydroxyacrolein. Then pyruvaldehyde is transformed into lactate in the presence of a base (eqn (2)).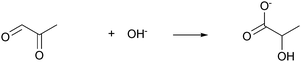 | (2) |
| Catalysts | H2/MPa | Glycerol/water/catalyst/g | Temp./K | Time/h | Conv. (%) | Products (selectivity (%)) | Ref. |
|---|---|---|---|---|---|---|---|
| a PD = propanediol, EG = ethylene glycol, PrOH = propanol. | |||||||
| Ru/TiO2, 5 wt% + LiOH | 3 | 1/4/0.102 + 0.024 | 443 | 12 | 89.6 | 1,2-PD (86.8), EG (7.5) | 32 |
| Ru/C, 5 wt% + CaO | 4 | 1.5/150/0.1 + 0.084 | 473 | 5 | 50 | 1,2-PD (46), lactate (32), EG (16) | 33 |
| Pt/C, 3 wt% + CaO | 4 | 1.5/150/0.35 + 0.084 | 473 | 5 | 40 | 1,2-PD (71), lactate (19), EG (9) | 33 |
| PtRu/C, 1.9 wt% Pt, 4.7 wt% Ru + NaOH | 4 | 1.5/150/0.1 + 4.8 | 473 | 5 | 100 | 1,2-PD (18), lactate (37), formate (37) | 34 |
| AuRu/C, 0.85 wt% Au, 5.0 wt% Ru + NaOH | 4 | 1.5/150/0.1 + 4.8 | 473 | 5 | 100 | 1,2-PD (12), lactate (38), formate (44) | 34 |
| Pt/hydrotalcite, 2 wt% Pt | 3 | 4/16/0.5 | 493 | 20 | 92.1 | 1,2-PD (93) | 35 |
2.4 Metal-oxide-modified noble-metal catalysts
As stated above, use of acid or base as a co-catalyst gives 1,2-PD as a main product. To obtain more valuable 1,3-PD, different approaches would be needed. Our group has been attempting the modification of the rhodium catalyst, which shows the highest hydrogenolysis activity in monometallic noble-metal catalysts in neutral conditions, with various early-transition-metal oxides (Table 4). Catalytic performance of modified Rh/SiO2 catalysts with various metals in the glycerol hydrogenolysis at 393 K is compared.36 The modification of Rh/SiO2 with Mn, V, and Zr species decreased the activity. In contrast, the modification of Rh/SiO2 with Re, Mo, and W species remarkably enhanced the activity of glycerol hydrogenolysis. Varying the amount of Re, Mo and W addition showed that the catalytic activity was maximum at M/Rh = 0.5, 0.06, and 0.13 (M = Re, Mo and W), respectively. Re addition showed the largest enhancing effect on catalytic activity and also increased the selectivity to 1,3-PD. The Rh–ReOx/SiO2 (Re/Rh = 0.5) exhibited 22 times higher glycerol conversion (79%) and 37 times higher 1,3-PD yield (11%) than Rh/SiO2. In addition, the C–C hydrogenolysis reactions were totally suppressed by the modification with Re. At longer reaction time, overhydrogenolysis of 1,2- and 1,3-PDs proceeded over Rh–ReOx/SiO2 (Re/Rh = 0.5) and high yield of PrOHs was obtained.37 The maximum yield of net PrOHs was 92%, and 1-PrOH was the major product (76% yield).| Catalyst | Substrate | Substrate/water/catalyst/g | Time/h | Conv. (%) | Products (selectivity (%)) | Ref. |
|---|---|---|---|---|---|---|
| a Conditions: loading amount of Rh or Ir, 4 wt%; H2, 8 MPa; 393 K. PD = propanediol, PrOH = propanol, THFA = tetrahydrofurfuryl alcohol, THPM = tetrahydropyran-2-methanol. b 373 K. | ||||||
| Rh–ReOx/SiO2 (Re/Rh = 0.5) | Glycerol | 4/16/0.15 | 5 | 79.0 | 1,3-PD (13.8), 1,2-PD (38.0), 1-PrOH (35.3), 2-PrOH (12.1) | 36 |
| Rh–MoOx/SiO2 (Mo/Rh = 0.06) | Glycerol | 4/16/0.15 | 5 | 44.1 | 1,3-PD (5.6), 1,2-PD (30.4), 1-PrOH (49.0), 2-PrOH (15.0) | 36 |
| Rh–WOx/SiO2 (W/Rh = 0.13) | Glycerol | 4/16/0.15 | 5 | 33.7 | 1,3-PD (11.3), 1,2-PD (43.2), 1-PrOH (32.9), 2-PrOH (9.8) | 36 |
| Rh–ReOx/SiO2 (Re/Rh = 0.5) | Glycerol | 4/0/0.15 | 24 | >99 | 1-PrOH (76), 2-PrOH (16), 1,2-PD (3), propane (5) | 37 |
| Ir–ReOx/SiO2 (Re/Ir = 1) | Glycerol | 4/1/0.15 | 36 | 81.0 | 1,3-PD (46.3), 1,2-PD (4.2), 1-PrOH (41.2), 2-PrOH (5.5) | 40 |
| Rh–ReOx/SiO2 (Re/Rh = 0.5) | THFA | 1/19/0.05 | 24 | 96.2 | 1,5-Pentanediol (80.1), 1-pentanol (15.9) | 38 |
| Rh–MoOx/SiO2 (Mo/Rh = 0.13) | THFA | 1/19/0.1 | 24 | 94.2 | 1,5-Pentanediol (90.3), 1-pentanol (8.7) | 39 |
