Dynamic atomic scale in situelectron microscopy in the development of an efficient heterogeneous catalytic process for pharmaceutical NSAIDS
N.
Raveendran Shiju†
a,
Kenta
Yoshida
bc,
Edward D.
Boyes
bde,
D.
Robert Brown
a and
Pratibha L.
Gai
*bcd
aDepartment of Chemical and Biological Sciences, University of Huddersfield, Huddersfield, HD1 3DH, UK
bThe York JEOL Nanocentre, The University of York, York YO10 5DD, UK
cDepartment of Chemistry, The University of York, York YO10 5DD, UK
dDepartment of Physics, The University of York, York YO10 5DD, UK
eDepartment of Electronics, The University of York, York YO10 5DD, UK. E-mail: pratibha.gai@york.ac.uk
First published on 16th March 2011
Abstract
In heterogeneous catalysis the identification of the active site and crucially its location to prevent unwanted sintering and deactivation during the transformation of the precursor to active catalyst require the integration of dynamic in situ imaging at the atomic level and reactivity studies. We report nanostructural and physico-chemical studies towards an efficient low temperature heterogeneous catalytic process for nonsteroidal anti-inflammatory drugs (NSAIDS) such as N-acetyl-p-aminophenol (paracetamol or acetaminophen) on tungstated zirconia nanocatalysts of only a few nanometres in size. We directly visualised, in real-time, the dynamic precursor transformation to the active catalyst, which is of great significance in heterogeneous catalysis, using double aberration-corrected in situelectron microscopy at the atomic level under controlled conditions. We quantified the observations with catalytic activity studies for the NSAIDS. The observations using negative defocus imaging in AC-TEM combined with HAADF-STEM have provided the direct evidence for the presence of surface WOx species with dimensions of ≤0.35 nm, nanoclusters and nanoparticles of WOx from up to 0.6 to 1 nm, located at grain boundaries on the surface of the zirconia nanoparticles. The observations illustrate that the nanoparticles (NPs) are disordered (distorted) tungsten trioxide. The correlation between the nanostructure and activity of catalysts with different W loadings indicate that surface WOx species, nanoclusters and distorted WO3 nanoparticles create Brønsted acid sites highly active for the low temperature N-acetyl-p-aminophenol reaction, with distorted WO3 NPs contributing to the activity. The results further elucidate that the anchoring of active sites at grain boundaries of the zirconia nanoparticles prevents undesirable coalescence of the active species and improves the catalyst stability and performance to make more product.
Introduction
The development of stable catalysts for heterogeneous catalysis is often hindered by migration and unwanted sintering of active species on the catalyst surface resulting in the deactivation of the catalyst.1 Identifying the location and stabilising the active site to prevent free active sites to migrate across the surface causing sintering are critical to designing improved catalytic materials for continuous operation. However despite several useful catalyst studies,2,3 they remain a challenge. This is because dynamic studies of the precursor transformation to active catalysts (catalyst activation) which is intensely important in heterogeneous catalysis, to determine the atomic structure and location of active sites, are limited. The lack of direct dynamic in situ imaging at the atomic level of the evolution and location of active sites during the formation of the active catalyst has hampered progress in designing durable catalysts for the development of efficient heterogeneous processes.Catalysts with strong acidic properties are of interest in key catalytic processes.4–6 The rearrangement of ketoximes to amides or lactams in the presence of acid catalysts is commonly known as the Beckmann rearrangement. Forming a caprolactam monomer is one of the most important applications of the Beckmann rearrangement from cyclohexanone oxime, as caprolactam is the feedstock in the production of Nylon 6.7–9N-acetyl-p-aminophenol (paracetamol or acetamenophen) is one of important non-steroidal anti-inflammatory pharmaceutical drugs (NSAIDS) commonly used by humans because of its analgesic property. Conventionally it is prepared by acetylation of p-aminophenol with acetic anhydridevia homogeneous catalysis. However mono acetylating the amine group can be difficult due to the possible oligomerisation of the hydroxyl aromatic amine resulting in poor selectivity. The preparation of N-acetyl-p-aminophenol involves reacting 4-hydroxyacetophenone to obtain the ketoxime (4-hydroxyacetophenone oxime) followed by the Beckmann rearrangement in the presence of an acid catalyst including fuming sulfuric acid.4,6 This process however produces unwanted wastes.
The use of solid acid catalysts can offer significant advantages over homogeneous acid catalysts for the synthesis of amides/lactams of pharmaceutical importance as in the case of caprolactam synthesis.7–9 Heterogeneous routes for the adsorption of 4-hydroxyacetophenone oxime and the rearrangement of the oxime, for example, over zeolite-H-beta,10 as well as microporous and mesoporous molecular sieves and delaminated zeolite catalysts11 have been reported. However these show low activity over time due to the difficulty in accessibility of reactants to active sites (because adsorbed species tend to block zeolite pores over time), and low surface area due to a large catalyst grain size (believed to be microns or more). Additionally, in these studies (e.g.ref. 7–12) nanostructural data to directly identifying and locating active sites in dynamic precursor transformations and catalyst grain sizes have not been reported. Studies of zirconia based catalysts with large grain sizes have been reported at elevated temperatures, including for industrial isomerization and alkylation processes (instead of the highly corrosive and pollutant liquid acids such as sulfuric acid), methanol dehydration and n-pentane isomerisation reactions.5,10–18 In these studies a number of active species have been proposed,12–18 including monotungstate and polytungstate (WOx) species in tungstated-ZrO2 systems15,16 and ZrOx containing Zr–WOx species17,18 based on their subtle contrast variations, with catalyst activity measured over short periods of usually less than an hour. However no consensus has yet emerged as to the precise nature of the active structures. In addition, in all these studies5,12–18 dynamic transformation of the precursor to active catalyst has not been reported making it difficult to identify the structural evolution of active sites and their location, which are crucial for the development of improved catalysts. Preparative procedures as well as reaction environments have a marked effect on the activity of the catalyst.1 To date, studies of the dynamic precursor transformation to active catalysts in tungstated zirconia nanosystems and their reactivity in NSAID reactions have not been reported.
In this study, we have investigated tungstated zirconia solid acid nanocatalysts (prepared by a variation of the impregnation method described by Hino and Arata)5 for the Beckmann rearrangement of 4-hydroxyacetophenone oxime to N-acetyl-p-aminophenol at low temperatures. The schematic is shown in Fig. 1. We prepared tungstated zirconia nanocatalysts of only a few nanometres (nm) in size from precursors, to catalyse the Beckmann rearrangement of oximes to the corresponding amides/lactams and the conversion of 4-hydroxyacetophenone to N-acetyl-p-aminophenol. We have used in situ aberration corrected transmission electron microscopy (in situ AC-TEM) in combination with energy filtered-AC TEM, AC high angular annular dark field Scanning TEM (AC HAADF-STEM) and energy dispersive X-ray spectroscopy (EDX) in the same instrument, and complemented the data with physico-chemical and catalytic activity studies, as described in the following sections.
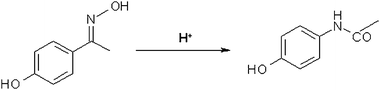 | ||
| Fig. 1 Beckmann rearrangement of 4-hydroxyacetophenone oxime to N-acetyl-p-aminophenol. | ||
The pioneering development of atomic resolution environmental TEM (atomic resolution-ETEM)19–21 for the direct visualisation of dynamic gas–solid reactions under controlled reaction conditions of gas pressures (with flowing gas) and temperatures at the atomic level has demonstrated that it is possible to study dynamic catalysis at the atomic scale, with time resolution consistent with contact times for gas molecules used in catalytic technologies (milliseconds). This atomic resolution-ETEM development as a Nanolaboratory has led to a new field of study for visualising gas–solid reactions at the atomic level under controlled reaction conditions, revealing new insights into fundamental reaction mechanisms. The atomic resolution-ETEM development19–21 has been adopted for commercial production by TEM manufacturer FEI and is used by academia and industry across the world,22 resulting in scientific, technological and economic benefits. The development of in situ aberration corrected electron microscopy23,24 under controlled reaction conditions allows the visualisation of dynamic reactions at a resolution better than 100 picometres (pm). The development of 4D electron microscopy25 has led to femtosecond time resolution. The use of a microelectromechanical system (MEMS) for a sample holder with a sample and gas enclosed in a cell has been reported for in situ examination.26
In this study, we directly demonstrate the dynamic transformation of the precursor to active catalysts for applications in NSAID reactions at low operating temperatures. Literature reports on tungstated zirconia describe polytungstate species with dimensions of about 0.25 nanometre (nm) to 0.35 nm as surface WOx species,12–18,27–31 which are similar to W3On−1 trimer species,27 in addition to monotungstate (single W atom) species. To be consistent with the literature reports on tungstated zirconia, we describe WOx species with dimensions ≤0.3 nm–0.35 nm as surface WOx species and species with larger dimensions than these (e.g. from up to 0.6 nm to ∼ 1 nm) as nanoclusters and nanoparticles (NPs) in this study. (On the other hand, it may be noted that in the chemical literature32 in general, a nanocluster refers to aggregates of atoms containing between 2 to 30 or more atoms. The term nanoparticle generally describes particles in the size range of >1 nm to 8 nm).
The dynamic studies show active sites containing surface WOx species of ≤0.3 nm to ∼0.375 nm consisting a few atoms and nanoclusters and NPs in the size range from up to 0.6 nm to ∼1 nm containing disordered (distorted) WO3 anchored primarily at grain boundaries on the surface of zirconia catalyst nanoparticles that are only a few nms in extent. Surface WOx species, nanoclusters and distorted WO3 NPs create active Brønsted acid sites. Catalytic data show that the surface WOx species and nanoclusters contribute significantly to the NSAID reaction with the larger (∼1 nm) distorted WO3 NPs enhancing this catalytic activity, leading to a marginal improvement.
The correlation of the catalysts containing these sites, with the reaction chemistry to N-acetyl-p-aminophenol shows very high selectivity (>90%) for the catalysts. Through comparative experiments we demonstrate that tungstated zirconia preparations reported in the literature with larger grain sizes of ≥30–50 nm on average and post-preparation procedures that deliberately increase the surface density of active species by co-impregnation of ZrOx and WOx on zirconia catalysts12–18 do not anchor active sites at grain boundaries of zirconia. Our observations indicate that the lack of anchoring of active species at grain boundaries of catalysts results in the migration and sintering of the species and the subsequent deactivation of the catalysts.
Experimental
Catalyst preparation and testing, X-ray diffraction and BET surface area measurements
We prepared two sets of tungstated zirconia described by Hino and Arata.5 They are hereafter referred to as WZA series and WZB series, respectively. The preparation procedures are described below:We prepared the first series of samples WZA, by the impregnation of zirconium oxohydroxide using an aqueous solution of ammonium metatungstate. Then the impregnated materials were dried and calcined. For the WZB series of samples, zirconium oxohydroxide was pre-calcined and then impregnated with required amount of aqueous solution of ammonium metatungstate. This sample was again subjected to the drying and calcination procedure similar to that of WZA. The WZA and WZB preparative procedures are therefore different and lead to different zirconia grain sizes and active species.
Details of synthesis procedures
Synthesis: we prepared the WZA series of tungstated zirconia nanocatalysts by calcination of the sample obtained by the impregnation of an aqueous solution of ammonium metatungstate (NH4)6H2W12O40 on zirconium oxohydroxide, ZrOx(OH)4−2x. In the synthesis, zirconium oxohydroxide was prepared by the controlled addition of aqueous zirconium oxychloride (ZrOCl2·8H2O, Sigma-Aldrich, UK) solution to an NH4OH (2 M)/NH4Cl (2 M) buffer solution to keep the pH at a constant value of 10.5. The solid was filtered and thoroughly washed with distilled water to remove the chlorides. Thus in the WZA series tungstated zirconia catalysts were prepared via the impregnation of zirconium oxohydroxide with an aqueous solution of ammonium metatungstate (Sigma-Aldrich, UK) to obtain W loadings in the range 5 to 35 wt%. The impregnated materials were dried at 333 K overnight and calcined for 4 h.For comparative purposes, we carried out catalyst preparations with the impregnation of ammonium metatungstate on zirconia obtained by the pre-calcination of zirconium oxohydroxide (hereafter referred to as WZB series). After the impregnation, these samples were calcined again at 973 K. For the WZB series, we prepared two series of samples: zirconium oxohydroxide was heated to 673 K first and then impregnated with the required amount of aqueous solution of ammonium tungstate and heated to 973 K (hereafter referred to as WZB1 series); and ammonium tungstate was impregnated on the ZrO2 support which was pre-formed by heating zirconium hydroxide at 973 K. After impregnation samples were heated again to 673 K (hereafter referred to as WZB2 series). The samples were subjected to a drying and calcination procedure similar to that of the WZA series.
The oximes were synthesized from the corresponding ketones by reaction with hydroxylamine hydrochloride in a mixture of ethanol and pyridine at 358 K, by the general method reported in the literature.33 The oximes were purified by recrystallization with ethanol. The rearrangement of oximes (Sigma-Aldrich, UK) was carried out in the liquid phase at 353 K to 423 K in a 50 ml glass reactor equipped with a condenser and a magnetic stirrer. Tetradecane (Koch-Light Laboratories, UK) was added as a GC internal standard. To monitor the reaction, 0.1 ml samples of the reaction mixture were taken periodically and analysed by gas chromatography (Perkin Elmer Clarus 500) using a 50 m BP1 capillary column and an FID detector. The N2 adsorption–desorption isotherms were measured at 77 K on a Micromeritics ASAP-2000 after evacuation at 473 K for 5 h. The surface area was calculated by the BET method. Powder X-ray diffraction patterns were collected on a Siemens Diffractometer (D5000) operated at 45 kV and 40 mA using Nickel filtrated Cu Kα radiation (1.5406 Å).
NH3 adsorption microcalorimetry and Fourier transform infrared (FTIR) spectroscopy
The system used for ammonia adsorption flow calorimetry has been described previously.34,35 It is based on a Setaram 111 DSC with an automated gas flow and switching system, with a mass spectrometer (Hiden HPR20) to sample the downstream gas flow. The sample (20 mg to 30 mg) was held on a glass frit in a vertical silica sample tube and activated at 423 K under a dried helium flow (5 ml min−1) for five hours. After activation, the sample temperature was maintained at 423 K and 1 ml pulses of the probe gas (1% ammonia in helium) at atmospheric pressure were injected at regular intervals into the carrier gas stream from a gas sampling valve. The ammonia concentration downstream of the sample was monitored continuously by mass spectroscopy. The pulse interval was chosen to ensure that the ammonia concentration in the carrier gas returned to zero to allow the DSC baseline to stabilise. The net amount of ammonia irreversibly adsorbed from each pulse was determined by comparing the MS signal with that recorded through a control experiment with a blank sample tube. The net heat released by each pulse was calculated from the thermal DSC curve. The trend in variation of the size of W-oxide groups can also be obtained from the optical absorption edge energy (AEE) values calculated using UV-Visible spectra of the samples.