| Rh–ReOx/SiO2 (Re/Rh = 0.5)b | THPM | 1/19/0.1 | 24 | 59 | 1,6-Hexanediol (88), 1-hexanol (9) | 41 |
| Rh–ReOx/SiO2 (Re/Rh = 0.5)b | 1,2-Hexanediol | 0.4/19.6/0.1 | 24 | 76 | 1-Hexanol (73), 2-hexanol (22) | 41 |
| Rh–ReOx/SiO2 (Re/Rh = 0.5) | 1,2-PD | 4/16/0.15 | 24 | 86.6 | 1-PrOH (74), 2-PrOH (19), propane (6) | 37 |
| Ir–ReOx/SiO2 (Re/Ir = 1) | 1,2-PD | 4/16/0.15 | 24 | 72 | 1-PrOH (85), 2-PrOH (10), propane (5) | 40 |
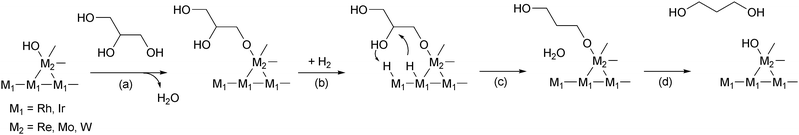
Rh/SiO2 and Rh–ReOx/SiO2 catalysts were characterized by CO adsorption, powder XRD, TEM, temperature-programmed reduction (TPR), XANES and EXAFS.36–38 The amount of CO adsorption, which means the surface area of noble-metal particles, decreased gradually with increasing Re amount. From the comparison between catalytic activity and the amount of CO adsorption, the additive effect of ReOx is not due to the increase of the surface Rh atom. XRD and TEM measurements showed that the average size of Rh particles in Rh–ReOx/SiO2 was almost the same (ca. 3 nm) as in Rh/SiO2. The TPR profile Rh–ReOx/SiO2 (Re/Rh = 0.5) gave a peak at lower temperature than the TPR peak of ReOx/SiO2, indicating that the presence of Rh promotes the reduction of Re species. The H2 consumption amount corresponded to the reduction of ReVII to average valence of 2.5 in addition to the total reduction of RhIII. The average valence of Re species can be also estimated by examining the area of the white line (the first peak) in the ReL3-edge XANES spectra, and was determined to be +2.2. These data indicate that Re species is not fully reduced to metallic state. The curve-fitting of ReL3-edge EXAFS of Rh–ReOx/SiO2 (Re/Rh = 0.5) after the reduction showed the presence of Re–O (CN = 1.4, R = 0.210 nm), Re–Rh (CN = 3.7, R = 0.265 nm) and Re–Re bonds (CN = 2.7; R = 0.272 nm). The Rh–Re bond was also detected by RhK-edge EXAFS (Rh–Re, CN = 1.8, R = 0.265 nm; in addition to Rh–Rh, CN = 10.1, R = 0.267 nm). Based on these data, the structure where Rh particles are covered with low-valent ReOx clusters is proposed for the Rh–ReOx/SiO2 catalyst (Fig. 1a). The presence of ReOx clusters much enhance the C–O hydrogenolysis activity of Rh metal surface and suppress the C–C hydrogenolysis activity.
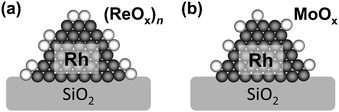 | ||
| Fig. 1 Structures of Rh–MOx/SiO2. (a) M = Re, (b) M = Mo. | ||
Rh–MoOx/SiO2 catalysts were also characterized by similar approaches. The catalyst with Mo/Rh = 0.13 was most characterized,39 while the catalysts with Mo/Rh = 0.6 and 0.13 show similar activities in glycerol hydrogenolysis.36 The average particle size of Rh–MoOx/SiO2 (Mo/Rh = 0.13) was determined to be 3.2 and 3.6 nm by TEM and XRD, respectively. These values were almost the same as those of Rh/SiO2 and Rh–ReOx/SiO2. The CO adsorption on Rh–MoOx/SiO2 (Mo/Rh = 0.13; CO/Rh = 0.29) was significantly lower than that of Rh/SiO2 (CO/Rh = 0.39) and almost the same as that of Rh–ReOx/SiO2 (Re/Rh = 0.13; CO/Rh = 0.28). The curve fitting result of MoK-edge EXAFS of Rh–MoOx/SiO2 (Mo/Rh = 0.13) after the reaction shows the presence of the Mo–O (CN = 1.1, R = 0.208 nm) and Mo–Rh (or –Mo) (CN = 3.0, R = 0.264 nm) bonds, while it is theoretically impossible to distinguish between Rh and Mo as a backscattering atom in the EXAFS analysis. The CN of the Mo–Rh (or –Mo) bond is comparable to that of the Re–Rh bond in Rh–ReOx/SiO2. This suggests that the MoOx species adsorbed on the surface of Rh metal particles are spatially well-defined and isolated (Fig. 1b). The structure difference between MoOx and ReOx on Rh metal particles can be related to the different optimum amount of the modifiers and the different selectivities of the modified catalysts.
Iridium lies just under rhodium in the periodic table. It has been known that iridium complexes, as well as rhodium counterparts, can activate the hydrogen molecule to give hydride complexes. We have very recently reported that the Ir–ReOx/SiO2 (Re/Ir = 1) catalyst prepared by a similar method to that for Rh–ReOx/SiO2 catalyzes the hydrogenolysis of glycerol to 1,3-PD.40 Although the reaction rate was lower, much higher selectivity to 1,3-PD was observed in the glycerol hydrogenolysis over Ir–ReOx/SiO2 (Re/Ir = 1) than over rhodium-based catalysts. The initial selectivity to 1,3-PD reached 67 ± 3%. The selectivity gradually decreased as the conversion increased because of the overhydrogenolysis of 1,3-PD to 1-PrOH. At 81% conversion of glycerol, the maximum yield of 1,3-PD of 38% was obtained. This value is higher than those of 1,3-PD yield in any reported catalytic systems. The Ir/SiO2 catalyst without ReOx modification showed almost no activity. The Ir–ReOx/SiO2 catalyst was characterized by similar approaches to Rh–MOx/SiO2 (M = Re and Mo). The TPR profile of Ir–ReOx/SiO2 (Re/Ir = 1) showed one intense H2 consumption signal at lower temperature than monometallic Ir/SiO2 or ReOx/SiO2. The consumption amount for Ir–ReOx/SiO2 (Re/Ir = 1) was 3.8 equiv. to Ir, indicating that Ir (initial valence, +4) and Re (initial valence, +7) were simultaneously reduced. The average particle size of Ir–ReOx/SiO2 (Re/Ir = 1) was determined to be 2.3 and 2.0 nm by TEM and XRD, respectively. The amount of CO adsorbed on Ir–ReOx/SiO2 (Re/Ir = 1) (0.07 equiv. to Ir) was lower than the number of surface Ir atoms calculated for a 2.3 nm Ir crystal (0.48 equiv. to total Ir). The ReL3-edge EXAFS detected the Re–O bond (CN = 1.2, R = 0.203 nm) as well as the Re–Ir (or –Re) bond (CN = 5.5, R = 0.269 nm). On the basis of all results and the analogy to rhodium counterparts, the structure where the oxidized low-valent Re clusters are attached to the Ir metal particles is proposed.
These catalysts can be applied to hydrogenolysis of other oxygen-containing substrates. Modified rhodium catalysts show excellent performance in the hydrogenolysis of tetrahydrofuryl alcohol (THFA).38,39THFA is one of the derivatives of furfural, which has been produced by acidic degradation of hemicellulose contained in agricultural raw materials abundantly. The hydrogenolysis of THFA over Rh–MOx/SiO2 (M = Mo, W, an Re) produced predominantly 1,5-pentanediol, which can be used as monomers for the production of polyesters and polyurethanes (eqn (3), n = 1). The initial selectivity to 1,5-pentanediol was more than 90%, and the maximum yields of 85% and 86% were obtained over Rh–MoOx/SiO2 and Rh–ReOx/SiO2, respectively. The Rh–WOx/SiO2 catalyst showed lower activity (about half activity of Mo or Re counterparts), while the selectivity to 1,5-pentanediol (85%) was comparable. On the other hand, the unmodified Rh/SiO2 catalyst showed much lower activity and gave 1,2-pentanediol as a main product (62% selectivity). The commercial Ru/C catalyst showed also much lower activity than modified Rh catalysts and the C–C hydrogenolysis was mainly catalyzed by the Ru/C. Hydrogenolysis of tetrahydrofuran without the –CH2OH group hardly proceeded over the same conditions using Rh–ReOx/SiO2. The ReOx-modified Rh catalyst was also applied to the hydrogenolysis of tetrahydropyran-2-methanol, which can be produced by the hydrogenation of the acrolein dimer (eqn (3), n = 2).41 The main product was 1,6-hexanediol, which is an important monomer for resin production. The initial selectivity to 1,6-hexanediol was around 90%. The modified catalysts were also effective to hydrogenolysis of 1,2-diols such as 1,2-PD to the corresponding mono-alcohols. The selectivity ratio of 1-ol/2-ol was 74/19 for 1,2-PD over Rh–ReOx/SiO2,37 73/22 for 1,2-hexanediol over Rh/ReOx/SiO2,41 and 85/10 for 1,2-PD over Ir–ReOx/SiO2.40 The reactivity of 1,2-PD was comparable to that of glycerol for both Rh–ReOx/SiO2 and Ir–ReOx/SiO2. On the other hand, mono-alcohols and other diols such as 1,3-PD and 1,6-hexanediol showed much lower reactivity to hydrogenolysis over these catalysts than glycerol and 1,2-diols. All these reactivities show that the C–O bond neighboring to a hydroxylated carbon is selectively dissociated in the hydrogenolysis over these modified catalysts.