UV-Visible
spectra were collected on Perkin Elmer lambda 35 spectrometer, using a labsphere reflectance spectroscopy accessory. The Kubelka-Munk function, F(R∞), for infinitely thick samples was used to convert reflectance measurements (Rsample) into equivalent absorption spectra using the reflectance of MgO as a reference (RMgO).34,35
| R∞ = Rsample/RMgO and F(R∞) = (1 − R∞)2/2R∞ |
FTIR spectroscopy of pyridine adsorption
The measurements were performed using a Nicolet Protégé 460 equipped with an evacuable furnace cell with KBr windows, containing a sample wafer. Initially, catalyst powder was pressed into a self-supported wafer, which was loaded into the IR chamber and heated up to 673 K overnight under reduced pressure of 10−3 mbar. After the cell was cooled down to 323 K the background spectrum was recorded. Spectra were collected as an average of 200 runs with 0.5 cm−1 definition. The pyridine adsorption was carried out slowly where the catalyst was equilibrated with pyridine vapour at 323 K. After 60 min evacuation, a spectrum was recorded. The sample was then heated stepwise, scanning with IR spectroscopy.In situ electron microscopy studies at the atomic level
Samples for electron microscopy studies were prepared by ultrasonically dispersing the as-synthesised tungstated zirconia nanocatalyst powders in alcohol and placing a small amount of the solution on holey carbon coated high purity copper or titanium grids and also on uncoated Ti grids for in situ and calibration studies.In the present study, we used our in-house development of modified double spherical aberration corrected JEOL 2200 FS TEM/STEM, operating at 200 kV. We modified the instrument with a larger gap (HRP) objective lens polepiece and gas tolerant pumping system23,24 in the Nanocentre at the University of York, to directly witness live precursor transformation, with a resolution better than 100 pm (0.1 nm). The in situ AC TEM studies were complemented by in situHAADF STEM. In high angle annular dark field—STEM,36,37 electrons that undergo Rutherford scattering in electron beam-sample interactions are collected and the image intensity is approximately proportional to Z2 where Z is the atomic number. This makes it ideal for studies of heterogeneous materials with components of different atomic numbers.
In our development of in situ AC-EM, the objective polepiece has a gap of ∼4.3 mm.23,24 This enables sample heating using a standard heating holder under controlled conditions where temperatures can be monitored with an accuracy of ±2 °C, as well as tilting of the sample for alignment into a correct crystal zone axis orientation. The instrument is fitted with an EDX detector for chemical composition analysis of the catalysts at the sub-nm scale. In situ studies using aberration corrected electron microscopy (AC-EM) at or better than 100 pm resolution were required to observe and image active sites at the atomic level during the dynamic transformation of the precursor containing residual water, to the active catalyst under controlled conditions of temperature and time in vacuum, with minimum electron beam delocalisation effects. This is because, in conventional HRTEM, the aberration in the imaging lens (spherical aberration) blurs images of nanoparticles of about 1 nm in size on supports, as the support contrast obscures the visibility of the nanoparticle.38 Therefore aberration correction has been crucial for observing the sub-nm species and nanoclusters in the in situ studies.
In our dynamic studies, temperatures were increased from room temperature (RT) to about 973 K and the onset of crystallization was observed at 758 K. Real time dynamic sequences were recorded at the relatively high temperatures used in this precursor study. In the in situ experiments, very low electron dose imaging techniques (operating below the threshold for the electron beam damage and atom displacement) were used as described previously.19–24,39–41In situ data were checked in calibration experiments where the electron beam was switched off during the in situ reaction (so the crystals are not under the beam) and the sample was exposed to the beam very briefly only to record the reaction end point.19–24,39–41 This control ensures a non-invasive characterisation. Real-time events were recorded on a CCD camera from around the same area of the sample under dynamic in situ conditions. These careful procedures were necessary as dynamic imaging at the atomic level at elevated temperatures on water containing precursors can be difficult.
Parallel catalytic activity measurements for liquid-phase Beckmann rearrangement were carried out ex situ at a low temperature of 403 K on larger amounts of fully crystallised catalyst samples (containing active structures), to correlate the nanostructure with activity.
Combination of methods
We have used electron microscopy in combination with chemical methods (XRD, UV-Vis and others) because it allows the direct visualisation of transformations locally at the atomic scale, which is difficult to obtain from the indirect chemical methods with data averaged over large areas.It may be noted that in electron microscopy (EM) studies, generally hundreds of particles are examined in pursuit of statistically valid sampling. EM (where sample is on 3 mm grids), XRD and other chemical techniques use relatively small amount of samples compared to hundreds of thousands of pounds to a tonne of catalysts in commercial reactors. Despite this fact, EM and chemical methods such as XRD, Raman, UV-Vis, TPR, etc. provide striking insights into the atomic structure, bonding and chemical properties. This is because even on the large commercial scale, the properties of heterogeneous solid catalysts still depend on detailed atomic structures of surfaces of multitude of small particles (grains) in the commercial catalyst systems and catalytic reactions occur at the atomic level. In situ dynamic electron microscopy at the atomic level enables the direct visualisation of the dynamic transformation of the precursor and the structural evolution of active species. In situEM studies of catalysts have been successfully correlated with structure–property relationships in technological reactors.41
Results
The tungsten content, BET surface area, and tungsten surface density (δ) of tungstated zirconia (WZA) samples are given in Table 1. The samples calcined at different temperatures produced solids with W surface densities ranging from 1.4 to 49.6 W at nm−2, with a 15WZA sample containing a W surface density of 7.3 at nm−2. Adsorption edge energies31 as a function of the surface W density are shown in the last column.| Sample | W content (wt%) | Calcination temperature/K | BET surface area/m2 g−1 | Nominal surface density, δa/W at nm−2 | Absorption edge energy/eV |
|---|---|---|---|---|---|
| a Calculated based on the amount of tungsten and BET surface area. b Calcination temperatures in K are indicated. | |||||
| ZrOH | 0 | — | 274.8 | — | |
| 5WZA | 5 | 973 | 45.3 | 3.6 | 3.57 |
| 10WZA | 10 | 973 | 61.4 | 5.3 | 3.45 |
| 15WZA | 15 | 973 | 67.7 | 7.3 | 3.35 |
| 25WZA | 25 | 973 | 50.5 | 16.2 | 3.10, 2.71 |
| 35WZA | 35 | 973 | 23.1 | 49.6 | 3.03, 2.71 |
| 10WZA373 b | 10 | 373 | 231.6 | 1.4 | 3.99 |
| 10WZA773 b | 10 | 773 | 100.4 | 3.3 | 3.61 |
| 10WZA1073 b | 10 | 1073 | 43.2 | 7.6 | 3.20, 2.87 |