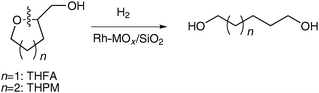 | (3) |
High 1,3-PD selectivities in the hydrogenolysis of aqueous glycerol have been also reported using the supported Pt catalyst with W or Re modifiers. Suzuki et al. from KAO Corporation patented the hydrogenolysis of glycerol over Al2O3-supported Pt–WO3 catalysts.44 High (≤67%) selectivity to 1,3-PD at 23% conversion of glycerol was achieved at 433 K. Qin et al. reported the hydrogenolysis of aqueous glycerol over the Pt/WO3/ZrO2 catalyst at 383–413 K.45 A 45.6% selectivity of 1,3-PD with a 1,3-PD/1,2-PD ratio of 17.7 was achieved at 70.2% conversion at 403 K. The authors explained that the high selectivity to 1,3-PD over Pt/WO3/ZrO2 is due to the catalysis of protic acid preferring the dehydration at the secondary OH over the primary OH in poly-ols. However, the difference of selectivity was not clearly explained between the Pt/WO3/ZrO2 system and other systems with the combination of noble metal and protic acid as stated in Section 2.2. The mechanism proposed for modified Rh and Ir catalysts as shown in Scheme 5 may involve these Pt–WO3 systems, leading the high 1,3-PD selectivities. Daniel et al. reported bimetallic Pt–Re catalysts supported on carbon that are effective at converting aqueous glycerol to both 1,3- and 1,2-PDs.46 The selectivity to 1,3-PD reached 34% at 20% conversion at 443 K. Characterizations with XRD, TEM, H2 chemisorption, XANES and EXAFS showed the formation of Pt–Re bimetallic particles with a partial positive charge (ca. +2) remained on the Re.
2.5 Nonconventional reaction media
While most studies of glycerol hydrogenolysis use aqueous or neat glycerol as a reaction medium, some reports have shown that the use of polar aprotic solvent with a high boiling point is effective to obtain a relatively high 1,3-PD selectivity. In addition, since both the hydrogenolysis products and co-produced water are relatively volatile compared to glycerol, the use of a solvent with very high boiling point may allow for the design of a continuous process, in which glycerol is added and products and water are removed by distillation.10 Che developed the glycerol hydrogenolysis in 1-methyl-2-pyrolidinone using syngas at 473 K in the presence of a homogeneous Rh(CO)2(acac) catalyst and a H2WO4 co-catalyst. 1,3-PD and 1,2-PD were produced with 20% and 23% yields, respectively.47 Chaminand et al. reported the hydrogenolysis of glycerol in sulfolane using the Rh/C catalyst and the H2WO4 co-catalyst.48 The 1,3-PD/1,2-PD ratio of 2 was obtained at 32% conversion and 6% total yield of PDs. Kurosaka et al. reported the glycerol hydrogenolysis in 1,3-dimethyl-2-imidazolidinone at 433 K over the Pt/WO3/ZrO2 catalyst.49 The yield of 1,3-PD was 24% (28% selectivity at 86% conversion). The main by-product in these systems is usually 1-PrOH. One explanation of high selectivities to 1,3-PD in these non-aqueous systems is the kinetic control of acid-catalyzed dehydration to either acetol or 3-hydroxypropanal (Scheme 6) in the initial stage of the dehydration–hydrogenation mechanism in Scheme 4: the protonation of the secondary OH in glycerol and subsequent dehydration produces a more stable cationic intermediate than the protonation of terminal OH produces. The deprotonation of the cationic intermediates produces more 3-hydroxypropanal than acetol, although thermodynamically 3-hydroxypropanal is less stable than acetol. The hydrogenation of 3-hydroxypropanal gives 1,3-PD, while the overdehydration of 3-hydroxypropanal produces acrolein and finally 1-PrOH. The key to obtain high selectivity to 1,3-PD may include the control of acid strength of the catalysts and basicity of the solvent to convert the 3-hydroxypropanal intermediate to 1,3-PD before the overdehydration to acrolein. In an aqueous medium, fast hydration leads to the interconversion between 3-hydroxypropanal and acetol, prohibiting the formation of less stable 3-hydroxypropanal.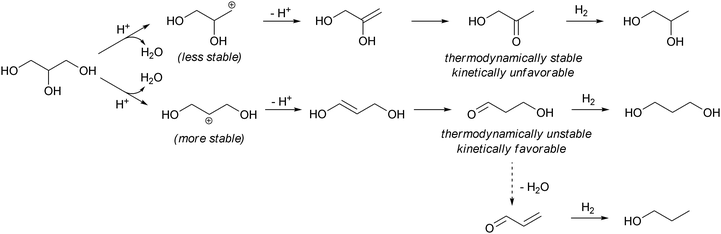 | ||
| Scheme 6 Elementary reactions in the dehydration of glycerol. | ||
Another explanation of 1,3-PD formation is the same mechanism as shown in Scheme 5, since all systems use reducible tungsten co-catalysts.