X-Ray diffraction patterns as a function of W concentration and the amorphous oxohydroxide are presented in Fig. 2. Samples calcined at 973 K were crystalline with a predominantly tetragonal zirconia phase, indicated by open circles in the figure. Additional peaks observed in the samples 25WZA and 35WZA in the 2θ range of 23–25° (indicated by stars (*) in the figure) could be indexed as 002, 020 and 200 reflections of monoclinic WO3. The double peak (doublet) observed at the 2θ value of 50° in 15WZA and 35WZA is not present in 10WZA. The doublet is indexed as due to WO3 and tetragonal-zirconia. Using excellent systematic chemical characterisation methods such as Raman spectroscopy in combination with TPR and XPS, it is reported that preparative routes influence catalytic properties of tungstated zirconia31 and at lower W loadings tungsten species are thought to be highly dispersed on the zirconia surface, with the formation of WOx microcrystals at higher loadings.31
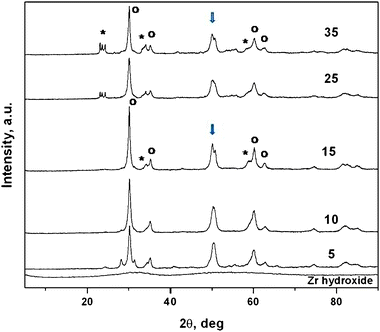 | ||
| Fig. 2 X-Ray diffraction patterns of tungstated zirconia catalysts as a function of W concentration, calcined at 973 K and amorphous zirconium oxohydroxide. Samples calcined at 973 K were crystalline with a predominant tetragonal zirconia phase (with a = 0.3607 nm; c = 0.5175 nm with the main 100 peak), indicated by open circles in the figure. Additional peaks observed in the samples 15WZA and 35WZA in the 2θ range of 23–25° (indicated by stars) could be indexed as monoclinic WO3. Furthermore the double peak (doublet) observed at a 2θ value of 50° in 15WZA and 35WZA is not present in 10WZA. The doublet is indexed as due to WO3 and tetragonal-zirconia. | ||
Catalytic tests for liquid phase Beckmann rearrangement
The WZA series were highly active and selective in the liquid-phase Beckmann rearrangement of oximes to the corresponding amides/lactams, as shown in Tables 2 and 3. Most notable example is the conversion of 4-hydroxyacetophenone oxime to N-acetyl-p-aminophenol (entry 2, Table 2). The reaction is highly selective and typically goes to completion within a few hours. The catalytic performance depends greatly on the tungsten loading and the calcination temperature, indicating a strong structural dependence of the activity (Table 3). Additionally, examples of the high selectivity and conversion of WZA nanocatalysts with low loadings of W as a function of calcination temperatures and the reaction temperatures are illustrated in Fig. 3(a) and (b), respectively. The reaction temperature of 403 K was selected based on the high conversion and selectivity for NSAIDs observed at this temperature (Fig. 3(b)). Table 3 shows that samples in the WZA series with high loadings of tungsten were less active than 15WZA and than lower loadings of 5WZA and 10WZA. The 35WZA catalyst sample (with a larger zirconia grain size) was found to have lower activity. 15WZA exhibited the highest selectivity and the highest surface area (Table 3). In contrast, tests on WZB samples showed much lower activity than the WZA series of samples for the Beckmann rearrangement. The 5WZB, 10WZB and 15WZB had selectivity to NSAIDS in the range of 63 to 66% initially and achieved about 47 ± 2% after only about 6 hours, which are lower than WZA samples. (Pure zirconia showed poor selectivity (of 20 ± 2%) for NSAIDS). To understand the differences in the performance of WZA and WZB series of samples, we employed a combination of electron microscopy and physico-chemical studies, which are described in the following sections.| Oxime | Product | Yield (%) | |
|---|---|---|---|
| Catalyst: 15WZA, reaction temperature = 403 K; solvent = benzonitrile; oxime = 0.9 mmol; catalyst = 20 mg; t = 6 h. | |||
| 1 |

|
|
90.0 |
| 2 |
|
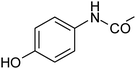
|
92.0 |
| 3 |
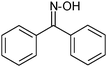
|
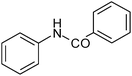
|
77 |
| 4 |
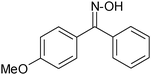
|
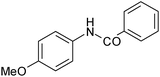
|
80 |
| Surface density/W at. nm−2 | Conversion of oxime (%) | Selectivity of paracetamol (%) |
|---|---|---|
| a Reaction temperature = 403 K; solvent = benzonitrile; oxime = 0.9 mmol; catalyst = 20 mg; t = 6 h. b 10WZA calcined at different temperatures. | ||
| 1.4b | 83.1 | 74.2 |
| 3.3b | 86.0 | 80.7 |
| 3.6 | 86.4 | 90.8 |
| 5.3 | 92.2 | 92.7 |
| 7.3 | 97.5 | 94.4 |
| 7.6b | 91.4 | 92.4 |
| 16.2 | 92.3 | 92.9 |
| 49.6 | 87.8 | 91.0 |
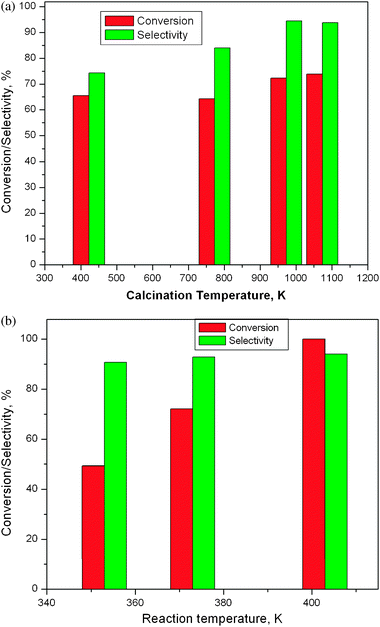 | ||
| Fig. 3 (a) Conversion of 4-hydroxyacetophenone oxime and selectivity of paracetamol as a function of calcination temperature of catalyst 10WZA. Reaction temperature = 403 K; t = 3 h; solvent = benzonitrile; oxime = 0.9 mmol; catalyst = 20 mg. (b) Conversion of 4-hydroxyacetophenone oxime and selectivity of paracetamol as a function of reaction temperature. Solvent = benzonitrile; time = 12 h; oxime = 0.9 mmol; catalyst = 20 mg. | ||
We used NH3 adsorption microcalorimetry to estimate the acid strengths of the WZA catalysts as a function W surface density and in turn the size of tungsten containing species. The heat of adsorption of NH3 measured by microcalorimetry as a function of W surface density is shown in Fig. 4(a), which is a measure of the strength of acid sites.34,35 It was found to be higher at lower loadings, linking directly the acidity to the size of the W-containing species on the surface. These acid sites are of predominantly Brønsted in nature as shown by FTIR using pyridine as the probe molecule. The band at 1545 cm−1, corresponding to the vibration of pyridinium ions shown in Fig. 4(b), has a higher area than the band at 1445 cm−1, characteristic of pyridine coordinatively bound to Lewis acid sites. For the WZB samples, the heat of adsorption of NH3 measured was much lower than that of NH3 for WZA, showing the weaker acidity of this sample, as shown in Fig. 5.
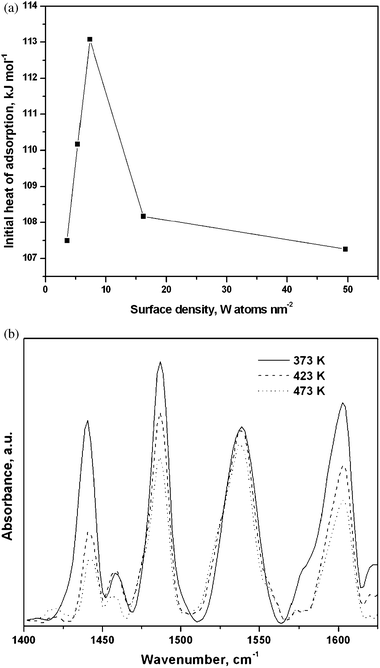 | ||
| Fig. 4 (a) Initial heat of NH3 adsorption measured by microcalorimetry as a function of W surface density. (b) FTIR spectra of adsorbed pyridine as a function of desorption temperatures. | ||
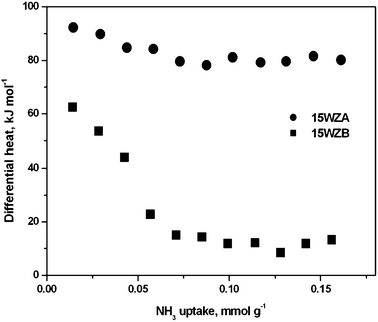 | ||
| Fig. 5 Heat of NH3 adsorption measured by microcalorimetry as a function of surface coverage of NH3 for series A and B samples containing 15% W, showing that WZA samples are superior. | ||
In situ AC-electron microscopy studies of dynamic precursor transformation
In order to identify active sites of the 15WZA catalyst (with a high surface area and W surface density), we have directly observed the dynamic transformation of the precursor containing water and the evolution of active catalysts at the atomic level, under controlled conditions from room temperature (RT) to 973 K, in the in situ AC TEM/STEM.An atomically resolved and time resolved sequence of the amorphous precursor undergoing dynamic transformation in real time is presented in Fig. 6. The same area of the sample was monitored to provide insights into the transformation at the atomic scale. The evolution of crystalline nanograins of zirconia in random crystallographic orientations and tungsten containing species at the grain boundaries of zirconia nanocrystals were observed (near the areas indicated by the arrow). Surface species with dimensions of about ≤0.3 nm form (exhibiting darker contrast (e.g. at P), indicated by white arrow in (b)), emerge and appear to be due to the scattering of the heavier tungsten in the oxide. The onset of crystallisation was observed from 758 K. A complete crystallisation of the sample was achieved at 973 K after several hours. A combination of AC HAADF-STEM and AC-TEM was employed to further analyse the species. In the WZA sample the presence of surface WOx species, nanoclusters and distorted WO3 NPs was observed.