3. First-row transition metal catalysts
As well as noble metals, Cu, Ni, and Co are known to be able to activate hydrogen molecules and are used as hydrogenation catalysts. Although the hydrogenation activity is generally lower than that of noble metals, the much lower prices and the higher resistance to poisoning by trace impurities make non-noble-metal catalysts practically important. Application of non-noble-metal catalysts to glycerol hydrogenolysis has been intensively investigated (Table 5). Copper is most often selected as an active component in the catalysts because of the high selectivity to 1,2-PD. Copper chromite, a conventional hydrogenation catalyst, shows good performance in the glycerol hydrogenolysis to 1,2-PD17 and is selected by current industrial projects. Nearly 90% selectivity at 65% conversion is obtained at 473 K. Cu–ZnO catalysts, which are industrially used in methanol synthesis, water gas shift reaction, and methanol steam reforming, are also well studied.50–53 Selectivities of ≥80% to 1,2-PD are reported at relatively low conversions. Small amount of EG (2–10% selectivity) is almost always formed as a by-product. Pre-reduction of Cu–ZnO catalysts by H2 stream is essential to avoid the aggregation of Cu particles in the reaction solutions and to obtain high selectivity to 1,2-PD. Meher et al. investigated the hydrogenolysis catalysis of Cu/Zn/Al mixed oxide catalysts prepared by calcination of the hydrotalcite-type precursor.52 A glycerol conversion of 48% with 93–94% selectivity toward propylene glycol was obtained over the optimized catalyst. The demethanolized crude glycerol (83% glycerol) from the biodiesel process could be used for the synthesis of 1,2-PD. Supported Cu catalysts on silica54–55 or alumina56 and the Cu–Al catalyst prepared by co-precipitation57 were reported to be active in the glycerol hydrogenolysis to 1,2-PD. Selectivities over 90% were reported at ≤80% conversions. The supported Cu–Zn catalyst on alumina was also reported,58 however, the performance (74% selectivity at 25% conversion) was not higher than Cu–ZnO without alumina. Yuan et al. prepared the Cu/MgO catalystviaco-precipitation method.59 The basic catalyst showed excellent 1,2-PD yield (96.4% selectivity at 94.2% conversion).| Catalyst | H2/MPa | Glycerol/water/catalyst/g | Temp./K | Time/h | Conv. (%) | Products (selectivity (%)) | Ref. |
|---|---|---|---|---|---|---|---|
| a PD = propanediol, EG = ethylene glycol, PrOH = propanol. b Weight ratio (total weight was not reported). c Flow system. d Molar ratio. e Gradient temperature. f Ratio by volume. | |||||||
| Copper chromite | 2.1 | 50/10/2.5 | 473 | 24 | 65.3 | 1,2-PD (89.6) | 17 |
| Cu–Zn–O (Cu/Zn = 1) | 4.2 | 15/65/1.2 | 473 | 12 | 22.5 | 1,2-PD (83.6), EG (10.7) | 50 |
| Cu–Zn–Al–O (Cu/Zn/Al = 1/1/4) | 1.38 | 20/80/5b | 473 | 24 | 48 | 1,2-PD (93.9), acetol (6) | 52 |
| Cu/SiO2 (31 wt%) | 9 | 64/16/4 | 473 | 12 | 73.4 | 1,2-PD (91.3), EG (3.6) | 54 |
| Cu/Al2O3 (17 wt%) | 3.6 | 32.5/32.5/0.65 | 473 | 10 | 34.6 | 1,2-PD + 1,3-PD (93.9) | 56 |
| Cu–Al–O (Cu/Al = 1) | 7 | 20/80/0.8 | 493 | 5 | 38 | 1,2-PD (91), EG (2.4), acetol (2.6) | 57 |
| Cu–ZnO/Al2O3 (50 wt% support) | 10 | 13/127/1 | 463 | 7 | 25 | 1,2-PD (74) | 58 |
| Cu–MgO (15 wt%) | 3 | 6/2/1 | 453 | 40 | 94.2 | 1,2-PD (96.4) | 59 |
| Cu–Zn–Al–O (32.3 wt% Cu) | 5 | 36/24/1.8 | 473 | 10 | 20.4 | 1,2-PD (80.1), EG (2.4) | 60 |
| Cu–Zn–Al–O (32.3 wt% Cu)c | 0.64 | H2/glycerol = 140d | 463 | WHSV = 0.08 h−1 | 96.2 | 1,2-PD (92.2) | 60 |
| Cu/Al2O3 (55.1 wt% CuO)c | 0.1 | H2/glycerol/water = 141/1/12d | 393–473e | WHSV = 0.066 h−1 | 100 | 1,2-PD (96.9), EG (1.4), acetol (0.4) | 62 |
| RANEY® Copperc | 1.4 | H2/20 wt% glycerol = 375/0.05f | 478 | LHSV = 0.043 h−1 | 100 | 1,2-PD (94), EG (1.6) | 63 |
| RANEY® Nickel | 1 | 8/0/2 | 463 | 44 | 97 | 1,2-PD (71), ethanol (19), CO2 (10) | 64 |
| Ni–Ce/active carbon | 5 | 37.5/112.5/6.95 | 473 | 6 | 90.4 | 1,2-PD (65.7), EG (10.7) | 67 |
| Ni–Pt/SiO2 (5 wt% Ni, 0.5 wt% Pt) | 8 | 1/19/1 | 453 | 48 | 79 | 1,2-PD (14), EG (39), methane (40) | 68 |
| Co nests | 5.2 | 4/36/0.05 | 493 | 7 | 35 | 1,2-PD (72) | 69 |
Very high 1,2-PD yields (∼90%) can be obtained in gas-phase hydrogenolysis of glycerol over copper-based catalysts. Huang et al. reported the hydrogenolysis of glycerol in both liquid and vapor phases using the co-precipitated Cu/Zn/Al2O3 catalyst.60 A selectivity of 92.2% at 96.2% conversion was achieved in the vapor phase at 463 K and 0.64 MPa H2. Using the same catalyst, the liquid-phase hydrogenolysis requires more severe conditions (473 K and 5 MPa H2) and showed lower selectivity to 1,2-PD (80.1% at 20.4% conversion). Sato et al. conducted the vapor-phase reaction of glycerol over the Cu/Al2O3 catalyst at ambient hydrogen pressure and gradient temperatures in the catalyst bed.61,62 Over 96% yield of 1,2-PD at complete conversion was achieved at gradient temperature between 393–473 K, while the yield of 1,2-PD was at most 80% at a constant temperature of 463 K. Schmidt et al. used a fixed bed RANEY® Copper catalyst for gas-phase hydrogenolysis of glycerol. They obtained 94% yield of 1,2-PD at 478 K and 1.4 MPa H2.63 The presence of chromium enhanced the C–C hydrogenolysis to ethanol and reduced the 1,2-PD yield.
The reaction mechanism of Cu-catalyzed hydrogenolysis of glycerol is under debate. Both the dehydrogenation–dehydration–hydrogenation route and the dehydration–hydrogenation route as shown in Schemes 3 and 4, respectively, have been proposed even on similar catalysts.53 Dasari et al. proposed the dehydration–hydrogenation route for the copper-chromite catalyst based on the data that relatively pure acetol was isolated from glycerol in the absence of hydrogen and the presence of catalyst at 473 K.17 On the other hand, Wang et al. proposed the former mechanism for the Cu–ZnO catalyst based on the data that besides acetol many kinds of products including 1,2-PD were formed on Cu–ZnO under N2.53 Huang et al. suggested that both routes can proceed over the sodium-containing CuO/SiO2 catalysts. The dehydration–hydrogenation route is predominant over sodium-free CuO/SiO2, while the formation of the by-product EG is likely to proceed mainly through the dehydrogenation–dehydration–hydrogenation route.55
Nickel-based catalysts including RANEY® Nickel and supported catalysts are also tested for glycerol hydrogenolysis.17,60,64–66 The main product is also 1,2-PD. However, the C–C hydrogenolysis to EG or ethanol is usually accompanied and the selectivity to 1,2-PD is generally lower than that of copper-based catalysts. Addition of secondary metals has been tested to change the activity and selectivity. The addition of cerium to Ni/active carbon decreased the reduction temperature of Ni, gave smaller particle size, and enhanced the catalytic activity.67 The addition of platinum to Ni/SiO2 enhanced the C–C hydrogenolysis activity to EG.68 Higher selectivity to EG (48%) than to 1,2-PD (27%) was obtained over Ni(5 wt%)–Pt(0.5 wt%)/SiO2. The maximum yield of EG reached 31%. Cobalt-based catalysts have been less studied. Liu et al. prepared various Co nanostructures such as Co nests, nanoflowers and nanowires and used them as hydrogenolysis catalysts of glycerol.69–721,2-PD was formed as a main product with moderate selectivity (57–72%) at 493 K.
These results show that first-row transition metal catalysts, especially Cu, are very effective to the production of 1,2-PD from glycerol, although the mechanism is not clear. The high yield of 1,2-PD means that these catalysts have much lower activity to overhydrogenolysis of 1,2-PD than glycerol hydrogenolysis.
4. Conclusions and outlook
Hydrogenolysis of glycerol is a complex reaction: there are many potential reaction routes and products. Among the potential products, 1,2-PD is most easily obtained, especially over Cu catalysts which have been used in the commercialized process. However, the Cu-based hydrogenolysis catalysts are specific to the very reaction of glycerol to 1,2-PD. To obtain other products such as 1,3-PD and propanols or to use other biomass-related substrates such as THFA, other types of hydrogenolysis catalysts should be developed. One approach is the combination of noble-metal catalysts with an acid. While this type of catalysts gave again 1,2-PD as an initial product, overhydrogenolysis products (PrOHs and propane) can be obtained in severe conditions. This type of catalysts can be applied to many substrates other than glycerol. In principle, any alcohol can be deoxygenated in appropriate conditions. To obtain one partial-hydrogenolysis product selectively, precise control of reaction conditions and catalyst properties such as acid strength will be needed. Another approach is the combination of noble-metal catalysts with low-valent metal oxide. In reductive conditions, group 4–7 metal becomes low-valent oxide and forms metallic bonds with the surface of noble-metal particles. The directly-bonded modifiers can affect the adsorption behaviors of reactants (especially substrate, and possibly H2) on the catalyst surface and change the electronic properties of noble metal. High 1,3-PD selectivity can only be obtained using this type of catalysts, at present. In addition, high propanol yield can be obtained since the hydrogenolysis activity for mono-alcohols is lower than for 1,2-diols or glycerol. This type of hydrogenolysis catalysts can be applied to some other biomass-derived multi-functionalized substrates. Very high selectivity to one sole product is achieved in some cases such as THFA hydrogenolysis. However, the yield of 1,3-PD or the 1-PrOH/2-PrOH ratio in the glycerol hydrogenolysis has still room for improvement. To improve the selectivity, the precious control of the catalyst structure will be needed: in addition to the selection of noble metal, modifier and support, the particle size of noble metal, the morphology of modifier metal oxide, and their uniformity are all important. The control of acidity/basicity of the catalyst may be also important in order to exclude the unexpected acid- or base-assisted hydrogenolysis routes. Although further developments of both the preparation and characterization techniques may be needed, highly selective catalysts for partial hydrogenolysis with broad substrate scope can be realized over the concept of noble-metal catalyst modified with low-valent metal oxide.References
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef.