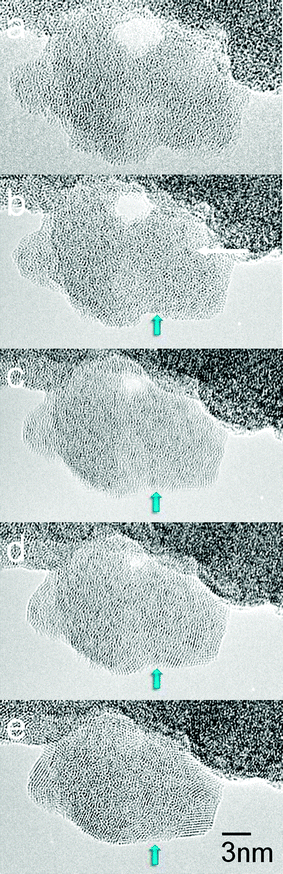 | ||
| Fig. 6 Real-time direct visualisation at the atomic level of the dynamic amorphous precursor to active catalyst transformation in the 15WZA sample using our modified double aberration-corrected in situtransmission EM as a function of time. Catalyst crystallisation commences at ∼485 °C. (a) Before heating (amorphous precursor); (b) after 5 minutes (min); (c) 10 min; (d) 15 min; and (e) 20 min. Crystalline nanoscale grains and surface species with dimensions of about ≤0.3 nm (darker contrast) form (indicated by white arrow in (b)). | ||
Surface WOx species with sizes of ≤0.35 nm to slightly larger species of ∼0.375 nm were primarily observed which are illustrated in Fig. 7(a) and (e), in addition to nanoclusters and WO3 NPs with size ranges of up to 0.6 nm to 1 nm. (The size measurements of WOx species were made on thin single crystal zirconia areas for accuracy (and not on overlapping zirconia support crystals which can obscure the contrast from the species)). The structure and composition of NPs were confirmed by measurements of lattice spacings42 and by sub-nm EDX, described in the following sections. Overlapping zirconia nanocrystals in random crystallographic orientations in the fully crystallised sample are illustrated in the AC-TEM image, Fig. 7(b).
 | ||
| Fig. 7 (a) AC HAADF-STEM image of 15WZA showing the presence of surface WOx species with dimensions of ≤0.3 nm (inside blue circles). (b) Atomic resolution AC-TEM image of the 15WZA sample (scale bar 36 mm = 15 nm). Darker species and clusters are just discernible. (c) shows a slightly negative defocused (under-focused) image of the crystal (near the area S) revealing the clusters. (c) Negative defocus imaging revealing uniformly distributed tungsten oxide clusters anchored at grain boundaries of zirconia nanocrystals (the same area (near S) as in (b) of 15WZA, and the magnification is the same as in (b)). The image is at 30 nm under focus and the WOx species and nanoclusters are in the size range of about ≤0.3 nm to 0.6 nm (arrowed); (d) a schematic diagram showing the species and clusters at grain boundaries of nano-zirconia. (e) Surface WOx species of ≤0.3 nm (e.g. at black arrow at S) and nanoclusters at grain boundaries of nano-zirconia (indicated by white arrows). Scale bar = 10 nm. | ||
For the same crystal, we employed slightly negative defocused imaging which revealed the presence of surface WOx species (at the sub-nm scale) with improved contrast (species with dark contrast and clusters indicated by arrows), shown in Fig. 7(c), which are not readily visible in the conventional AC image at the zero defocus shown in Fig. 7(b). The species have sizes primarily in the range of ≤0.35 nm to ∼0.375 nm and some up to 0.6 nm to 1 nm, uniformly distributed and located at grain boundaries of nano-zirconia, as illustrated in Fig. 7(c). Fig. 7(d) shows a schematic diagram showing the WOx species at grain boundaries of nano-zirconia. Fig. 7(e) demonstrates a further example of surface WOx species of ≤0.35 nm (indicated for example, by a black arrow at S), species of ≥0.375 nm and larger nanoclusters, and anchoring of these species at grain boundaries of nano-zirconia (indicated by white arrows).
To analyse the structure and chemical composition of the observed species, we have used the combination of atomic resolution imaging and EDX in the same AC HAADF-STEM. Z contrast imaging illustrates WOx species of ≤0.3 nm−0.35 nm (shown in Fig. 7(a)), and nanoclusters and NPs up to ∼0.6 nm to 1 nm in size (generally consistent with the AC-TEM lattice images in Fig. 7(c) and (e)). Some of them are also shown in Fig. 8(a) and (b), indicating surface tungstate species (circled and in blue region arrowed) in (a); and the intensity profile from the region after noise filtering (b). A sub-nm cluster of ∼0.5 nm observed is indicated by the blue circle in the image shown in Fig. 8(c). An EDX spectrum in STEM for a NP located at the edge of zirconia nanograin (e.g. E in Fig. 8(d)) is shown in Fig. 8(c), confirming the presence of W and oxygen.
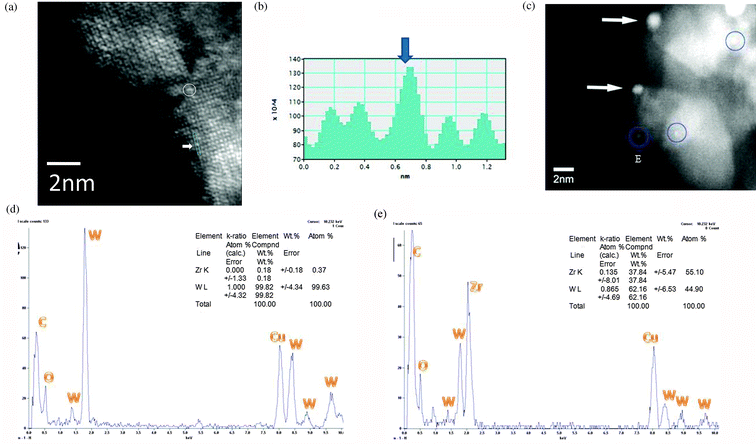 | ||
| Fig. 8 AC-HAADF-STEM images of tungsten oxide species and clusters in the 15WZ catalyst in the double AC-TEM/STEM are shown in (a), (b) and (c). (a) indicates surface WOx species (circled) and tungsten atom contrast in the blue rectangle region marked on (a) at the white arrow, with the corresponding intensity profile from the region after noise filtering. Sub-nanometre clusters are circled in (c). (d) EDX composition spectrum from a WO3 nanocluster at the edge of zirconia nanocrystal, confirming the composition with W and O (Cu is a support grid used to support the catalyst). (e) EDX composition spectrum from WO3 nanocluster on nano-zirconia with contribution from the underlying zirconia. | ||
The combination of atomic resolution imaging with better than 0.1 nm resolution and sub-nm scale EDX of clusters located at the edge of the catalyst surface is more accurate for composition analyses of the nanoclusters. This is because if EDX alone is used for clusters lying on zirconia, the electron probe may extend to zirconia areas around the cluster contributing to the cluster composition, thus complicating analyses of the active site as shown in Fig. 8(d): this is likely to be the case for the Zr–W–Ox active species proposed inconclusively in literature reports.
The atomically resolved structure of a NP (about 1 nm) located at the edge of the nano-zirconia surface is revealed in the inset in Fig. 9 and is directly identified as disordered (distorted/defective and multiply twinned) (110) crystal of cubic WO3 based on the lattice imaging at the atomic resolution and sub-nm scale chemical analysis, consistent with the structure of ultrafine tungsten trioxide.43 Here, the cluster is in (110) crystallographic orientation and the (111) planar spacing is indicated. All the NPs examined in this size range were distorted WO3. Our observations at the atomic scale indicate that the NPs do not form solid solution with zirconia and are distorted.
 | ||
| Fig. 9 Atomic resolution AC-TEM image of a sub-nanometre cluster of faceted and multiply twinned disordered WO3 in 15WZA (shown by white/blue arrow; and magnified in the inset). (111) plane of the cluster is indicated by a black arrow. | ||
The optical diffractogram (Fast Fourier Transform (FFT)) obtained from the zirconia nanocrystals could be indexed based on the tetragonal zirconia structure with a = 0.3607 nm and c = 0.5175 nm, consistent with our XRD data (Fig. 2) and literature reports.44 The crystallite size of the larger zirconia nanocrystals was estimated to be 10 nm or more.
Many of the nanocrystals were observed to be in [001] crystal zone axis orientation as indicated by the selected area FFT/optical diffractogram, and a few in 〈110〉 zone axis orientation as shown in Fig. 10(a) and (b), respectively. We then used dark-field TEM using weak diffracted 200, 020, 220 and −220 reflections in an effort to enhance the grain boundary contrast to determine zirconia nanocrystal sizes more accurately. A dark field image in 200 reflection indicating the presence of small grains, and its schematic are shown in Fig. 11(a) and (b), respectively. Many seemingly single grains contain nanocrystals with very low angle grain boundaries between them as described below. Significantly, the images reveal several crystallites less than 5 nm in diameter in the larger zirconia nanocrystals. The average size of such ZrO2 crystallites (nanoparticles or nanocrystals) was estimated as 2.6 nm (with sample variance σ2 = 0.334 nm2). Ultra-fine grained materials exhibit high mechanical strengths45,46 and this could be the reason for the robustness of WZA nanoscale catalysts.
 | ||
| Fig. 10 (a) Aberration corrected-TEM (AC-TEM) image of the 15WZA sample showing nano-zirconia grains in 〈001〉 zone axis orientation. Inset shows the optical diffractogram (OD). (b) AC-TEM image of the 15WZA sample with nano-zirconia grains in 〈110〉 zone axis orientation. The inset shows the corresponding optical diffractogram. | ||
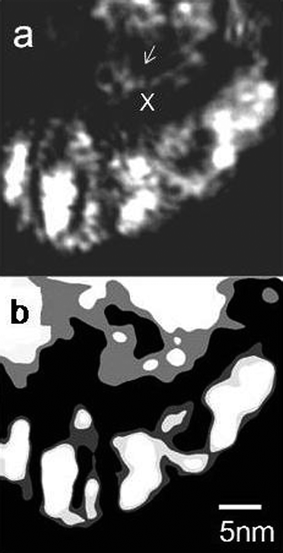 | ||
| Fig. 11 Dark-field TEM images of the 15WZA catalyst using weak 200 Bragg reflection, indicating smaller nano-zirconia grain sizes (indicated, e.g. by arrow at x). A schematic of the nanograins is shown in (b). The nanograins have low angle grain boundaries with only a couple of degrees rotation, appearing as ‘single’ grains (shown in Fig. 12). | ||
 | ||
| Fig. 12 (a) Nanograins of zirconia and a FFT pattern of the 15 wt% WZA catalyst. (b) Enlarged area of the square area in (a) shows nanograins and a schematic diagram showing mosaic crystals and low angle grain boundary with only a couple of degrees rotation. | ||
Many of the nanograins examined had very low angle grain boundaries as shown in Fig. 12(a) and (b), where (a) shows nanograins of zirconia (with a FFT pattern) of 15WZA catalysts. (b) The enlarged area of the square area in (a) and (b) shows a schematic diagram showing mosaic crystals and low angle grain boundary rotated by only a couple of degrees with respect to each other. This results in minimum rotation in electron diffraction patterns and therefore it is possible to get good quality ODM and electron nanodiffraction from such grains. Nanodiffraction can be carried out at the sub-nm level with advanced STEM instruments.47
WZA catalysts with lower W loadings (5WZA and 10WZA) also exhibited surface WOx species, but did not show distorted WO3 NPs. Fig. 13(a) shows a HAADF-STEM image of the nanostructure of 10WZA consisting of primarily WOx species of about 0.3 nm to 0.35 nm and some larger clusters. When the 35WZA catalyst sample was examined in a similar way, much larger WO3 crystallites up to 20 nm (and in a few cases up to 53 nm) in size and zirconia grain sizes of 12–30 nm were observed. Fig. 13(b–d) show nanostructural data of 35WZA using AC-HAADF STEM, where (b) and (c) show zirconia grain sizes and WO3 crystals, respectively, and (d) shows the lattice of zirconia.
 | ||
| Fig. 13 HAADF-STEM images: (a) the nanostructure of 10WZA with WOx species. Nanostructure of 35WZA is shown in (b–d): (b) and (c) show large zirconia grain sizes of 12–30 nm; (b) also shows much larger WO3 nanocrystals of ∼20 nm in size (particle at (iii) indicated by a blue arrow) with some up to 53 nm. These were confirmed by EDX. (d) shows the zirconia lattice. | ||
Comparative nanostructural studies
The direct observation of 15WZA nanocatalysts clearly illustrates the segregation of surface species and clusters to grain boundaries on the nano-zirconia surface as illustrated in Fig. 7. When the 15WZB catalyst was examined in a similar way, a number of differences were apparent in the size and morphology of the catalyst particles compared to 15WZA. The most striking differences were the lack of tungsten oxide species at grain boundaries of zirconia particles and larger grain sizes (≥30–50 nm) sizes of zirconia particles in the 15WZB sample.The results for comparative studies are summarised in Fig. 14 and compared with WZA samples. The figure shows low-magnification TEM images (at ×120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 magnification or 120 K) of: (a) the 15WZA sample, (b) WZB1 sample; and (c) WZB2. High-magnification ACTEM images (at a magnification of 400 K) shown in Fig. 14(d) to (i) are respectively from regions indicated by a square in the figures (a) to (c). (d–f): conventional just focused images; and (g–i): 30 nm under-focused images. The images clearly reveal that grain sizes are large in WZB samples (up to an order of magnitude higher compared to WZA) and tungsten oxide species and nanoclusters are randomly distributed and not at grain boundaries in WZB samples. Our observations indicate that this leads to sintering of the species after a few hours. Whereas in the WZA series, zirconia grains are on the (sub) nm scale (nanograins) with grain boundary segregation of tungsten oxide species (a, d, g). Examination of the WZA catalysts after several hours of the liquid phase NSAID reaction has shown that the active structures are still anchored at grain boundaries, suggesting that they are related to the state of the catalyst under the reaction and that the anchoring at grain boundaries prevents sintering of the active species.
000 magnification or 120 K) of: (a) the 15WZA sample, (b) WZB1 sample; and (c) WZB2. High-magnification ACTEM images (at a magnification of 400 K) shown in Fig. 14(d) to (i) are respectively from regions indicated by a square in the figures (a) to (c). (d–f): conventional just focused images; and (g–i): 30 nm under-focused images. The images clearly reveal that grain sizes are large in WZB samples (up to an order of magnitude higher compared to WZA) and tungsten oxide species and nanoclusters are randomly distributed and not at grain boundaries in WZB samples. Our observations indicate that this leads to sintering of the species after a few hours. Whereas in the WZA series, zirconia grains are on the (sub) nm scale (nanograins) with grain boundary segregation of tungsten oxide species (a, d, g). Examination of the WZA catalysts after several hours of the liquid phase NSAID reaction has shown that the active structures are still anchored at grain boundaries, suggesting that they are related to the state of the catalyst under the reaction and that the anchoring at grain boundaries prevents sintering of the active species.