- M. F. Demirbas and M. Balat, Energy Convers. Manage., 2006, 47, 2371–2381 CrossRef CAS.
- C.-H. Zhou, J. N. Beltramini, Y.-X. Fan and G. Q. Lu, Chem. Soc. Rev., 2008, 37, 527–549 RSC.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass. Volume I – Results of Screening for Potential Candidates from Sugars and Synthesis Gas, U. S. Department of Energy, 2004 Search PubMed.
- D. T. Johnson and K. A. Taconi, Environ. Prog., 2007, 26, 338–348 CrossRef CAS.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. D. Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS.
- M. Schlaf, Dalton Trans., 2006, 4645–4653 RSC.
- G. P. da Silva, M. Mack and J. Contiero, Biotechnol. Adv., 2009, 27, 30–39 CrossRef.
- http://www.cargill.com/news-center/news-releases/2007/NA3007798.jsp .
- J. D. Unruh and D. Pearson, Kirk–Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, 2000 DOI:10.1002/0471238961.1618151621141821.a01.
- J. E. Logsdon and R. A. Loke, Kirk–Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, 2000 DOI:10.1002/0471238961.0919151612150719.a01.
- S. Rebsdat and D. Mayer, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2000 DOI:10.1002/14356007.a10_101.
- C. Montassier, J. C. Ménézo, L. C. Hoang, C. Renaud and J. Barbier, J. Mol. Catal., 1991, 70, 99–110 CrossRef CAS.
- M. A. Dasari, P.-P. Kiatsimkul, W. R. Sutterlin and G. J. Suppes, Appl. Catal., A, 2005, 281, 225–231 CrossRef CAS.
- T. Miyazawa, S. Koso, K. Kunimori and K. Tomishige, Appl. Catal., A, 2007, 318, 244–251 CrossRef CAS.
- J. Wang, S. Shen, B. Li, H. Lin and Y. Yuan, Chem. Lett., 2009, 38, 572–573 CrossRef CAS.
- E. P. Maris and R. J. Davis, J. Catal., 2007, 249, 328–337 CrossRef CAS.
- I. Furikado, T. Miyazawa, S. Koso, A. Shimao, K. Kunimori and K. Tomishige, Green Chem., 2007, 9, 582–588 RSC.
- E. D'Hondt, S. Van de Vyver, B. F. Sels and P. A. Jacobs, Chem. Commun., 2008, 6011–6012 RSC.
- D. Roy, B. Subramaniam and R. V. Chaudhari, Catal. Today, 2010, 156, 31–37 CrossRef CAS.
- Y. Kusunoki, T. Miyazawa, K. Kunimori and K. Tomishige, Catal. Commun., 2005, 6, 645–649 CrossRef CAS.
- T. Miyazawa, Y. Kusunoki, K. Kunimori and K. Tomishige, J. Catal., 2006, 240, 213–221 CrossRef CAS.
- T. Miyazawa, S. Koso, K. Kunimori and K. Tomishige, Appl. Catal., A, 2007, 329, 30–35 CrossRef CAS.
- M. Balaraju, V. Rekha, P. S. Sai Prasad, B. L. A. Prabhavathi Devi, R. B. N. Prasad and N. Lingaiah, Appl. Catal., A, 2009, 354, 82–87 CrossRef CAS.
- A. Alhanash, E. F. Kozhevnikova and I. V. Kozhevnikov, Catal. Lett., 2007, 120, 307–311.
- E. S. Vasiliadou, E. Heracleous, I. A. Vasalos and A. A. Lemonidou, Appl. Catal., B, 2009, 92, 90–99 CrossRef CAS.
- I. Gandarias, P. L. Arias, J. Requies, M. B. Güemez and J. L. G. Fierro, Appl. Catal., B, 2010, 97, 248–256 CrossRef CAS.
- D. Taher, M. E. Thibault, D. Di Mondo, M. Jennings and M. Schlaf, Chem.–Eur. J., 2009, 15, 10132–10143 CrossRef CAS.
- J. Feng, J. Wang, Y. Zhou, H. Fu, H. Chen and X. Li, Chem. Lett., 2007, 36, 1274–1275 CrossRef CAS.
- E. P. Maris, W. C. Ketchie, M. Murayama and R. J. Davis, J. Catal., 2007, 251, 281–294 CrossRef CAS.
- E. P. Maris and R. J. Davis, J. Catal., 2007, 249, 328–337 CrossRef CAS.
- Z. Yuan, P. Wu, J. Gao, X. Lu, Z. Hou and X. Zheng, Catal. Lett., 2009, 130, 261–265 CrossRef CAS.
- Y. Shinmi, S. Koso, T. Kubota, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2010, 94, 318–326 CrossRef CAS.
- Y. Amada, S. Koso, Y. Nakagawa and K. Tomishige, ChemSusChem, 2010, 3, 728–736 CrossRef CAS.
- S. Koso, I. Furikado, A. Shimao, T. Miyazawa, K. Kunimori and K. Tomishige, Chem. Commun., 2009, 2035–2037 RSC.
- S. Koso, N. Ueda, Y. Shinmi, K. Okumura, T. Kizuka and K. Tomishige, J. Catal., 2009, 267, 89–92 CrossRef CAS.