 | ||
| Fig. 14 AC-TEM images of the various preparations of tungstated zirconia for comparative purposes: (a, (d, g) are for the 15WZA sample; and (b, e, h) and (c, f, i) are for the WZB series of samples using the conventional preparations. Details are as follows: Low-magnification TEM images (×120K) of (a) the 15WZA sample, (b) WZB1 and (c) WZB2. The regions indicated by squares in (a, b, c) are magnified in (d to i), as follows: high-magnification AC-TEM images (×400K) (d, e, f) are at just focus; (g, h, i) are at 30 nm under focused. The images clearly reveal that grain sizes are very large in WZB samples (compared to WZA) and tungsten oxide species and nanoclusters are randomly distributed and not at grain boundaries of the grains in WZB samples, leading to sintering of the clusters. Whereas in the WZA series zirconia has nanograins with grain boundary segregation of tungsten oxide species and nanoclusters (a, d, g). The anchoring of the species at grain boundaries prevents unwanted sintering. | ||
As the observations in Fig. 14 illustrate, WOx species are randomly distributed in WZB and sinter faster leading to lower activity within a couple of hours. Fig. 15(g) shows an enlarged image of WZA shown in Fig. 14(g); and Fig. 15(e) shows an enlarged image of the conventionally impregnated catalyst shown in Fig. 14(e), illustrating the differences in zirconia grain sizes. In WZA preparations, zirconia grains are only a few nms compared to grain sizes of ∼30 nm–50 nm or more in WZB catalysts. W-oxide species sintered faster in WZB catalysts in Fig. 14(e), growing to a several nanometres in about 2.5 hours (shown in the inset), and to larger NPs after several hours. This observation confirms that anchoring of active species to grain boundaries of zirconia nanograins prevents their sintering, leading to more robust catalysts. This is confirmed by our correlation experiments which have shown very high selectivity for NSAIDS for the samples over several hours (Table 3), and sizes of sub-nm WOx species tend to remain about the same after the long periods, essentially without sintering. This indicates the importance of the nano-scale sizes for both the active sites and zirconia particles for optimum catalytic performance.
 | ||
| Fig. 15 Enlarged images of the WZA catalyst shown in Fig. 14(g) and WZB catalysts in (e), respectively, illustrating the difference in zirconia grain sizes. In our preparation (g), zirconia grains are only a few nms compared to 50 nm or more in conventional catalysts (e). In (e), W-oxide clusters (e.g. arrowed) sintered faster in the conventional catalyst, growing to a few nanometres (∼5 nms) in about 2.5 hours (inset A, using HAADF-STEM), and many nms after several hours. | ||
Discussion
The calcination temperature plays a key role on the structural properties as described in Table 1. The table shows that at a given tungsten loading, the structure depends on the calcination temperature, confirmed by the XRD data. For example, the sample with 10% W loading (10WZA) heated at 373 K was amorphous. When the sample 10WZA was calcined at 773 K, tetragonal zirconia was crystallised, while the calcination at 1073 K led to the formation of WO3 crystallites.The UV-Vis absorption data presented in Table 1 provide important insights into active species. They indicate that edge energies of e.g. 10WZA, 15WZA and 35WZA samples decrease with increasing W surface density. Previous studies31 have suggested that the absorption edge energy is sensitive to the domain size of WOx species with highly dispersed nature of WOx domains (species) at lower surface densities. Thus the decrease in adsorption edge energy at higher W surface densities indicates that the average size of the WOx domain also increases and WO3 particles form. The increasing domain size of WOx species in going to bulk structures may enhance the electron delocalization. Based on the UV-Vis data29 it is reported that critical WOx coverage for the formation of WOx species is possible below a surface density of 2.9 W at nm−2 and above the monolayer coverage clusters/NPs are expected. Thus, edge energies described in Table 1 suggest that WOx species are expected to predominate for 10WZA with 1.4 W nm−2 and coexisting with an increased WOx domain size and WO3 NPs with increasing surface density. These findings are consistent with the XRD data presented in Fig. 2 and discussed below.
XRD patterns form the WZA series presented in Fig. 2 show that the double peak (doublet) observed at a 2θ value of 50° in 15WZA and 35WZA is not present in 10WZA indicating that it has essentially no WO3 crystals. The doublet is indexed as due to WO3 and tetragonal-zirconia. This indicates that 10WZA samples are expected to have surface WOx species and that the onset of WO3 crystallites occurs at a W surface density of 7.3 W nm−2 and higher. These findings are consistent with Raman, TPR and XPS studies.31 Our in situ studies on precursor transformation and HAADF–STEM have shown projected surface WOx species less than 0.3 nm to 0.35 nm, nanoclusters and NPs. The surface WOx species are present in both the 10WZA and 15WZA samples and significantly, distorted WO3 nanoparticles (NPs) present in the 15WZA are not observed in 10WZA. This is consistent with the XRD data in Fig. 2.
As discussed in the preceding sections, 10WZA showed amorphous, tetragonal crystalline and the formation of WO3 crystallites at calcination temperatures of 373 K, 773 K and 1073 K, respectively. Table 1 and Fig. 3 show that the 10WZA sample calcined at 373 K with a W surface density of 1.4 W nm−2 exhibits high conversion of 83% and selectivity of ∼74% in the NSAID catalytic reaction. The sample calcined at 773 K shows ∼86% conversions and ∼81% selectivity and that at 1073 K shows about 91% conversion and 92% selectivity for the reaction. The WZA catalysts with higher W nm−2 density with distorted WO3 NPs yield 86–97% conversion and 80–94% selectivity. These data indicate that the surface WOx species are participating significantly in the catalytic reaction, with WO3 NPs contributing to the catalytic activity leading to a marginal improvement.
Enhanced activity of 15WZA consisting of WO3 NPs is consistent with literature reports that larger WO3 NPs (in the region of 0.8 nm–1 nm) may be beneficial to catalytic reactions,48 because of the larger number of WOx units (approximately 10–15 WOx units) than the surface WOx species (containing 2–6 WOx units)27 and help to stabilise catalytically important acidic sites in the reaction. The surface WOx species (of ≤0.3 nm–0.35 nm) are believed to have 2–6 WOx units as reported in the literature,27 or may be less. It is difficult to estimate the number of the units because of the disordered arrangement of atoms and differences in W–O–W bond distances. Larger species/clusters (0.5 nm–0.8 nm) have more WOx units (10 to 15). (Other simulations reported in the literature indicate that nanoclusters of ∼0.5 nm (e.g. shown in Fig. 8(b)) contain approximately 10 tungsten and oxygen atom units49 and fewer atoms in 0.3 nm clusters50).
Microcalorimetry data (Fig. 4a) confirm the XRD and UV-Vis findings. The enhanced heat of adsorption of 6.5 kJ mol−1, representing a ∼6% increase, is observed for the sample with a surface density of 7.3 W nm−2. Thus, the heats of adsorption indicate that dispersed surface WOx species and nanoclusters impact significantly on the reaction and distorted WO3 NPs enhance the catalytic activity to some extent. This is quantified by the XRD, UV-Vis and electron microscopy data as discussed in the preceding sections. It may be noted that the heat of adsorption is not a stand-alone technique to compare the activity of the catalysts. It can give a trend on comparing a series of catalysts, and a quantitative comparison is only possible with complementary data.
We believe that the presence of anion vacancies at zirconia nano-grain boundaries introduced during the precursor transformation leads to a change in surface energy which is compensated by the segregation of the tungsten oxide species and nanoclusters to grain boundaries, leading to the minimization of the surface energy, heat of adsorption (segregation) and the stabilisation of the species and nanoclusters. The results are consistent with adsorption properties of oxides on oxide supports such as tungstated zirconia used as auto-exhaust catalysts and segregation effects in ceramic grain boundaries.51,52
The WZB catalyst series also initially exhibits ∼65% conversion even though WO3 nanoparticles are largely absent in this catalyst system. This suggests that surface WOx species are contributing to the NSAID reaction. This also indicates that distorted WO3 NPs observed in 15WZA enhance the catalytic activity for the reaction to some extent. It seems likely therefore that distorted WO3 contribute to the activity, with significant participation of surface WOx species and nanoclusters in the overall catalytic reaction.
Conclusions
We have carried out dynamic studies of the precursor transformation of tungstated zirconia nanocatalysts and quantified them by chemical studies for pharmaceutically important NSAIDs by the Beckmann rearrangement of oximes on the nanocatalysts at low temperatures. Using double aberration corrected in situelectron microscopy studies at the atomic level, we have directly identified active sites containing surface WOx species, nanoclusters and distorted WO3 NPs and their location at grain boundaries of zirconia nanoparticles. Our results provide a method for improving the stability and performance of the catalysts by anchoring the active sites at grain boundaries of the catalyst nanoparticles. They further provide a clean alternative to the current homogeneous process for NSAIDS, with important implications for the development of improved pharmaceuticals.Acknowledgements
We thank the University of York, Yorkshire Forward/ERDF and JEOL for sponsoring the York-JEOL Nanocentre at the University of York; for a fellowship at Huddersfield (NRS); and Japan Society for Promotion of Science (JSPS)/Japan Fine Ceramic Center for a fellowship (KY). Ian Wright is thanked for assistance with sample preparation.References
- G. Ertl, H. Knozinger and J. Weitkamp, Handbook of Heterogeneous Catalysis, Wiley-VCH, Weinheim, 1997 Search PubMed.
- Y. Ono, Catal. Today, 2003, 81, 3 CrossRef CAS.
- M. Ghiaci, H. Aghaei, V. Rives, M. A. Vicente, I. Sobrados and J. Sanz, Catal. Commun., 2009, 10, 1486 CrossRef CAS.
- K. G. Davenport and C. B. Hilton, US Patent, 4 524 217, 1985 Search PubMed.
- M. Hino and K. Arata, J. Chem. Soc., Chem. Commun., 1988, 1259 RSC.
- J. R. Fritch, D. A. Aguilla, T. Horlenko and O. S. Fruchey, EP, 469 742, 1991 Search PubMed.
- G. Bellussi and C. Perego, Industrial catalytic aspects of the synthesis of monomers for nylon production, CATTECH, 2000, 4, 4 CrossRef CAS.
- G. Dahlhoff, J. P. M. Niederer and W. F. Hoelderich, Catal. Rev. Sci. Eng., 2001, 43, 381 CrossRef CAS , and references therein.
- H. Ichihashi, M. Ishida, A. Shiga, M. Kitamura, T. Suzuki, K. Suenobu and K. Sugita, Catal. Surv. Asia, 2003, 7, 261 CrossRef CAS , and references therein.
- Y.-M. Chung and H. K. Rhee, J. Mol. Catal. A: Chem., 2000, 159, 389 CrossRef CAS.
- M. J. Climent, A. Corma and S. Iborra, J. Catal., 2005, 233, 308 CrossRef CAS.
- D. G. Barton, G. D. Meitzner, G. Fuentes and E. Iglesia, J. Catal., 1999, 181, 57 CrossRef CAS.
- D. G. Barton, M. Shtein, R. Wilson, S. Soled and E. Iglesia, J. Phys. Chem. B, 1999, 103, 630 CrossRef CAS.
- C. D. Baertsch and S. L. Soled, J. Phys. Chem., 2001, 105, 1320 Search PubMed.
- E. I. Ross-Medgaarden, W. Knowles, T. Kim and M. S. Wong, J. Catal., 2008, 256, 108 CrossRef CAS , and references therein.
- C. Angeles-Chavez, M. A. Cortes-Jácome, E. Torres-Garcia and J. A. Toledo-Antonioa, J. Mater. Res., 2006, 21, 807 CrossRef CAS.
- W. Zhou, E. Ross-Medgaarden, C. Kiley, E. Wachs, M. Wong and W. Knowles, Nat. Chem., 2009, 1, 722 CrossRef CAS , and references therein.
- N. Soltanidis, W. Zho, A. Psaras, J. Gonzalez and M. Wong, J. Am. Chem. Soc., 2010, 132, 13462 CrossRef CAS.
- E. D. Boyes and P. L. Gai, Ultramicroscopy, 1997, 92, 119.
- P. L. Gai, et al. , Science, 1995, 267, 661 CrossRef CAS.
- P. L. Gai, Adv. Mater., 1998, 10, 1259 CrossRef CAS.
- P. L. Gai, E. D. Boyes, P. Hansen, S. Helveg, S. Giorgio and C. Henry, MRS Bull., 2011, 32, 1044.
- P. L. Gai and E. D. Boyes, Microsc. Res. Tech., 2009, 72(3), 153 CrossRef CAS.
- P. L. Gai and E. D. Boyes, J. Phys.: Conf. Ser., 2010, 241, 012055 CrossRef.
- A. H. Zewail, Science, 2010, 328, 187 CrossRef CAS (invited article and references therein).
- J. Creemar, S. Helveg, G. Hoveling and S. Ullman, Ultramicroscopy, 2008, 108, 993 CrossRef CAS.
- X. Huang, H. Zhai, J. Li and L. S. Wang, J. Phys. Chem. A, 2006, 110, 85 CrossRef CAS.
- S. Kuba, et al. , J. Catal., 2003, 216, 353 CrossRef.
- E. I. Ross-Medgaarden, et al. , J. Catal., 2008, 256, 108 CrossRef CAS.
- J. Macht and E. Iglesia, Phys. Chem. Chem. Phys., 2008, 10, 5331 RSC.
- A. Martínez, G. Prieto, M. A. Arribas, P. Concepción and J. F. J. Sánchez-Royo, J. Catal., 2007, 248, 288 CrossRef CAS.
- A. M. Argo, J. F. Odzak, F. S. Lai and B. C. Gates, Nature, 2002, 415, 623 CrossRef CAS.
- A. I. Vogel, Vogel's Text Book of Practical Organic Chemistry, Longman, London, 4th edn, 1978 Search PubMed.
- N. R. Shiju, M. AnilKumar, W. F. Hoelderich and D. R. Brown, J. Phys. Chem. C, 2009, 113, 7735 CrossRef CAS.
- N. R. Shiju, H. M. Williams and D. R. Brown, Appl. Catal., B, 2009, 90, 451 CrossRef CAS.
- M. Isaacson, D. Kopf, M. Uylaut, N. Parker and A. V. Crewe, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 1802 CrossRef CAS.
- A. Howie, J. Microsc., 1979, 117, 11.
- P. L. Gai, M. Goringe and J. C. Barry, J. Microsc., 1986, 142, 9 CAS.
- P. L. Gai-boyes, Catal. Rev. Sci. Eng., 1992, 34, 1 CAS.
- P. L. Gai, Top. Catal., 2002, 21, 162.
- P. L. Gai and E. D. Boyes, Electron Microscopy in Heterogeneous Catalysis, Institute of Physics Publ, Bristol and Philadelphia (UK and USA), 2003 Search PubMed.
- N. R. Shiju, K. Yoshida, D. R. Brown, E. D. Boyes and P. L. Gai, Microscopy and Microanalysis Proceedings, August, 2009.
- M. Kurumuda, O. Kido, T. Sato and C. Kaito, J. Cryst. Growth, 2005, 275, e1673 CrossRef.
- M. Benaissa, J. G. Santiesteban, G. Díaz, C. D. Chang and M. José-Yacamán, J. Catal., 1996, 161, 694 CrossRef CAS.
- P. L. Gai, K. Zhang and J. R. Weertman, Scr. Mater., 2007, 56, 25 CrossRef CAS.
- H. Geiter, Prog. Mater. Sci., 1991, 33, 233.
- J. M. Cowley and F. Sundell, Ultramicroscopy, 1997, 68, 1 CrossRef CAS.
- G. Barton, et al. , J. Catal., 1999, 181, 57 CrossRef CAS.
- W. Wunderlich and M. Takahashi, Ceram. Trans., 2002, 133, 189 (ed. M. Matsui, American Ceramic Society), and references therein.
- K. Kayama, J. Electron Microsc., 1977, 26, 1.
- S. Takashima, Patent, WO/2006/112374, Toyota, 2006 Search PubMed.
- Y. Lei and Y. Ito, J. Am. Ceram. Soc., 2002, 85, 2359 CrossRef CAS.
Footnote |
| † Current address: Department of Chemistry, Van't Hoff Laboratory, University of Amsterdam, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2011 |