- Y. Nakagawa, Y. Shinmi, S. Koso and K. Tomishige, J. Catal., 2010, 272, 191–194 CrossRef CAS.
- K. Chen, S. Koso, T. Kubota, Y. Nakagawa and K. Tomishige, ChemCatChem, 2010, 2, 547–555 Search PubMed.
- Y. Nakagawa, K. Uehara and N. Mizuno, Inorg. Chem., 2005, 44, 14–16 CrossRef CAS.
- V. W. Day, W. G. Klemperer and C. Schwartz, J. Am. Chem. Soc., 1987, 109, 6030–6044 CrossRef CAS.
- N. Suzuki, Y. Yoshikawa, M. Takahashi and M. Tamura, World Pat. WO 2007129560 to KAO Corporation, 2007 Search PubMed.
- L.-Z. Qin, M.-J. Song and C.-L. Chen, Green Chem., 2010, 12, 1466–1472 RSC.
- O. M. Daniel, A. DeLaRiva, E. L. Kunkes, A. K. Datye, J. A. Dumesic and R. J. Davis, ChemCatChem, 2010, 2, 1107–1114 Search PubMed.
- T. M. Che, US Pat. US 4642394 to Celanese Corp, 1987 Search PubMed.
- J. Chaminand, L. Djakovitch, P. Gallezot, P. Marion, C. Pinel and C. Rosier, Green Chem., 2004, 6, 359–361 RSC.
- T. Kurosaka, H. Maruyama, I. Naribayashi and Y. Sasaki, Catal. Commun., 2008, 9, 1360–1363 CrossRef CAS.
- S. Wang and H. Liu, Catal. Lett., 2007, 117, 62–67 CrossRef CAS.
- M. Balaraju, V. Rekha, P. S. Sai Prasad, R. B. N. Prasad and N. Lingaiah, Catal. Lett., 2008, 126, 119–124 CrossRef CAS.
- L. C. Meher, R. Gopinath, S. N. Naik and A. K. Dalai, Ind. Eng. Chem. Res., 2009, 48, 1840–1846 CrossRef CAS.
- S. Wang, Y. Zhang and H. Liu, Chem.–Asian J., 2010, 5, 1100–1111 CAS.
- Z. Huang, F. Cui, H. Kang, J. Chen, X. Zhang and C. Xia, Chem. Mater., 2008, 20, 5090–5099 CrossRef CAS.
- Z. Huang, F. Cui, H. Kang, J. Chen and C. Xia, Appl. Catal., A, 2009, 366, 288–298 CrossRef CAS.
- L. Guo, J. Zhou, J. Mao, X. Guo and S. Zhang, Appl. Catal., A, 2009, 367, 93–98 CrossRef CAS.
- R. B. Mane, A. M. Hengne, A. A. Ghalwadkar, S. Vijayanand, P. H. Mohite, H. S. Potdar and C. V. Rode, Catal. Lett., 2010, 135, 141–147 CrossRef CAS.
- A. Brandner, K. Lehnert, A. Bienholz, M. Lucas and P. Claus, Top. Catal., 2009, 52, 278–287 CrossRef CAS.
- Z. Yuan, J. Wang, L. Wang, W. Xie, P. Chen, Z. Hou and X. Zheng, Bioresour. Technol., 2010, 101, 7088–7092 CrossRef CAS.
- L. Huang, Y.-L. Zhu, H.-Y. Zheng, Y.-W. Li and Z.-Y. Zeng, J. Chem. Technol. Biotechnol., 2008, 83, 1670–1675 CrossRef CAS.
- S. Sato, M. Akiyama, K. Inui and M. Yokota, Chem. Lett., 2009, 38, 560–561 CrossRef CAS.
- M. Akiyama, S. Sato, R. Takahashi, K. Inui and M. Yokota, Appl. Catal., A, 2009, 371, 60–66 CrossRef CAS.
- S. R. Schmidt, S. K. Tanielyan, N. Marin, G. Alvez and R. L. Augustine, Top. Catal., 2010, 53, 1214–1216 CrossRef CAS.
- A. Perosa and P. Tundo, Ind. Eng. Chem. Res., 2005, 44, 8535–8537 CrossRef CAS.
- J. Zhao, W. Yu, C. Chen, H. Miao, H. Ma and J. Xu, Catal. Lett., 2009, 134, 184–189.
- W. Yu, J. Xu, H. Ma, C. Chen, J. Zhao, H. Miao and Q. Song, Catal. Commun., 2010, 11, 493–497 CrossRef CAS.
- W. Yu, J. Zhao, H. Ma, H. Miao, Q. Song and J. Xu, Appl. Catal., A, 2010, 383, 73–78 CrossRef CAS.
- N. Ueda, Y. Nakagawa and K. Tomishige, Chem. Lett., 2010, 39, 506–507 CrossRef CAS.
- Q. Liu, X. Guo, Y. Li and W. Shen, Langmuir, 2009, 25, 6425–6430 CrossRef CAS.
- Q. Liu, X. Guo, Y. Li and W. Shen, J. Phys. Chem. C, 2009, 113, 3436–3441 CrossRef CAS.
- Q. Liu, X. Guo, Y. Li and W. Shen, Mater. Lett., 2009, 63, 1407–1409 CrossRef CAS.
- Q. Liu, X. Guo, T. Wang, Y. Li and W. Shen, Mater. Lett., 2010, 64, 1271–1274 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |